

Abstract
Compensation is a critical process for the unbiased analysis of flow cytometry data. Numerous compensation strategies exist, including the use of bead‐based products. The purpose of this study was to determine whether beads, specifically polystyrene microspheres (PSMS) compare to the use of primary leukocytes for single color based compensation when conducting polychromatic flow cytometry. To do so, we stained individual tubes of both PSMS and leukocytes with panel specific antibodies conjugated to fluorochromes corresponding to fluorescent channels FL1‐FL10. We compared the matrix generated by PSMS to that generated using peripheral blood mononuclear cells (PBMC). Ideal for compensation is a sample with both a discrete negative population and a bright positive population. We demonstrate that PSMS display autofluorescence properties similar to PBMC. When comparing PSMS to PBMC for compensation PSMS yielded more evenly distributed and discrete negative and positive populations to use for compensation. We analyzed three donors' PBMC stained with our 10‐color T cell subpopulation panel using compensation generated by PSMS vs.PBMC and detected no significant differences in the population distribution. Panel specific antibodies bound to PSMS represent an invaluable valid tool to generate suitable compensation matrices especially when sample material is limited and/or the sample requires analysis of dynamically modulated or rare events. © 2015 The Authors. Cytometry Part A Published by Wiley Periodicals, Inc.
Keywords: compensation, polystyrene microspheres, flow cytometry, immunophenotyping, polychromatic, beads, leukocytes
With the advent of fluorescein and rhodamine centered two‐color flow cytometry, eliminating spectral overlap using compensation has become a necessary requirement for data analysis 1. Perhaps the most well‐known two‐color flow cytometry analysis requiring compensation is fluorescein isothiocyanate (FITC) and phycoerythrin (PE). As the number of fluorochromes used in multicolor flow cytometry increases, so does the need for appropriate compensation 2, 3, 4, 5. Consequently, a multitude of theories and strategies exist for suitable compensation 6, 7, 8, 9, 10. Of consensus is that single color compensation controls are required for experimental setup 11, 12. Single color compensation that hinges upon the use of cells from sample material can be problematic as more often than not sample material may be limited and heterogeneous. Importantly, cells display a wide variance in background fluorescence 13. Widely accepted is the use of beads coated with antibody capture sites. This allows for a bright binding of even the most dynamically regulated antigens. Beads have a smaller error in their distribution of background fluorescence, allowing for precise spillover computation. However, many commercially available beads introduce false negative background and do not recognize a diverse amount of host isotypes 14.
Polystyrene microspheres (PSMS) are antibody‐capture beads made of polystyrene, a petroleum based plastic made of the monomer styrene. PSMS are 3.0–3.4 μm in size. Recognizing all mouse and rat isotypes, most hamster isotypes and rabbit polyclonal IgG, PSMS can be used for single color compensation in situations where cell samples are limited. Negative, uncoated PSMS provide background fluorescence similar to unstained cells across the different excitation/emission wavelength combinations. Many clinical studies use precious sample material as single color controls for compensation such that their consumption in compensation could limit the analytical power of flow cytometry. We sought to determine whether PSMS can be used as a substitute for cells for compensation of spectral overlap of 10 fluorochromes in flow cytometry analysis. To do so, we compared single color controls from PSMS incubated with antibodies from our 10‐color panel to single color controls from primary human leukocytes (peripheral blood mononuclear cells, PBMC) incubated with the same antibodies. We then applied the compensation matrices generated from PSMS and PBMC, respectively to three donors' PBMC.
Materials and Methods
Experiment Overview
In this work, we compare PSMS to cells to be used for compensation for immunophenotyping of primary human leukocytes in a clinical study. We designed in our laboratory a T cell subpopulation panel that would allow us to stratify T cell subsets based on cell surface markers to ultimately be used for sorting and RNA extraction. We designed this panel based on the following parameters: antigens of interest, antibody with conjugated fluorochrome availability and the expected brightness/frequency of antigen on the target cell population. As this panel will ultimately be used for patient material, specifically PBMC and tumor‐infiltrating lymphocytes, it was important that we develop a system that will limit the use of our sample material.
Flow Sample and Specimen Description
T cell subpopulation panel
Cell surface markers were chosen based on their ability to discriminate subsets in the T cell population of humans. The panel consists of the following antibodies: TCRαβ FITC (Becton Dickinson, Franklin Lakes, NJ), TCRγδ PE (Becton Dickinson, Franklin Lakes, NJ), CD25 ECD (IOTest, Beckman Coulter, Brea, CA), CD4 PERCP (Becton Dickinson, Franklin Lakes, NJ), CCR6 PE‐Cy7 (Biolegend, San Diego, CA), CD45RO APC (Becton Dickinson, Franklin Lakes, NJ), IL‐23R AF700 (R&D Systems, Minneapolis, MN), CD3 APC‐AF750 (IOTest, Beckman Coulter, Brea, CA), CD8 Pacific Blue (IOTest, Beckman Coulter, Brea, CA), or CD45 Krome Orange (IOTest, Beckman Coulter, Brea, CA), corresponding to fluorescent channels FL1‐FL10, respectively, for the generation of a compensation matrix.
PSMS preparation and staining
Each bottle of PSMS (VersaComp Antibody capture beads, Beckman Coulter, Brea, CA) contains ∼10 × 106 PSMS/ml. To generate single color controls one 50 μl drop (5 × 105) of coated PSMS and one 50 μl drop (5 × 105) of uncoated PSMS for a total of 1 × 106 PSMS were added per tube. Each tube was stained per manufacturer's recommendation for ≤1 × 106 cells with a different antibody from the T cell subpopulation panel for 20 min at room temperature in the dark. Tubes containing PSMS were washed in two milliliters of phosphate buffered saline (PBS; Sigma‐Aldrich, St Louis, MO) containing 1% fetal calf serum (FCS; HyClone™, Thermo Scientific, Logan, UT) and centrifuged at 400g for 5 min. Excess buffer was then decanted leaving the PSMS pellet in a residual volume of 100 μl. Tubes were briefly vortexed and then assayed for expression of cell surface marker antibodies.
PBMC preparation and staining
PBMC were isolated from donors' blood using a density gradient medium (Lymphoprep; Greiner Bio‐One, NC). After which the cells were washed in PBS (PBS; Sigma‐Aldrich, St. Louis, MO) and frozen in medium containing 50% FCS (HyClone™, Thermo Scientific, Logan, UT), 40% RPMI 1640 (Sigma‐Aldrich, St Louis, MO), and 10% DMSO (Sigma‐Aldrich) and placed at −80°C in a Nalgene “Mr. Frosty” freezing container (Thermo Scientific, Waltham, MA). Prior to staining, cells were thawed in a 37°C water bath, followed by two washes with PBS for 5 min at 400g. Cells were then aliquotted into 1 × 106 cells per condition. PBMC were prepared and stained individually (single color tubes) with antibodies from the T cell subpopulation panel as per manufacturer's test recommendations.
Instrument and Data Analysis Details
Flow cytometry analyzer and data analysis software
All samples were run on the Gallios Flow Cytometer with the following 3 laser, 10 color configuration: Blue (488 nm laser) – fluorescent channel 1 (FL1): 525/30 band pass filter/window width (BP), FL2: 575/30 BP, FL3: 620/30 BP, FL4: 670 long pass filter (LP), FL5: 775 LP, Red (638 nm laser) ‐ FL6: 660/20 BP, FL7: 725/20 BP, FL8: 775 LP, and Violet (405 nm laser) ‐ FL9: 450/50 BP, FL10: 550/40 BP, Beckman Coulter, Brea, CA). All samples were run with the same voltage settings. PSMS were identified on the basis of forward and side scatter area (FSC‐A and SSC‐A), FSC‐A vs. time of flight (singlet identification) and back‐gating for the antigen of interest for each channel. PBMC, specifically lymphocytes, were identified based on back‐gating of the CD45 and CD3 positive cells with intermediate size (FSC‐A) and low granularity (SSC‐A). Upon selection of these gates the rest of the data were then analyzed post‐acquisition using the Kaluza software (Beckman Coulter, Brea, CA). The two compensation matrices generated from 1 PSMS and individual T cell subpopulation markers antibodies and 2 PBMC and individual T cell subpopulation markers were applied post acquisition in Kaluza on three donors' PBMC stained with all of the antibodies in the T cell subpopulation panel.
Statistical analysis
Percent gated, median and standard deviation were calculated using Kaluza. Statistical analysis was performed with GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA, www.graphpad.com). Differences were tested by Student's t test as appropriate. All reported P values are two‐tailed.
Results
PSMS Share Similar Physical Properties to PBMC
To investigate whether the use of PSMS would be capable of replacing cell sample for single color compensation setup, it was necessary to characterize the microspheres. PSMS were prepared and stained with single color conjugates reflecting the markers of interest in the T cell subpopulation panel, specifically: TCRαβ FITC, TCRγδ PE, CD25 ECD, CD4 PERCP, CCR6 PE‐Cy7, CD45RO APC, IL‐23R AF700, CD3 APC‐AF750, CD8 Pacific Blue, and CD45 Krome Orange. Finally, PBMC were prepared and stained with individual markers of the T cell subpopulation panel. Using the same cytometer voltages, compensation was manually determined stepwise moving from FL1‐FL10 on a Gallios 3 laser/10 detector flow cytometer for PSMS stained with markers in the T cell subpopulation panel. Compensation was then cleared and manually optimized using PBMC stained with individual markers in the T cell subpopulation panel. In particular, we compared each fluorescent channel pairwise to the nine other channels. Upon analyzing the bivariate plots of the fluorochrome combinations x and y medians were adjusted such that they were on average within +/− 0.2 standard deviations.
PSMS can be located using the same parameters as PBMC. The same photomultiplier tube (PMT) voltages were kept constant; 3–3.4 microns in size PSMS are just smaller than lymphocytes (4–10 microns; 15). Figure 1A shows the results of ungated acquisition of PSMS and PBMC using the same settings. Aggregated PSMS and dead cells and debris respectively were excluded on the basis of forward and side scatter (Fig. 1A).
Figure 1.
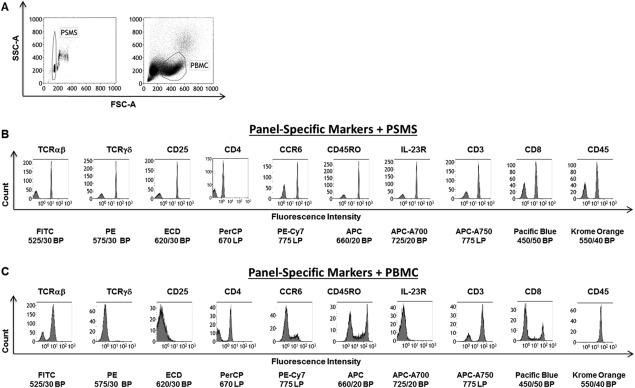
PSMS share similar physical properties to PBMC yet have a better discrete distribution of negative and positive populations. A: Left: Bivariate plot of forward and side scatter area of ungated acquisition of PSMS. Gate is placed on singlets of PSMS. Right: Forward versus side scatter plot of ungated PBMC. Region circled reflects lymphocytes on the basis of size and granularity. B: PSMS were stained with TCRαβ FITC, TCRγδ FITC, CD25 ECD, CD4 PERCP, CCR6 PE‐Cy7, CD45RO APC, IL‐23R AF700, CD3 APC‐AF750, CD8 PacBlu, or CD45 Krome Orange. C: Single color PBMCs were prepared and stained with with TCRαβ FITC, TCRγδ FITC, CD25 ECD, CD4 PERCP, CCR6 PE‐Cy7, CD45RO APC, IL‐23R AF700, CD3 APC‐AF750, CD8 PacBlu, or CD45 Krome Orange. The same voltage settings were used during acquisition. All plots are gated on PSMS or PBMC based on light scatter.
PSMS Yield More Discrete Negative and Positive Populations across All 10 Fluorescent Channels
Figure 1B displays single parameter histograms yielded from analysis of PSMS stained with panel antibodies. The same is shown for PBMC (Fig. 1C). Based on these single parameter histograms plots percent negative and percent positive populations were selected for each fluorescent channel. The exact regions included to make these calculations along with representative data can be found in Supporting Information Figures 1 and 2, for PSMS and PBMC, respectively. The percent gated are summarized in Table 1. On average PSMS possessed more even distribution of percent positive and negative events as coated and uncoated PSMS, respectively, ensured the presence of both populations. Compensating using PBMC was more challenging due to the dynamic range of expression of the antigen on PBMC. This was especially true for staining corresponding to fluorescent channels FL2 (ECD: 620/30 BP), 3 (PE 575/30 BP), and 7 (APC‐A700 725/20 BP), corresponding to the antibodies TCRγδ PE, CD25 ECD, and IL‐23R AF700, respectively. Gamma delta T cells represent a very small fraction of the PBMC population; hence identifying a population to use for compensation can be difficult. This is amplified when dealing with hard to obtain specimens for which sample is limited. Markers CD25 (FL3) and IL‐23R (FL7) displayed no clear degree of separation between positive and negative cells, but instead have a gradient of expression (Fig. 1C). These are extremely difficult to compensate as one may not be able to determine whether such a result is due to poor titration/antibody function or if the sample being used to set up compensation is dim/negative for that particular antigen. On the contrary, PSMS stained with the same antibodies consistently yielded discrete positive and negative positive populations with high enough frequency to be readily used for compensation (Fig. 1B, Table 1).
Table 1.
Population distribution PSMS vs. PBMC
| PSMS | PBMC | |||
|---|---|---|---|---|
| % Negative | % Positive | % Negative | % Positive | |
| FL1: FITC 525/30 BP | 44.76 | 55.24 | 21.05 | 78.95 |
| FL2: PE 575/30 BP | 38.69 | 61.31 | 98.58 | 1.42 |
| FL3: ECD 620/30 BP | 41.5 | 58.5 | 98.59 | 1.41 |
| FL4: PERCP 670 LP | 38.61 | 61.39 | 50.24 | 49.76 |
| FL5: PE‐Cy7 775 LP | 49.15 | 50.85 | 77.9 | 22.1 |
| FL6: APC 660/20 BP | 30.21 | 69.79 | 48.41 | 51.59 |
| FL7: APC‐A700 725/20 BP | 31.76 | 68.24 | 99.68 | 0.31 |
| FL8: APC‐A750 775 LP | 34.97 | 65.03 | 21.19 | 78.83 |
| FL9: PacBlue 450/50 BP | 41.35 | 58.65 | 69.3 | 30.62 |
| FL10: Krome Orange 550/40 BP | 38.94 | 61.06 | 0.27 | 99.73 |
Background Autofluorescence of PSMS Are Comparable to PBMC
Next we compared the autofluorescent properties of PSMS when compared to PBMC (Table 2). To assess this we calculated the median of the negative population of univariate histogram plots for each of the fluorescent channels. The geometric mean of fluorescence intensity is often used as a representative number for a population of cells. However, median provides a more accurate reflection of the fluorescent intensity of a population as a whole as most flow cytometry data is not normally distributed. Hence we chose the median fluorescence intensity (MedFI) as a way to quantify autofluorescence. For PSMS the autofluorescence of the negative population for PSMS ranged from 0.14 to 2.05. PBMC MedFI ranged from 0.12 to 3.13. Paired two tailed t test indicated that there was no significant difference between the autofluorescence between PSMS and PBMC for FL1‐ FL10 (Table 2). With a range equivalent to or lower than PBMC, PSMS do not contribute to the compensation spillover calculation; allowing one to apply the compensation matrix to PBMC.
Table 2.
Autofluorescence of PSMS vs. PBMC
| X Median of % negative | ||
|---|---|---|
| PSMS | PBMC | |
| FL1: FITC 525/30 BP | 0.23 | 0.52 |
| FL2: PE 575/30 BP | 0.76 | 0.93 |
| FL3: ECD 620/30 BP | 0.29 | 0.28 |
| FL4: PERCP 670 LP | 0.14 | 0.12 |
| FL5: PE‐Cy7 775 LP | 2.05 | 1.14 |
| FL6: APC 660/20 BP | 1.35 | 3.13 |
| FL7: APC‐A700 725/20 BP | 0.77 | 0.46 |
| FL8: APC‐A750 775 LP | 1.59 | 1.97 |
| FL9: Pacific Blue 450/50 BP | 0.73 | 0.84 |
| FL10: Krome Orange 550/40 BP | 0.45 | 1.62 |
Lastly before applying our matrices to three donors for analysis, we assessed a parameter referred to as the stain index (SI). SI takes into account the spread of the percent negative histogram peak, unlike simpler calculations like Signal:Noise (S:N) that just look at the fold increase of positive over negative. The formula for SI is the difference between the X median of the positive and negative population peaks on a histogram plot divided by two times the standard deviation of the negative peak. On average, the SI was higher for PSMS compared to PBMC (Table 3). The one exception was FL4, corresponding to the dim fluorochrome PerCP (Table 3).
Table 3.
Stain index
| PSMS | PBMC | ||
|---|---|---|---|
| FL1: FITC 525/30 BP | 50.8 | 14.1 | |
| FL2: PE 575/30 BP | 44.4 | 22.3 | |
| FL3: ECD 620/30 BP | 78.4 | 4.3 | |
| FL4: PERCP 670 LP | 1.4 | 18.3 | |
| FL5: PE‐Cy7 775 LP | 51.2 | 10.1 | |
| FL6: APC 660/20 BP | 61.4 | 49.2 | |
| FL7: APC‐A700 725/20 BP | 38.9 | 9.0 | |
| FL8: APC‐A750 775 LP | 28.0 | 40.3 | |
| FL9: Pacific Blue 450/50 BP | 28.9 | 89.9 | |
| FL10: Krome Orange 550/40 BP | 20.1 | 24.8 | |
|
| |||
PSMS Vs. PBMC Yield Two Different Compensation Matrices
As a result of the two compensation series, two separate compensation matrices were generated (Figs. 2A and 2B). There were minimal differences in compensation between the two matrices. The highest compensation variability often occurs between these pairs: FL2 (PE) vs. FL3 (ECD), FL3 (ECD) vs. FL4 (PerCP), and FL4 (PerCP) vs. FL5 (PE‐Cy7). To better understand the differences in these compensations matrices, a sample of PBMC were processed and stained with all the antibodies in the T cell subpopulation panel. The representative dot plots of PBMC stained with the multicolor T cell subpopulation panel using the Kaluza® software are shown in Figures 2C and 2D.
Figure 2.
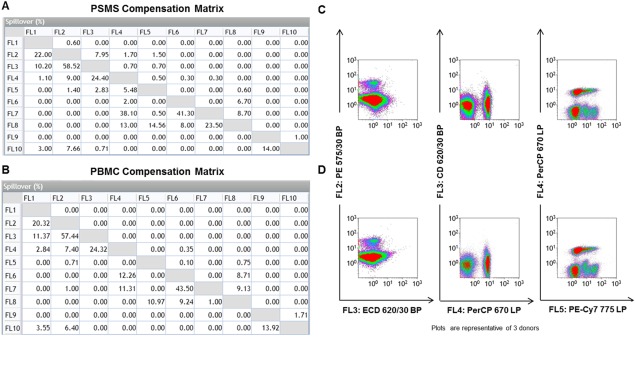
PSMS vs. PBMC generate two different compensation matrices. A: The compensation matrix generated for PSMS stained with panel specific markers and (B) single‐color PBMC stained with panel specific markers is displayed. C: Compensation between FL2 vs. FL3, FL3 vs. FL4, and FL4 vs. FL5 is highlighted. C: PBMC stained with panel specific markers were given the compensation matrix generated by PSMS stained with panel specific antibodies post‐acquisition in Kaluza analysis. D: The same PBMC stained with panel specific markers were given the compensation matrix generated by the single color PBMC stained with panel specific antibodies postacquisition in Kaluza analysis (row D). Compensation between FL2 vs. FL3, FL3 vs. FL4, and FL4 vs. FL5 is highlighted. All plots are gated on PBMC based on light scatter. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
PSMS Are Able to Effectively Compensate Data from Three Donors Stained with Our 10‐Color T Cell Subpopulation Panel
We next analyzed the effect these two different compensation matrices had on a variety of T cell subpopulations. After exhaustive study over the entirety of the T cell subpopulation panel, no obvious differences were found regardless of which compensation matrix was applied to the PBMC stained with the multicolor T cell subpopulation panel. In Figure 3, the following cell populations were evaluated: TCRαβ, TCRγδ, CD4 T cells, CD8 T cells, and IL‐23R+ CD3+ cells (Figs. 3A and 3B). Regardless of which series of compensation tubes were used for single color setup, the PBMC stained with the T cell subpopulation displayed no evidence of significant over‐ or under‐compensation.
Figure 3.
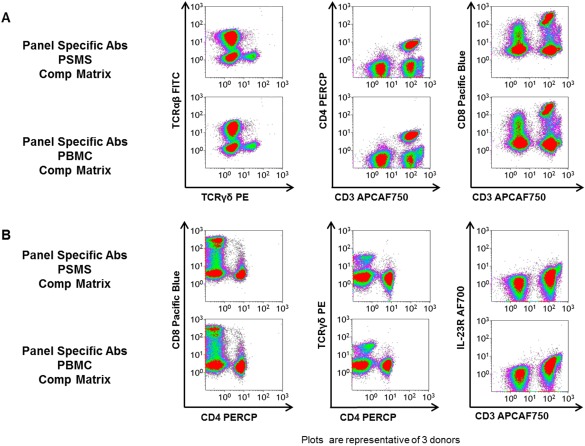
Two different compensation matrices generated similar visuals of the data. PBMC stained with panel specific markers were given the compensation matrix generated by the PSMS stained with panel specific antibodies postacquisition in Kaluza analysis (rows 1 and 3). The same PBMC stained with panel specific markers were given the compensation matrix generated by the single color PBMC stained with panel specific antibodies post‐acquisition in Kaluza analysis (rows 2 and 4). All plots are gated on PBMC based on light scatter. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To determine the effects of compensating with PSMS versus PBMC we studied three donors in which we compared the data on a global scale looking at the percent negative and positive populations (Fig. 4A), the MedFI of both of these populations (Fig. 4B) and SI (Fig. 4C). For all three parameters (autofluorescence, population distribution, and SI) no major significant differences were detected when using either PSMS or PBMC compensation matrices (Figs. 4A–4C). The only significant difference detected was the X MedFI of the negative fraction of FL9 (Pacific Blue 450/50 BP), on average the X MedFI of data using the PBMC based compensation was 1.07 lower than data compensated using PSMS. While this difference was observed, it is important to note that this difference did not affect the population distribution of the sample set (Fig. 4A). There were large differences in the standard deviation of FL6 (APC 660/20 BP) and FL8 (Pacific Blue 450/50 BP) MedFI of the positive cell population. However, this result was independent of the compensation used and reflects donor variability. Perhaps the most striking difference between PSMS and PBMC compensation matrices were between FL7 (APC‐A700 725/20 BP) and FL8 (APC‐A750 775 LP). This is important to note as FL7 corresponds to IL23R APC‐A700 staining. This staining using PBMC did not yield a distinct positive and negative fraction thus making compensation difficult.
Figure 4.
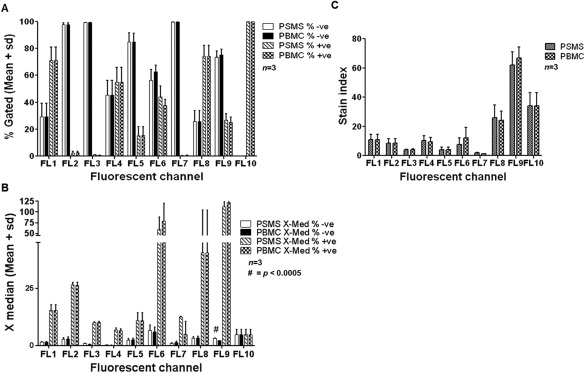
Analysis of three donors indicate that data obtained from PSMS vs. PBMC generated compensation matrices are similar. A: Mean and standard deviation of negative and positive populations for each T cell panel marker using compensation matrices generated from PSMS and PBMC. B: X‐Median (Median fluorescence intensity) of three donors PBMC stained with panel specific markers given the compensation matrix generated by PSMS and PBMC. C: SI of PBMC from three donors using PSMS and PBMC matrices.
PSMS Bind Intracellular Staining Antibodies as Well as Isotypes from Different Species
While we focused on cell surface markers for our T cell subpopulation panel, we also tested the ability of PSMS to bind antibodies designed for intracellular staining as this could serve as another useful tool for PSMS. Intracellular staining requires permeabilization of the cell membrane and often 4 h. of stimulation for detection. We stained PSMS with both human and murine antibodies against the immunomodulatory cytokines, IFN‐gamma, IL‐2, and IL‐10 (Fig. 5A). The antibodies used consisted of both different host species as well as isotypes (Fig. 5B). Results demonstrate that PSMS were able to bind all of these antibodies and serve as a compensation option not only for rare samples and dynamically expressed events, but also in lieu of intracellular stained cells (Fig. 5A).
Figure 5.
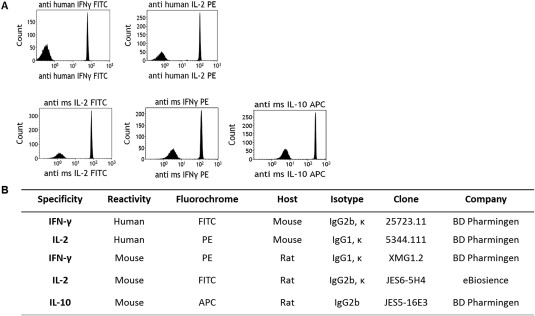
PSMS bind antibodies for intracellular staining as well as isotypes from different species. A: PSMS stained with intracellular staining antibodies anti human IFN‐γ FITC and IL‐2 PE (top panel) and anti‐mouse IL‐2 FITC, IFN‐γ PE, and IL‐10 APC (lower panel). B: Antibody description chart.
Discussion
Our findings demonstrate that PSMS closely reflect the background autofluorescence of primary human leukocytes. As seen in Figure 1, PSMS and PBMC had a similarly defined negative peak. Second, using panel‐specific antibodies is appropriate for staining PSMS. PSMS were able to bind a variety of antibodies with different hosts and isotypes (Supporting Information Fig. 3). No significant differences the compensation percentages required were similar across both matrices. Furthermore, the two compensation matrices resulted in no obvious over‐ or under‐compensation of critical cell populations. Altogether, we conclude that the use of PSMS is a valid tool for single color compensation setup.
The PSMS used to conduct these experiments (VersaComp antibody capture beads, Beckman Coulter, Brea, CA) are just one example of a number of products on the market. Similar products include the ABC Bead kit and ArC™ Amine Reactive Compensation Bead Kit (Invitrogen) and Ultracomp eBeads (eBioscience) and have previously been used for compensation 16, 17.
PSMS similar to all methods are not without concerns for compensation. No method is without limitations. There are inherent differences within the machine and day to day variability which often leads to a spreading effect even in properly compensated data. Further, there are interactions that can sometimes not be predicted from single color controls. To address these issues, it may be necessary to use fluorescence minus one stained cells in order to identify true populations versus artifacts 14, 18. PSMS are similar to leukocytes in size. Many bead based platforms and tools for flow cytometry are optimized for leukocytes. When assaying larger cells, such as tumor cells it may be important to use cells initially to define appropriate ranges for PMT voltages and then use these cytometer setting to run the PSMS 14. PSMS tend to stain as a bright as or brighter than the antigen of interest. This is ideal; however, this will need to be verified. In cases where PSMS are less bright than cells sample it will need to be initially tested whether this has an effect on compensation before routinely using PSMS. Of particular concern in our panel was the low SI observed by PSMS bound to CD4 PERCP in FL4: 670 LP. Fortunately, this difference did not have a significant impact on the percentages of populations within our sample set (Fig. 4A). To test what could be the source of this difference, we first examined lot to lot variation. Upon using a new vial and lot of CD4 PerCP (Becton Dickinson, Franklin Lakes, NJ) the SI increased from 1.4 to 60.7 (Supporting Information Fig. 4). Other options include using a different fluorochrome. We selected PerCP as it was included as a cocktail mixture in an intracellular staining panel (panel not shown); however, PE Cy5.5 is an alternate option, which stains more brightly on PSMS (data not shown).
For our 10‐color T cell subpopulation panel, we used only direct flow cytometry in which all antibodies were directly conjugated to a particular fluorochrome. With smaller panels it is possible, with careful selection of species reactivity, order of antibody addition and washes to use indirect flow cytometry staining. Consequently, we decided to test whether PSMS could be used for compensation of samples stained using indirect flow cytometry, in which a primary unconjugated Ab and a secondary fluorochrome conjugated antibody are used. To test this, we stained both PSMS and PBMC with either the secondary conjugated antibody, rat anti‐mouse IgG1 PE (RAM IgG1 PE, Becton Dickinson, Franklin Lakes, NJ) or mouse (IgG1) anti‐human CD28 primary unconjugated antibody (Becton Dickinson, Franklin Lakes, NJ) followed by RAM IgG1 PE secondary antibody. As we expected PSMS were able to bind the secondary alone. We also observed a sharp clean signal from the use of a primary and secondary with our PSMS. This signal as anticipated was amplified when compared with using secondary alone. The MedFI of the %positive peak and SI of PSMS with secondary only or primary and secondary were 129.31, 136.6, and 249.27, 282.3, respectively. As expected our human T cells stained with secondary alone served as a negative control. When using primary and secondary antibodies T cells stained nicely for CD28. More importantly, PSMS stained with primary and secondary antibodies were as bright as T cells (Supporting Information Fig. 5).
With single color controls recorded compensation can often be applied post‐acquisition. A feature which is rarely utilized is the use of auto‐compensation to generate a compensation matrix from single color controls to be applied to a data set. We decided to test how the compensation matrices would differ when using the Kaluza 1.2 auto‐compensation features (Supporting Information Fig. 6). We opted for the positive‐negative autocompensation gating setting to conduct our analysis. While the final compensation applied to the mixed tubes from both PSMS and PBMC single color controls were easy to generate the process did require some manual adjustments. In particular, it was necessary that we manually adjust the “dim vs. bright” or % negative vs. % positive regions. It is also important to note that while we had difficulty manually compensating the data in FL7: IL23R APC‐A700 725/20 BP using PBMC, due to the very small population of IL23R positive cells, autocompensation values of channels FL7 vs. FL8 were closer to the results we saw when using manual compensation and PSMS (Supporting Information Fig. 6).
Conclusion
Other multiparameter based cytometry platforms, such as mass cytometry (CyTOF; 19, 20, 21, 22, 23) exist. However, to date flow cytometry remains the gold standard for multiparameter single cell analysis. In the clinical setting flow cytometry plays a critical role in immunophenotyping especially in the context of diagnosis of hematological malignancies, such as leukemia and lymphoma. PSMS provide a resource for compensation when it is unfeasible to run single color controls of cell sample. PSMS are a useful tool for rare or dynamically expressed events. Further PSMS can be used to set up compensation for intracellular staining platforms. Bead‐based platforms for compensation have been in existence since the late 1990s 18. Perhaps more widespread recognition of such tools will aid in proper compensation in multicolor flow analysis.
Supporting information
Supporting Information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Acknowledgments
This research was supported by the Alliance for Cancer Gene Therapy (ACGT, Inc), Alex's Lemonade Stand Pediatric Cancer Foundation (ALSF) and Stand Up To Cancer St. Baldrick's Pediatric Dream Team Translational Research Grant (SU2C‐AACR‐DT1113). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. This research is also supported in part by Award Numbers T32GM088129 and 5T32HL092332 from the National Institute Of General Medical Sciences and National Heart, Lung and Blood Institute, respectively. This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (AI036211, CA125123, and RR024574). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute Of General Medical Sciences, National Heart, Lung and Blood Institute or the National Institutes of Health.
Literature Cited
- 1. Herzenberg LA, Parks D, Sahaf B, Perez O, Roederer M, Herzenberg LA. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin Chem 2002;48:1819–1827. [PubMed] [Google Scholar]
- 2. Stewart CC, Stewart SJ. Four color compensation. Cytometry 1999;38:161–175. [DOI] [PubMed] [Google Scholar]
- 3. McLaughlin BE, Baumgarth N, Bigos M, Roederer M, De Rosa SC, Altman JD, Nixon DF, Ottinger J, Oxford C, Evans TG, et al. Nine‐color flow cytometry for accurate measurement of T cell subsets and cytokine responses. Part I: Panel design by an empiric approach. Cytometry A 2008;73A:400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McLaughlin BE, Baumgarth N, Bigos M, Roederer M, De Rosa SC, Altman JD, Nixon DF, Ottinger J, Li J, Beckett L, et al. Nine‐color flow cytometry for accurate measurement of T cell subsets and cytokine responses. Part II: Panel performance across different instrument platforms. Cytometry A 2008;73A:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Njemini R, Onyema OO, Renmans W, Bautmans I, De Waele M, Mets T. Shortcomings in the application of multicolour flow cytometry in lymphocyte subsets enumeration. Scand J Immunol 2014;79:75–89. [DOI] [PubMed] [Google Scholar]
- 6. Bagwell CB, Adams EG. Fluorescence spectral overlap compensation for any number of flow cytometry parameters. Ann N Y Acad Sci 1993;677:167–184. [DOI] [PubMed] [Google Scholar]
- 7. Bayer J, Grunwald D, Lambert C, Mayol JF, Maynadie M. Thematic workshop on fluorescence compensation settings in multicolor flow cytometry. Cytometry B Clin Cytom 2007;72B:8–13. [DOI] [PubMed] [Google Scholar]
- 8. Novo D, Gregori G, Rajwa B. Generalized unmixing model for multispectral flow cytometry utilizing nonsquare compensation matrices. Cytometry A 2013;83A:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sugar IP, Gonzalez‐Lergier J, Sealfon SC. Improved compensation in flow cytometry by multivariable optimization. Cytometry A 2011;79A:356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Rodijnen NM, Pieters M, Hoop S, Nap M. Data‐driven compensation for flow cytometry of solid tissues. Adv Bioinformatics 2011;2011:184731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tung JW, Heydari K, Tirouvanziam R, Sahaf B, Parks DR, Herzenberg LA, Herzenberg LA. Modern flow cytometry: A practical approach. Clin Lab Med 2007;27:453–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Rosa SC, Herzenberg LA, Herzenberg LA, Roederer M. 11‐color, 13‐parameter flow cytometry: Identification of human naive T cells by phenotype, function, and T‐cell receptor diversity. Nat Med 2001;7:245–248. [DOI] [PubMed] [Google Scholar]
- 13. Hulspas R, O'Gorman MR, Wood BL, Gratama JW, Sutherland DR. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry B Clin Cytom 2009;76B:355–364. [DOI] [PubMed] [Google Scholar]
- 14. Roederer M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry 2001;45:194–205. [DOI] [PubMed] [Google Scholar]
- 15. Polliack A, Lampen N, Clarkson BD, De Harven E, Bentwich Z, Siegal FP, Kunkel HG. Identification of human B and T lymphocytes by scanning electron microscopy. J Exp Med 1973;138:607–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zimmerlin L, Donnenberg VS, Donnenberg AD. Rare event detection and analysis in flow cytometry: Bone marrow mesenchymal stem cells, breast cancer stem/progenitor cells in malignant effusions, and pericytes in disaggregated adipose tissue. Methods Mol Biol 2011;699:251–273. [DOI] [PubMed] [Google Scholar]
- 17. Mittag A, Tarnok A. Basics of standardization and calibration in cytometry–a review. J Biophotonics 2009;2:470–481. [DOI] [PubMed] [Google Scholar]
- 18. Zhang YZ, Kemper C, Bakke A, Haugland RP. Novel flow cytometry compensation standards: Internally stained fluorescent microspheres with matched emission spectra and long‐term stability. Cytometry 1998;33:244–248. [DOI] [PubMed] [Google Scholar]
- 19. Lai L, Ong R, Li J, Albani S. A CD45‐based barcoding approach to multiplex mass‐cytometry (CyTOF). Cytometry A 2015;87:369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yao Y, Liu R, Shin MS, Trentalange M, Allore H, Nassar A, Kang I, Pober JS, Montgomery RR. CyTOF supports efficient detection of immune cell subsets from small samples. J Immunol Methods 2014;415:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen G, Weng NP. Analyzing the phenotypic and functional complexity of lymphocytes using CyTOF (cytometry by time‐of‐flight). Cell Mol Immunol 2012;9:322–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheung RK, Utz PJ. Screening: CyTOF‐the next generation of cell detection. Nat Rev Rheumatol 2011;7:502–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bjornson ZB, Nolan GP, Fantl WJ. Single‐cell mass cytometry for analysis of immune system functional states. Curr Opin Immunol 2013;25:484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.