

Abstract
In this study we demonstrate that molecular fragments, which can be readily coupled via a simple, in situ RO—C=OR bond-forming reaction, can subsequently undergo metal insertion–decarboxylation–recombination to generate Csp2–Csp3 bonds when subjected to metallaphotoredox catalysis. In this embodiment the conversion of a wide variety of mixed anhydrides (formed in situ from carboxylic acids and acyl chlorides) to fragment-coupled ketones is accomplished in good to high yield. A three-step synthesis of the medicinal agent edivoxetine is also described using this new decarboxylation–recombination protocol.
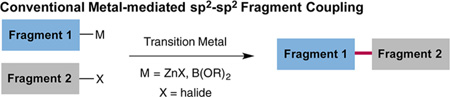
Metal-catalyzed intermolecular C–C bond formation has long been established as the predominant technology for fragment coupling in chemical synthesis. In particular, organometallic nucleophiles and organic halide/pseudohalide electrophiles have become the mainstay coupling partners for the transition metal-catalyzed production of hetero Csp2–Csp3 bonds in a highly efficient and selective fashion (eq 1).1 Moreover, the allylation and benzylation of enolates via the decarboxylative formation of π-allyl systems from β-keto-allyl esters have long been established as an important variant of the classic Tsuji–Trost mechanism (eq 2).2 The recent merger of photoredox and transition metal catalysis (termed metallaphotoredox catalysis) has gained momentum as a strategy for unique cross-coupling protocols,3 mainly due to the capacity to employ naturally occurring functional groups as traceless activation handles and the ability to achieve fragment couplings that readily build challenging Csp2–Csp3 bonds. In this context, our lab recently disclosed a light-enabled decarboxylative cross-coupling strategy that employs a diverse range of carboxylic acids in lieu of organometallic nucleophiles in combination with Ni catalysis.4 These methodologies utilize abundant and easily accessible starting materials to build a diverse array of Csp2–Csp3 bonds at room temperature while producing CO2 as a traceless byproduct.
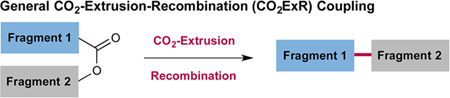
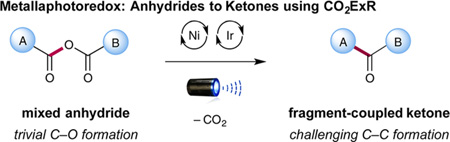
Recently, we became interested in establishing a heretofore unknown fragment-coupling reaction that employs a CO2 extrusion–recombination strategy (CO2ExR) that in a general sense bears the hallmarks of the classic Tsuji allylation reaction. Specifically, we hoped to demonstrate that two fragments that can be readily coupled via a simple C–O bond-forming step (e.g., in situ formation of an anhydride, ester, carbamate, etc.) might subsequently undergo metal insertion–decarboxylation–recombination under metallaphotoredox conditions to enable the production of relatively complex C–C bonds (e.g., Csp2–Csp3, Csp3–Csp3, eq 3). While the strategy of CO2ExR has long been established in organometallic catalysis for enolate allylation or benzylation,2,5 we hoped this new metallaphotoredox mechanism would provide an expansion in the types of organic fragments or motifs (e.g., nucleophiles and electrophiles) that can be linked via a simple RO—C=OR bond-forming step prior to CO2ExR.6 As an initial proof of concept, we chose to examine a protocol that would selectively combine and convert acid chlorides and carboxylic acids to fragment-coupled ketones via the intermediacy of a mixed anhydride (formed in situ, eq 4).
 |
(Eq 1) |
 |
(Eq 2) |
 |
(Eq 3) |
 |
(Eq 4) |
Here we present the successful implementation of these ideals and disclose the first application of this expanded CO2ExR concept toward the production of fragment coupled ketones and Csp2–Csp3 bonds.
Design Plan
As shown in Scheme 1, our proposed mechanism begins with oxidative insertion of Ni0 complex 3 to acid anhydride 1 (generated in situ from carboxylic acid and acyl chloride coupling partners) to form the corresponding acylcarboxylate-NiII complex 4.7 Concurrently, IrIII photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (7) [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine] is known to undergo photoexcitation in the presence of visible light to yield the corresponding *IrIII complex 8. This long-lived excited state (τ = 2.3 µs)8 possesses a high oxidizing power ( V vs SCE in MeCN)8 and should rapidly accept an electron from the NiII anhydride-insertion species 4, thereby inducing oxidative decarboxylation to forge the corresponding alkyl acyl NiIII complex 5.9 Rapid reductive elimination should then deliver ketone product 2 and the corresponding NiI species 6. Finally, completion of both catalytic cycles would occur simultaneously via single electron transfer (SET) between the highly reducing IrII complex 9 ( V vs SCE in MeCN)8 and the transient NiI species 6 to reconstitute the ground state of photocatalyst 7 and Ni0 catalyst 3 ( V vs SCE in DMF).10
Scheme 1.

Mechanism of CO2 Extrusion–Recombination
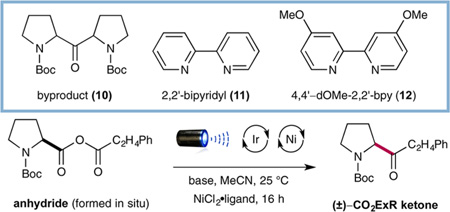
Studies toward the proposed CO2ExR of mixed anhydrides began with the coupling of hydrocinnamoyl chloride and Boc-L-proline in the presence of photocatalyst 7, NiCl2·glyme, 2,2′-bipyridyl (11), Cs2CO3, and blue LEDs as the light source (Table 1). As a critical design element, we recognized that in situ formation of the requisite anhydride would eliminate the need for an intermediate isolation step, thereby rendering the overall transformation operationally simple. To our delight, our initial experiment furnished the desired fragment-coupled ketone in a promising 40% yield (Table 1, entry 1) albeit with 20% yield of undesired homodimeric ketone 10. We recognized that production of this latter symmetrical dialkyl ketone likely arises from anhydride metathesis (metal or base catalyzed) prior to the oxidative decarboxylation step. Indeed, variation of the reaction base from Cs2CO3 to 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) provided a significant increase in the yield of the desired ketone (entry 2, 70% yield). Moreover, we were pleased to find that implementation of more electron-donating ligand 12 on the NiIICl2·Ln complex afforded the desired fragment-coupled ketone in 84% yield while limiting the formation of symmetrical ketone 10 to <3% yield (entry 3). The necessity of each catalytic component was demonstrated via a series of control experiments (entries 4–7, 0% yield). Finally, the direct implementation of purified anhydride gave comparable results (entry 8, 73% yield), demonstrating that the in situO—C=O formation step does not impact the efficiency of the subsequent C–C fragment coupling.
Table 1.
Optimization of Anhydride CO2 Extrusion–Recombinationa
 | |||||
|---|---|---|---|---|---|
| entry | conditions | base | ligand | byproduct 10b | ketone |
| 1 | as shown | Cs2CO3 | 11 | 20% | 40% |
| 2 | as shown | DBU | 11 | 14% | 70% |
| 3 | as shown | DBU | 12 | 3% | 84% |
| 4 | no photocatalyst | DBU | 12 | 0% | 0% |
| 5 | no Ni catalyst | DBU | – | 0% | 0% |
| 6 | no base | – | 12 | 0% | 0% |
| 7 | no light | DBU | 12 | 0% | 0% |
| 8c | as shown | DBU | 12 | 5% | 73% |
Reactions performed using photocatalyst 7 (1 mol%), NiCl2·glyme (5 mol%), bipyridine ligand (5 mol%), hydrocinnamoyl chloride (0.10 mmol), N-Boc-l-proline (0.13 mmol), and base (0.13 mmol). Yields determined by GC analysis using an internal standard.
Major byproduct.
Anhydride was synthesized and isolated prior to reaction.
With the optimized conditions in hand, we began to explore the scope of this transformation with respect to the acyl chloride component. As shown in Table 2, CO2ExR is successful with a range of α-methylene-bearing acid chlorides (14–18, 77–86% yield). Moreover, various carbo- and heterocyclic-substituted anhydrides may be employed to rapidly generate ketones that incorporate three- through six-membered rings at the α-carbonyl position (19–25, 65–85% yield). Notably, many of these ketones would not be readily accessible using conventional ketone forming technologies such as Weinreb amide-Grignard additions.11,12 We have found that acid chlorides that contain sterically demanding groups, such as tert-butyl or neopentyl moieties, deliver appreciable levels of efficiency (26 and 27, 50 and 32% yield). Finally, CO2ExR provides a new strategy for the production of aryl-substituted ketones as exemplified by the formation of adduct 28 in good yield.
Table 2.
Mixed Anhydride CO2 Extrusion−Recombination: Scope of Acyl Chloride and Carboxylic Acid Components
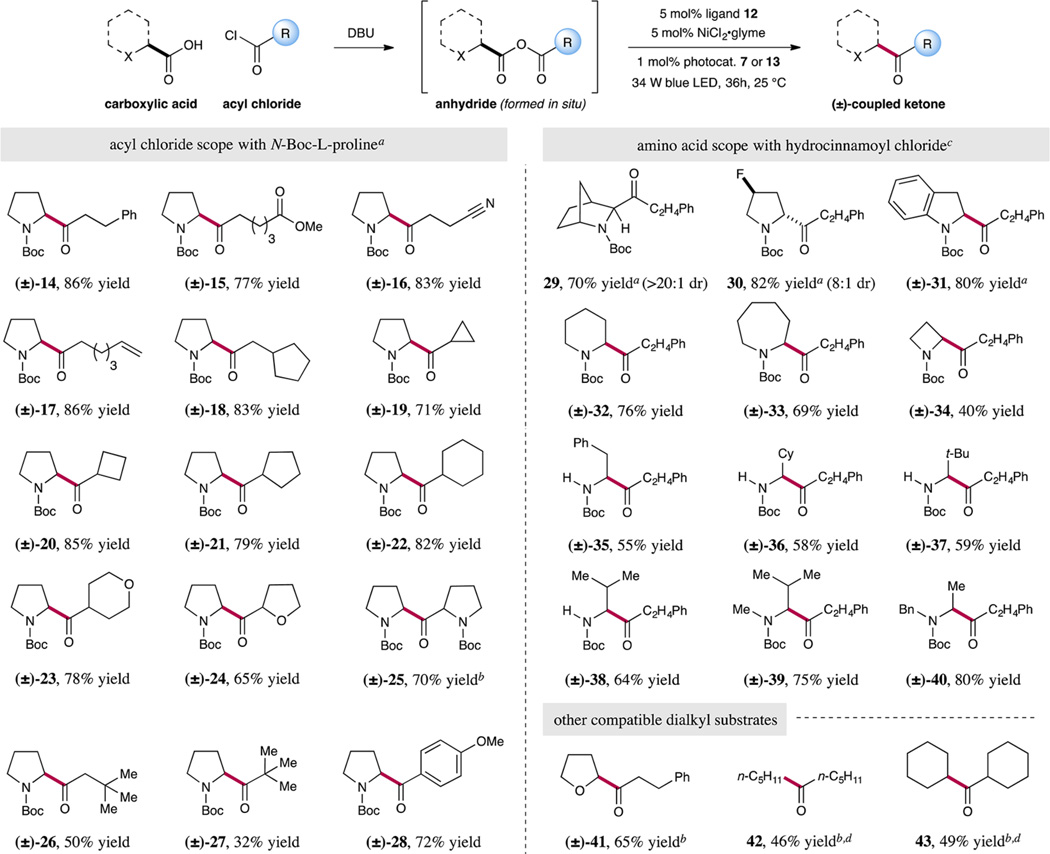 |
Reactions performed using photocatalyst 7 (1 mol%) in acetonitrile, NiCl2·glyme (5 mol%), ligand 12 (5 mol%), acyl chloride (0.10 mmol), N-Boc-l-proline (0.13 mmol), and DBU (0.13 mmol). Yield of isolated product.
See Supporting Information for experimental procedure.
Reactions performed using photocatalyst 13 (1 mol%) in dioxane using hydrocinnamoyl chloride (0.10 mmol), amino acid (0.10 mmol), and DBU (0.13 mmol). Yield of isolated product.
GC yield.
We next examined the generality of anhydride CO2ExR with respect to the carboxylic acid component. Given their widespread availability, abundance, and diverse structural complexity, we were delighted to find that α-amino acids were exemplary substrates for this new decarboxylation fragment-coupling protocol (Table 2). For example, various five-membered ring amino acids rapidly undergo CO2ExR with hydrocinnamoyl chloride to form the corresponding of α-amino ketone adducts in good to excellent yield (29–31, 70–82% yield). Interestingly, the bridged bicyclic ketone 29 was formed as a single diastereomer while maintaining good levels of reaction efficiency (70% yield). For amino acid systems beyond proline derivatives, we found that the photocatalyst Ir[dF(OMe)ppy]2 (dtbbpy)PF6 (13) [dF-(OMe)ppy = 2-(2,4-difluorophenyl)-5-(methoxy)pyridine] was more effective (products 32–40). As an example of the useful levels of complexity that can be generated in this new CO2ExR protocol, direct access to six- and seven-membered cyclic α-amino ketones is readily accomplished (32 and 33, 76 and 69% yield). We were also pleased to find that smaller strained rings, such as azetidine 34, can be tolerated, albeit in slightly diminished yield (40% yield). Gratifyingly, this transformation does not appear to be overly influenced by the steric constraints of the amino acid substrate (e.g., valine and tert-leucine systems are readily employed, 35–38, 55–64% yield). Indeed, N-alkylated N-Boc acids were found to exhibit superior efficiency over N-H-bearing substrates (cf. 38, 39, and 40). We speculate that the presence of additional electron-donating groups on the nitrogen group might lower the barrier to SET in the oxidative decarboxylation step, resulting in improved rates of formation of the requisite NiIII species (i.e., 5, Scheme 1). Beyond amino acids, we were pleased to find that α-oxy and aliphatic acids can be utilized in this new coupling protocol to forge the corresponding ketones in moderate to good yield (41–43, 46–65% yield).13,14
During the examination of the substrate scope, an interesting rearrangement was observed when β-cyclopropyl anhydride 44 (formed in situ) was subjected to this decarboxylation– recombination strategy. As shown in eq 5, the product obtained was not the expected β-cyclopropyl ketone (formed in <3% as determined by 1H NMR and GC analysis), but instead homoallylic ketone 45 (formed in 82% yield). Control experiments have demonstrated that this rearrangement occurs during the CO2ExR pathway (see Supporting Information). Moreover, labeling experiments involving 13C-labeled amino acids have shown that CO2 loss occurs mainly from the proline subunit with 85% of the acid chloride carbonyl being retained in the ketone product (eqs 5 and 6).15 Given that the Ni(0) anhydride-insertion step (Scheme 1, 3→4) is likely reversible and nonregioselective (outside of small electronic and steric perturbations), we presume that the relative activation barriers of the two possible decarboxylation steps most likely dictate the observed 85:15 ratio of isotopically labeled products. This would seem reasonable given that proline carboxylate undergoes oxidation–decarboxylation much faster than the corresponding hydrocinnamate, due to the relative stabilities of the resulting radical intermediates.16 As such, we presume that cyclopropyl ring-opened product 47 must arise from a NiII-mediated decarbonylation–recarbonylation process, wherein formation of NiII complex 49 enables cyclopropyl ring opening prior to recarbonylation.17 Given the wealth of previously reported Ni-catalyzed carbonylation and decarbonylation reactions,18,19 we feel this mechanism best supports the results of our labeling experiments.
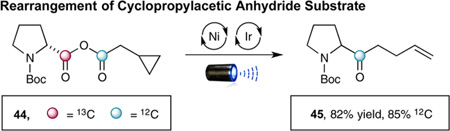 |
(Eq 5) |
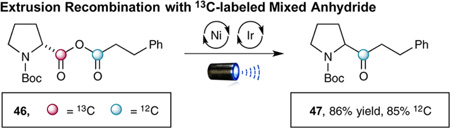 |
(Eq 6) |
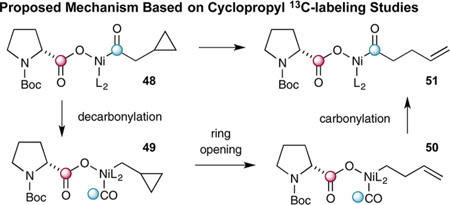 |
(Eq 7) |
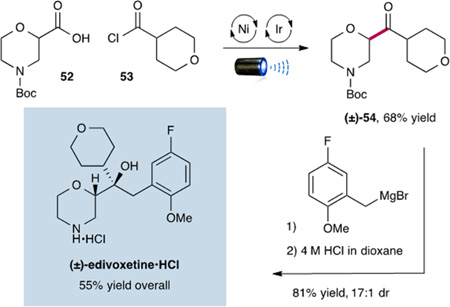
Finally, we applied our CO2ExR technology to a three-step synthesis of (±)-edivoxetine·HCl, a medicinal agent in development for the treatment of ADHD.20 As shown, commercial acid 52 and acyl chloride 53 were readily coupled using the optimized metallaphotoredox conditions to generate ketone 54 in good yield (68%). The synthesis was thereafter completed via a Grignard addition, followed by HCl-mediated Boc removal to afford (±)-edivoxetine·HCl in 55% yield over three steps.
 |
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by NIHGMS (R01 GM078201-05) and gifts from Merck and Amgen.
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Experimental procedures, structural proofs, and spectral data for all new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.(a) de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd ed. Weinheim: Wiley; 2004. [Google Scholar]; (b) Johansson Seechurn CCC, Kitching MO, Colacot TJ, Snieckus V. Angew. Chem. Int. Ed. 2012;51:5062. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 2. Shimizu I, Yamada T, Tsuji J. Tetrahedron Lett. 1980;21:3199. Tsuda T, Chujo Y, Nishi S, Tawara K, Saegusa T. J. Am. Chem. Soc. 1980;102:6381. (c) For a review on decarboxylative allylation and benzylation, see: Weaver JD, Recio A, Grenning AJ, Tunge JA. Chem. Rev. 2011;111:1846. doi: 10.1021/cr1002744. Liu Y, Han S-J, Liu W-B, Stoltz BM. Acc. Chem. Res. 2015;48:740. doi: 10.1021/ar5004658.
- 3.(a) Osawa M, Nagai H, Akita M. Dalton Trans. 2007:827. doi: 10.1039/b618007h. [DOI] [PubMed] [Google Scholar]; (b) Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J. Am. Chem. Soc. 2011;133:18566. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ye Y, Sanford MS. J. Am. Chem. Soc. 2012;134:9034. doi: 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rueping M, Koenigs RM, Poscharny K, Fabry DC, Leonori D, Vila C. Chem.-Eur. J. 2012;18:5170. doi: 10.1002/chem.201200050. [DOI] [PubMed] [Google Scholar]; (e) Sahoo B, Hopkinson MN, Glorius F. J. Am. Chem. Soc. 2013;135:5505. doi: 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]; (f) Fabry DC, Zoller J, Raja S, Rueping M. Angew. Chem. Int. Ed. 2014;53:10228. doi: 10.1002/anie.201400560. [DOI] [PubMed] [Google Scholar]; (g) Shu X, Zhang M, He Y, Frei H, Toste FD. J. Am. Chem. Soc. 2014;136:5844. doi: 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zoller J, Fabry DC, Ronge MA, Rueping M. Angew. Chem. Int. Ed. 2014;53:13264. doi: 10.1002/anie.201405478. [DOI] [PubMed] [Google Scholar]; (i) Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Lang SB, O’Nele KM, Tunge JA. J. Am. Chem. Soc. 2014;136:13606. doi: 10.1021/ja508317j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Yoo W-J, Tsukamoto T, Kobayashi S. Angew. Chem. Int. Ed. 2015;54:6587. doi: 10.1002/anie.201500074. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Noble A, McCarver SJ, MacMillan DWC. J. Am. Chem. Soc. 2015;137:624. doi: 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Jana R, Trivedi R, Tunge JA. Org. Lett. 2009;11:3434. doi: 10.1021/ol901288r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rodríguez N, Manjolinho F, Grünberg MF, Gooßen LJ. Chem.-Eur. J. 2011;17:13688. doi: 10.1002/chem.201102584. [DOI] [PubMed] [Google Scholar]; (c) Hossian A, Singha S, Jana R. Org. Lett. 2014;16:3934. doi: 10.1021/ol5017349. [DOI] [PubMed] [Google Scholar]; (d) Pfister KF, Grünberg MF, Gooßen LJ. Adv. Synth. Catal. 2014;356:3302. [Google Scholar]
- 6.Example of in situ generation of allyl ester for decarboxylative allylation of arylglyoxylic acid was reported by Gooßen and co-workers: Grünberg MF, Gooßen LJ. J. Organomet. Chem. 2013;744:140.
- 7.(a) Uhlig E, Nestler B. Z. Chem. 1981;21:451. [Google Scholar]; (b) Fischer R, Walther D, Kempe R, Sieler J, Schonecker B. J. Organomet. Chem. 1993;447:131. [Google Scholar]; (c) Kajita Y, Kurahashi T, Matsubara S. J. Am. Chem. Soc. 2008;130:17226. doi: 10.1021/ja806569h. [DOI] [PubMed] [Google Scholar]; (d) Ochi Y, Kurahashi T, Matsubara S. Org. Lett. 2011;13:1374. doi: 10.1021/ol200044y. [DOI] [PubMed] [Google Scholar]; (e) Zhao C, Jia X, Wang X, Gong H. J. Am. Chem. Soc. 2014;136:17645. doi: 10.1021/ja510653n. [DOI] [PubMed] [Google Scholar]
- 8.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem. Mater. 2005;17:5712. [Google Scholar]
- 9.Oxidation of either nickel carboxylate complex or free carboxylate is operable. See SI for details.
- 10.Durandetti M, Devaud M, Perichon J. New J. Chem. 1996;20:659. [Google Scholar]
- 11.Selected examples: Gao X, Liu Y, Kwong S, Xu Z, Ye T. Org. Lett. 2010;12:3018. doi: 10.1021/ol101021v. Ma N, Yao Y, Zhao B-X, Wang Y, Ye W-C, Jiang S. Chem. Commun. 2014;50:9284. doi: 10.1039/c4cc02575j.
- 12.Dieter RK, Sharma RR, Yu H, Gore VK. Tetrahedron. 2003;59:1083. [Google Scholar]
- 13.Early examples of formation of symmetrical ketones from corresponding anhydrides: Easterfield TH, Taylor CM. J. Chem. Soc. Trans. 1911;99:2298. Grün A, Ulbrich E, Krczil F. Angew. Chem. 1926;39:421. Man EH, Hauser CR. J. Am. Chem. Soc. 1950;72:3294.
- 14.Substrates with slow oxidative decarboxylation allow competitive anhydride metathesis, which leads to formation of homocoupling adducts. Extensive studies are ongoing to suppress this pathway for unsymmetrical aliphatic anhydride substrates.
- 15.With inverse isotopic labeling of 44 (carboxylate = 12C hydrocinnamyl chloride = 13C) we observed an 85:15 ratio of 13C:12C ketone product.
- 16.(a) Bockman TM, Hubig SM, Kochi JK. J. Am. Chem. Soc. 1996;118:4502. [Google Scholar]; (b) Bockman TM, Hubig SM, Kochi JK. J. Org. Chem. 1997;62:2210. doi: 10.1021/jo9617833. [DOI] [PubMed] [Google Scholar]
- 17.Rearrangement can occur via different mechanisms: Pinke PA, Stauffer RD, Miller RG. J. Am. Chem. Soc. 1974;96:4229. Nakamura I, Yamamoto Y. Adv. Synth. Catal. 2002;344:111. Masarwa A, Marek I. Chem.-Eur. J. 2010;16:9712. doi: 10.1002/chem.201001246. Newcomb M. Kinetics of Radical Reactions: Radical Clocks. In: Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. 1st ed. Weinheim: Wiley; 2001. Biswas S, Weix DJ. J. Am. Chem. Soc. 2013;135:16192. doi: 10.1021/ja407589e.
- 18.Examples of decarbonylation in Ni complexes: Otsuka S, Nakamura A, Yoshida T, Naruto M, Ataka K. J. Am. Chem. Soc. 1973;95:3180. Yamamoto T, Ishizu J, Kohara T, Komiya S, Yamamoto A. J. Am. Chem. Soc. 1980;102:3758. Malelckis A, Sanford MS. Organometallics. 2014;33:3831.
- 19.Examples of carbonylation in Ni complexes: Bauld NL. Tetrahedron Lett. 1963;4:1841. Chiusoli GP, Cassar L. Angew. Chem. Int. Ed. Engl. 1967;6:124. Ocafrain M, Devaud M, Troupel M, Perichon J. J. Chem. Soc. Chem. Commun. 1995:2331. Dolhem E, Barhdadi R, Folest J, Nédelec JY, Troupel M. Tetrahedron. 2001;57:525. Wotal AC, Ribson RD, Weix D. J. Organometallics. 2014;33:5874. doi: 10.1021/om5004682. Hoshimoto Y, Ohata T, Sasaoka Y, Ohashi M, Ogoshi S. J. Am. Chem. Soc. 2014;136:15877. doi: 10.1021/ja509171a.
- 20.Campell GI, Cases-Thomas MJ, Man T, Masters JJ, Eugenine Rudyk HC, Walter MW. Appl. 2007/0083046 A1. U.S. Patent. 2007
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.