

Abstract
ThyX is an essential thymidylate synthase that is mechanistically and structurally unrelated to the functionally analogous human enzyme, thus providing means for selective inhibition of bacterial growth. To identify novel compounds with anti-bacterial activity against the human pathogenic bacterium Helicobacter pylori, based on our earlier biochemical and structural analyses, we designed a series of eighteen 2-hydroxy-1,4-naphthoquinones (2-OH-1,4-NQs) that target HpThyX. Our lead-like molecules markedly inhibited the NADPH oxidation and 2′-deoxythymidine-5′-monophosphate-forming activities of HpThyX enzyme in vitro, with inhibitory constants in the low nanomolar range. The identification of non-cytotoxic and non-mitotoxic 2-OH-1,4-NQ inhibitors permitted testing their in vivo efficacy in a mouse model for H. pylori infections. Despite the widely assumed toxicity of naphthoquinones (NQs), we identified tight-binding ThyX inhibitors that were tolerated in mice and can be associated with a modest effect in reducing the number of colonizing bacteria. Our results thus provide proof-of-concept that targeting ThyX enzymes is a highly feasible strategy for the development of therapies against H. pylori and a high number of other ThyX-dependent pathogenic bacteria. We also demonstrate that chemical reactivity of NQs does not prevent their exploitation as anti-microbial compounds, particularly when mitotoxicity screening is used to prioritize these compounds for further experimentation.
Keywords: naphthoquinone, anti-microbial agents, Helicobacter pylori, thymidylate synthase ThyX
1. Introduction
De novo synthesis of 2′-deoxythymidine-5′-monophosphate (dTMP or thymidylate) is essential for cellular survival. Consequently, inhibiting the methylation reaction of 2′-deoxyuridine-5′-monophosphate (dUMP) to dTMP by thymidylate synthases (TS) provides a powerful means for controlling the growth of eukaryotic or bacterial cells. This is illustrated by the development of several chemotherapeutic agents that target thymidylate biosynthesis. For instance, fluoropyrimidines (e.g. 5-fluorouracil and capecitabine) and antifolates (e.g. methotrexate and pemetrexed), which target human TS, are successful drugs used in cancer chemotherapy [1]. Moreover, methotrexate and trimethoprim target dihydrofolate reductase (DHFR) that is also required for efficient thymidylate synthesis in many eukaryotes, including pathogenic parasites and bacteria [2,3].
Human TS belongs to the ThyA family of enzymes (EC 2.1.1.45) that uses N5,N10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate) in a reductive methylation reaction [4]. In this reaction, tetrahydrofolate (H4folate) reduces the methylene moiety after its transfer to the uracil ring, thus resulting in the formation of dihydrofolate (H2folate). The second distinct family of TS, ThyX proteins (EC 2.1.1.148; flavin-dependent thymidylate synthase), uses a non-covalently bound flavin adenine dinucleotide (FAD) cofactor to facilitate hydride transfer from NAD(P)H [5–8]. Consequently, the end product of ThyX catalysis is H4folate, explaining why ThyX-containing bacteria (≈30% of all bacterial species) do not require DHFR FolA that recycles H2folate to H4folate in actively dividing thyA-carrying cells [9,10].
Several observations have established essential ThyX proteins as highly relevant drug targets [11–13]. They are found in a large number of human pathogenic bacteria (electronic supplementary material, table S1), including Helicobacter pylori (HpThyX) and Mycobacterium tuberculosis (MtbThyX), but are absent in humans. Moreover, the structure and mechanisms of ThyA and ThyX proteins are strikingly different, a fact that greatly facilitates the development of specific ThyX inhibitors that do not act on human TS [11–14]. The key feature of the active site of ThyX proteins is its location in a large and flexible cavity at the interface of three subunits of the ThyX homotetramer. This location allows surface exposure of the N5 atom of the flavine isoalloxazine ring that, at the millisecond time scale, is involved in hydride transfer [15]. As the hydride transfer to ThyX-bound FAD from NAD(P)H is either strictly dependent on or activated by the nucleotide substrate dUMP, this renders ThyX inhibitors that are competitive with respect to dUMP selective within the bacterial cell. Indeed, we previously identified non-substrate-based, tight-binding ThyX inhibitors that inhibited growth of genetically modified Escherichia coli cells carrying thyX. The observed pattern of inhibition mimics a genetic knockout of TS [11], indicating selective in vivo targeting. The co-crystal structure of one such inhibitor—2-hydroxy-3-(4-methoxybenzyl)-1,4-naphthoquinone (the molecule C8-C1)—revealed binding within the conserved active site, partially overlapping with the dUMP-binding pocket. In addition to our inhibitor studies on ThyX proteins, several dUMP analogues have also been described that inhibit MtbThyX at micromolar concentrations [12,13,16]. Importantly, recent studies have also indicated that 5-fluoro-dUMP, a metabolite of fluorouracil, selectively inhibits ThyX in living cells of M. tuberculosis [17].
The fact that naphthoquinones (NQs) inhibit ThyX proteins is of great interest, as biological activities of these compounds are widely reported. For instance, the anti-cancer activity of plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), a natural naphthoquinone derivative isolated from Plumbago or Dyospiro sp., has been observed in cell cultures, as well as in animal models [18,19]. This molecule and dyospirin (a dimeric analogue of plumbagin) have also shown anti-microbial activity against different pathogens, including M. tuberculosis [20–22]. Moreover, atovaquone (2-(trans-4-(P-chlorophenyl)cyclohexyl)-3-hydroxy-1,4-naphthoquinone), a well-known 2-OH-1,4-NQ (Malarone, GlaxoSmithKline), targets the respiratory electron transfer chain, and is clinically used in anti-pneumocystis, anti-toxoplasmosis and anti-malarial treatments [23]. Recently, NQ-based inhibitors of DNA gyrase with a novel mechanism of action have also been described [24]. Despite these remarkable observations, the further use of NQs in biomedical applications has been hindered by their redox activity and widely assumed toxicity [25–28].
The first flavin-dependent TS to be biochemically characterized was the ThyX enzyme from H. pylori [9]. This spiral-shaped, Gram-negative bacterium infects the gastric mucosa of about half of the world's population, and is associated with chronic gastritis, peptic ulcers and gastric carcinoma [29]. Here, we report on the identification of the new 2-OH-1,4-NQ derivatives with relatively low cyto- and mitotoxicity. These molecules display a potent inhibition of H. pylori ThyX activity. Some of these ThyX inhibitors are well tolerated, and one of them has shown modest but significant activity in an animal model of infection. We expect that our results will not only significantly speed up thymidylate synthase-based anti-microbial discovery approaches, but will also increase the interest in biological activities of NQs.
2. Material and methods
2.1. Chemicals
The 2-OH-1,4-NQ derivatives designed and used in this study (figure 1a) were synthetized by Roowin (Riom, France). Purity of the compounds (more than 95%) was confirmed by HPLC analyses using detection at 254 nm. 1H NMR and mass spectrometry (ESI+) were used to confirm the conformity of the synthetized molecules. Aqueous solubility (logSW) of the compounds was estimated using the Yalkowsky formula logSW = 0.8–0.01(MP − 25) − logP, with MP being an experimentally determined melting point of the compound. Concentrated stock solutions (10 mg ml−1) of the different compounds were prepared in dimethyl sulfoxide (DMSO). For mouse infection and treatment, the compounds were dissolved in 2% β-cyclodextrine. All other chemicals were purchased from Sigma-Aldrich: ampicillin (A0166), amphotericin B (A4888), atovaquone (A7986), β-cyclodextrine (C4767), DMSO (D2650), FAD (F6625), NADPH (N7505), polymyxin B (P4932), rotenone (R8875), dUMP (D3876) and vancomycin (V2002). CH2H4folate was provided by Eprova, Merck.
Figure 1.

(a) Chemical structures of 2-OH-1,4-NQ derivatives tested in this study. Asterisk refers to the fact that molecule 010-G has a chloride (and not hydroxyl) at position 2. (b) Chemical structure of 2-(trans-4-(4-chlorophenyl)cyclohexyl)-3-hydroxy-1,4-naphthoquinone (atovaquone). (c) Predicted logS values (aqueous solubility) of the different drugs versus their molecular weight (g mol−1). The four molecules selected for in vitro testing (figure 4) and for mouse experiments (figure 6) are indicated above their symbol (filled squares). Atov, atovaquone.
2.2. Helicobacter pylori strains and growth conditions
Helicobacter pylori strains used in this study were 26695 and the mouse-adapted strain SS1 [30,31]. Helicobacter pylori strains were grown on Blood Agar Base 2 (Oxoïd) plates supplemented with 10% defibrinated horse blood, or in Brain Heart Infusion liquid medium (Oxoïd), supplemented with 8% decomplemented fetal bovine serum (FBS; Invitrogen) with an antibiotic–fungicide mix consisting of vancomycin (final concentration 12.5 µg ml−1), polymyxin B (0.31 µg ml−1) and amphotericin B (2.5 µg ml−1). Helicobacter pylori was grown at 37°C under microaerophilic conditions obtained using the CampyGen system (Oxoïd).
2.3. Cytotoxicity and mitotoxicity of 2-OH-1,4-NQ compounds
Cytotoxicity of the 2-OH-1,4-NQ derivatives was assessed by measuring lactate dehydrogenase (LDH) release following manufacturer's instructions (Cytotoxicity Detection Kit; Roche Applied Sciences). Briefly, AGS cells (human gastric adenocarcinoma cell line; ATCC Catalog no. CRL-1739TM) were cultured in Ham's F-12 K medium containing 1% of FBS. A total of 3 × 104 cells were added per well in a sterile 96-well tissue culture plate. Cells were then treated with different doses of 2-OH-1,4-NQ compounds ranging from 0.78 to 50 μg ml−1. After a 24 h incubation at 37°C (5% CO2, 90% humidity), the microplates were centrifuged at 250g for 10 min, and the supernatants were carefully removed and transferred into optically clear 96-well microplates (Greiner Bio-One). The dye solution containing iodotetrazolium chloride and sodium lactate was then added to each well to quantify the amount of LDH released into the extracellular medium. LDH was quantified by measuring the A490 using a PowerWave Microplate Spectrophotometer (BioTek).
Mitochondrial toxicity (mitotoxicity) was assessed by measuring resazurin (7-hydroxy-3H-phenoxazin-3-one 10-oxide) reduction by following absorption changes at 570 nm (Mitochondrial Viability Assay; Abcam). Resazurin is an indicator dye that reports on oxidation–reduction reactions taking place in the mitochondria of living cells. AGS cells (≅2.5 × 105cells well−1) cultured in a DMEM galactose/glutamine-supplemented medium were seeded in sterile white-walled 96-well clear-bottom microplates and treated for 4 h with different doses of 2-OH-1,4-NQ compounds ranging from 0.78 to 50 μg ml−1. Addition of the stain solution was followed by a further 4 h incubation at 37°C (5% CO2, 90% humidity) and A570 was measured. Rotenone, an inhibitor of the mitochondrial respiratory chain complex I, was used as a positive control.
2.4. Anti-microbial susceptibility testing: disc diffusion method and minimal inhibitory concentration values
The in vitro anti-bacterial activity of 2-OH-1,4-NQ compounds was assessed against the 26695 and SS1 strains of H. pylori. DMSO was used as control in all experiments.
Disc diffusion tests were performed in triplicate, including a DMSO-only control on each plate, as specified in the electronic supplementary material. Minimal inhibitory concentrations (MICs) were determined using a broth microdilution test in 24-well microtitre plates. For test inocula, an overnight preculture of H. pylori strains 26695 and SS1 was diluted to an OD600 of approximately 0.1 and 0.5 ml of this suspension was transferred to each well. Ten microlitres of twofold serial dilutions of each compound in DMSO, ranging in concentration from 0.625 to 20 µg ml−1, were added and the covered plates were incubated for 24 h at 37°C with orbital shaking at 140 r.p.m. under microaerophilic conditions. The MIC was determined as the lowest compound concentration resulting in full growth inhibition after 24 h of incubation. All the tests were done in triplicate, including cell-free and DMSO-only controls.
To determine the bactericidal activity of C8-C1 against H. pylori, liquid BHI medium was inoculated with an overnight culture of H. pylori 26695 at an initial OD600 of 0.1. C8-C1 or chloramphenicol, used as bacteriostatic control, were added after 4 h of culture time at final concentrations of 5 and 30 µg ml−1. To determine the viable counts of surviving bacteria (colony forming units, CFU), aliquots of culture were then plated in triplicate on blood agar plates after 3 and 24 h exposure.
2.5. Protein production and purification
Escherichia coli strain BL21 (fhuA2 [lon] ompT gal [dcm] ΔhsdS), carrying HpthyX on plasmid pGL2 under the control of a pBAD promoter, was grown at 37°C on solid or liquid Luria Bertani medium [9]. This strain was used for overproduction and purification of HpThyX tagged with six histidine residues [32]. The protein was purified by affinity chromatography on a 5 ml resin HiTrap TALON column (GE Healthcare) using a linear imidazole gradient. Imidazole was removed using a PD-10 column (Bio-Rad). Concentrated fractions were pooled and stored at −80°C in 30 mM HEPES (pH 8.0), NaCl 300 mM, glycerol 10% (v/v). The concentration of purified proteins was determined by the Bradford method (Bio-Rad). A450 values were used to detect FAD bound to purified ThyX proteins.
2.6. Thyx activity measurements
HpThyX activity was assessed by measuring either deprotonation of [5-3H]dUMP or NAPDH oxidation activities [11,32].
In the tritium release (deprotonation) assays, typical reactions contained 10 mM MgCl2, 10% (v/v) glycerol, 500 µM FAD, 2 mM NADPH, 1 mM CH2H4Folate and 10 mM β-mercaptoethanol in 50 mM HEPES (pH 8). Different concentrations of dUMP were also included in the reaction mixtures. The specific activity of tritiated [5-3H]dUMP (diammonium salt) stock was 15–30 Ci mmol−1 (Moravek Biochemicals, CA, USA). 2-OH-1,4-NQ derivatives were prepared at 100 µM in 1% DMSO. Reactions were initiated by adding the enzyme (10 µM) and were stopped after 20 min incubation at 37°C.
NADPH oxidation assays were performed at 37°C in 96-well plates (Greiner Bio-One). One hundred microlitres of reaction mixture contained HEPES 50 mM (pH 8.6), NaCl 150 mM, FAD 50 µM, β-mercaptoethanol 1.43 mM, NADPH 500 µM and 10 µM of purified HpThyX. dUMP and 2-OH-1,4-NQ concentrations were varied across 12.5–200 µM and 0.3–100 µM, respectively. Microtitre plates were prepared and transferred to the microplate reader Chameleon II (Hidex). The reactions were started by automatically injecting NADPH into individual wells and ThyX activity was determined by following a decrease in absorbance at 340 nm. A molar extinction coefficient of 6220 M−1 cm−1 at 340 nm (ε340) was used to quantify NADPH oxidation. Samples with added DMSO and enzyme-free reactions were used as positive and negative controls, respectively.
2.7. Thyx docking methodology
The H. pylori ThyX protein structure (PDB code 3AH5 [33,34]) was processed by Pymol software [35] to remove water molecules and the C:dUMP cofactor. Polar hydrogen atoms were added and atomic partial charges were assigned using the Pymol Vina plugin [36]. The residues A:Arg197, C:Arg109 and C:Tyr110 were chosen to be flexible during docking performed with the Vina software [37]. A cubic search volume of 25 × 25 × 25Å centred on A:FAD N5 atom was defined and the lowest predicted energy conformation was kept for analysis.
2.8. Mouse infection and treatment
NMRI-specific pathogen-free mice (Charles River Laboratories) were orogastrically inoculated with 108 CFU of the H. pylori mouse-adapted strain SS1, prepared in 100 μl of peptone broth. Four groups of six to eight mice were infected by H. pylori strain SS1. Earlier experiments have established that after one week, colonization of mice is fully established [38]. A negative control group of five mice was inoculated with peptone broth alone and was not colonized by H. pylori. One week after infection, three groups of mice were treated orogastrically three times a day with 500 µl of either compound 010-C, 010-E or 010-I dissolved in 2% β-cyclodextrin at 0.25 mg ml−1 (0.375 mg mouse d−1) during one week. As a control, the fourth group of mice was treated orogastrically three times a day by the same amount of 2% β-cyclodextrin (17.6 mM), the vehicle of the compounds. Administered particles had a hydrodynamic radius of 180–200 nm and their size and monodispersity were measured using dynamic light scattering (Malvern Instruments, Zetasizer Nano-S instrument). This average size is in agreement with the average size of β-cyclodextrin particles that self-aggregate in water at a concentration of 12 mM [39]. After treatment, viable H. pylori cells, colonizing the mouse stomach, were enumerated by culture of serial dilutions of homogenized tissue on blood agar plates containing bacitracin (200 μg ml−1) and nalidixic acid (10 μg ml−1) as in [40].
3. Results
3.1. Optimization and custom-synthesis of new 2-OH-1,4-NQ compounds
To identify novel compounds targeting the ThyX enzyme of H. pylori (HpThyX), we designed a series of 2-OH-1,4-NQ derivatives using the commercial molecule C8-C1 (or its resynthesized version 007-A) as starting point (figure 1). This molecule was identified earlier as selective ThyX inhibitor with cellular activity against genetically modified E. coli strains [11]. Mass spectrometry and 1H NMR analyses confirmed the molecular structures of the NQ derivatives indicated in figure 1a. All molecules tested in further experiments were more than 95% pure based upon HPLC analyses using detection at 254 nm. Molecular weight and predicted logP values of these molecules ranged from 230 to 375 Da and 1.85 to 5.2, respectively (electronic supplementary material, table S2). Aqueous solubility, a key factor determining the ADME-Tox properties of small molecules, was estimated using the Yalkowsky formula (see Material and methods) relying on experimentally determined melting points and calculated logP values. These calculations predict that the aqueous solubility of the compounds varies from ≈40 µM to ≈2 mM (figure 1c). It is of note that most of these molecules are predicted to be considerably more soluble than the closely related atovaquone, a commercially available anti-malarial compound targeting the mitochondrial cytochrome bc1 complex (figure 1b,c).
3.2. Helicobacter pylori growth is inhibited by 2-OH-1,4-NQ derivatives targeting HpThyX
Using semi-quantitative deprotonation assays that detect proton release from [5-3H]dUMP during ThyX catalysis, the effect of these 2-OH-1,4-NQ compounds against HpThyX was investigated. Table 1 shows that all molecules substantially inhibited ThyX activity at micromolar concentrations. The susceptibility of H. pylori to these compounds was determined using the disc diffusion method (electronic supplementary material, figure S2) and via the determination of MICs (table 1; broth microdilution test). Overall, both tests gave similar results, indicating anti-pylori activity against both strains 26695 (wild-type strain) and SS1 (mouse-adapted strain) at micromolar concentrations (typical MIC-values ranging from 0.625 to 10 µg ml−1). When H. pylori liquid cultures were exposed to C8-C1 for up to 24 h, no viable cells were recovered after replating on solid media lacking this compound (figure 2), indicating that the anti-microbial activity of the C8-C1 compound is bactericidal. Expectedly, chloramphenicol behaved as bacteriostatic agent under these conditions (figure 2). The molecule 010-G, where a hydroxyl group at the position 2 was replaced with a chlorine, was very active in liquid cultures (MIC ∼ 0.625 µg ml−1), but inactive in disc diffusion tests, due to limited diffusion of the compound from its filter.
Table 1.
In vitro and biological activity of 2-OH-1,4-NQ compounds against HpThyX and H. pylori, respectively.
| minimal inhibitory concentration (MIC) |
|||
|---|---|---|---|
| molecule | inhibition against ThyX (% remaining activity relative to control) | strain 26695 (µg ml−1) (µM) | strain SS1 (µg ml−1) (µM) |
| 007-A (C8C1) | 38.5 | 10 (33.98) | 10 (33.98) |
| 007-B | 60.2 | 10 (26.68) | 10 (26.68) |
| 007-C | 62.5 | 5 (13.01) | 5 (13.01) |
| 007-D | 45.7 | ≥20 (62.03) | 20 (62.03) |
| 007-E | 66.3 | 10 (35.42) | 10 (35.42) |
| 007-F | 60.6 | 5 (15.06) | 10 (15.06) |
| 007-G | 61.8 | 20 (56.16) | 5 (14.04) |
| 010-A | 49.5 | 10 (33.48) | 5 (16.74) |
| 010-B | 74.3 | 10 (33.30) | 10 (33.30) |
| 010-C | 76.4 | 10 (39.34) | 10 (39.34) |
| 010-D | 36.4 | 10 (37.00) | 10 (37.00) |
| 010-E | 77.7 | 5 (14.27) | 5 (14.27) |
| 010-F | 74.5 | 10 (31.58) | 5 (15.79) |
| 010-G | 21.1 | 0.625 (2.07) | 0.625 (2.07) |
| 010-H | 57.8 | 20 (64.45) | 10 (32.23) |
| 010-I | 64.3 | 10 (43.42) | 10 (43.42) |
| 010-J | 49.7 | 2.5 (8.49) | 2.5 (8.49) |
| 010-K | 75.1 | 20 (61.67) | 10 (30.84) |
Figure 2.
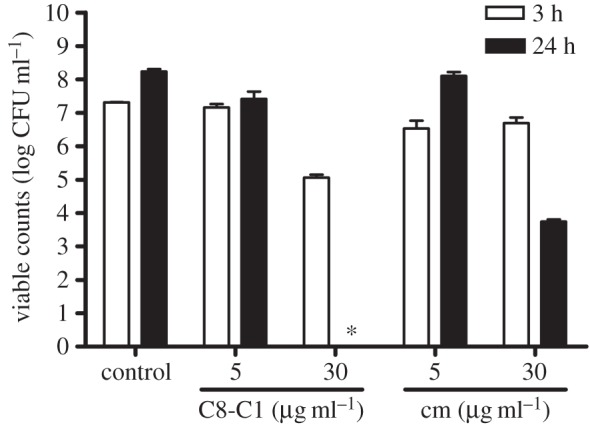
Effect of C8-C1 on the in vitro growth of H. pylori strain 26695. Chloramphenicol, a well-known bacteriostatic antibiotic, was used as control. CFUs were determined after 3 h or 24 h exposure. The asterisk indicates that no growth was observed.
3.3. In vitro cytotoxicity and mitotoxicity tests
NQs are known to possess biological activities against different cell types, prompting us to investigate the possible cytotoxicity of the synthetized compounds, using atovaquone as control. The cytotoxicity of the compounds was determined by measuring LDH release from AGS cells, a human gastric adenocarcinoma cell line, as is detailed in Material and methods. These experiments revealed that the majority of the compounds are either non-cytotoxic in the concentration range tested or are less cytotoxic than atovaquone, which at 25 µg ml−1 had a cytotoxic effect of 48 ± 5% (figure 3a; electronic supplementary material, figure S3A). This result is similar to what has been described previously for atovaquone in human hepatic HL-7702 cells [26]. The molecule 010-G with a potent anti-microbial activity was found to be cytotoxic and was not studied further in this study (electronic supplementary material, figure S3).
Figure 3.
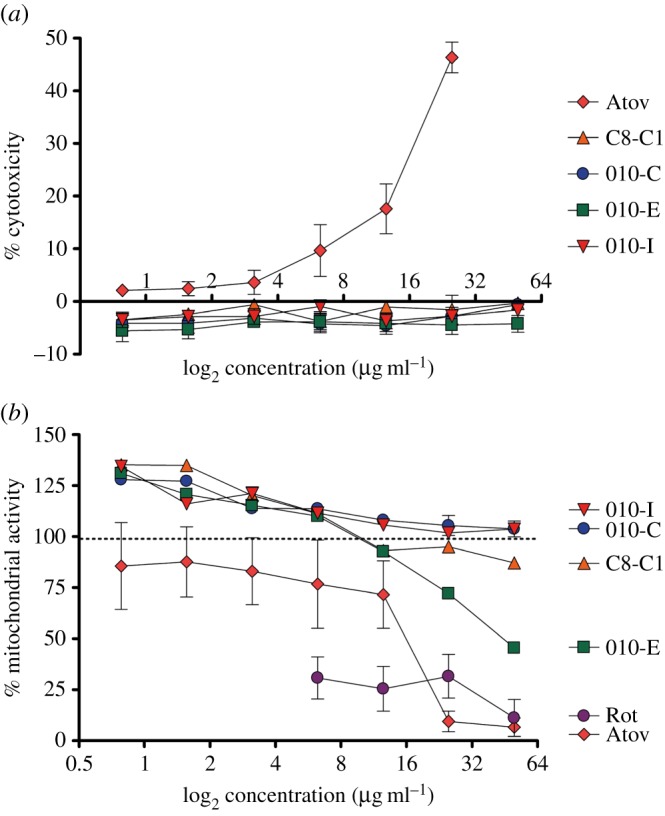
(a) Cytotoxic and (b) mitotoxic effects of 2-OH-1,4-NQ compounds. Atovaquone (Atov) and rotenone (Rot) were used as positive controls.
The mitotoxicity of these compounds was assessed by measuring NADPH/NADH-dependent resazurin reduction by the mitochondria of living AGS cells (figure 3b). Rotenone and atovaquone, which target complexes I and III of the mitochondrial respiratory chain, respectively, were used as positive controls. These experiments revealed that the cytotoxic compounds identified above target the mitochondrial respiratory chain (electronic supplementary material, figure S3B), whereas the non-cytotoxic inhibitors were also found to be non-mitotoxic (figure 3).
3.4. In vitro testing of 2-OH-1,4-NQ derivatives against ThyX from Helicobacter pylori
To investigate the inhibitory potential of the molecules 007-A (C8-C1), 010-C, 010-E and 010-I against HpThyX in more detail, we used a quantitative tritium release assay (see Material and methods). These molecules were chosen for more detailed studies because they were found to be non-cytotoxic and non-mitotoxic in our assays. We first showed that these molecules suppressed the dTMP-forming activity at a compound concentration of 100 μM using 10 μM of ThyX enzyme (figure 4a). Using the same set of compounds in NADPH oxidation assays, our results revealed that IC50 values varied linearly as a function of the dUMP concentration (figure 4b). The observed linear correlation with a positive slope is indicative of the molecules tested acting as tight-binding competitive inhibitors with respect to dUMP. From the slope of these curves, we estimate Ki-values of 1000, 367, 258 and 28 nM for the molecules 010-E, C8-C1, 010-I and 010-C, respectively. Thus, our chemical series allowed a 10-fold improvement of the inhibitor affinity towards H. pylori ThyX proteins in comparison to the starting molecule C8-C1. Overall, our data indicate that the relatively small size and high lipophilicity of the R substitution at position 3 favours optimal inhibition of H. pylori ThyX. Molecular modelling of the inhibitory binding in the active site of HpThyX (figure 5) shows that binding in the vicinity of the catalytically crucial N5-atom of the FAD cofactor is highly feasible.
Figure 4.
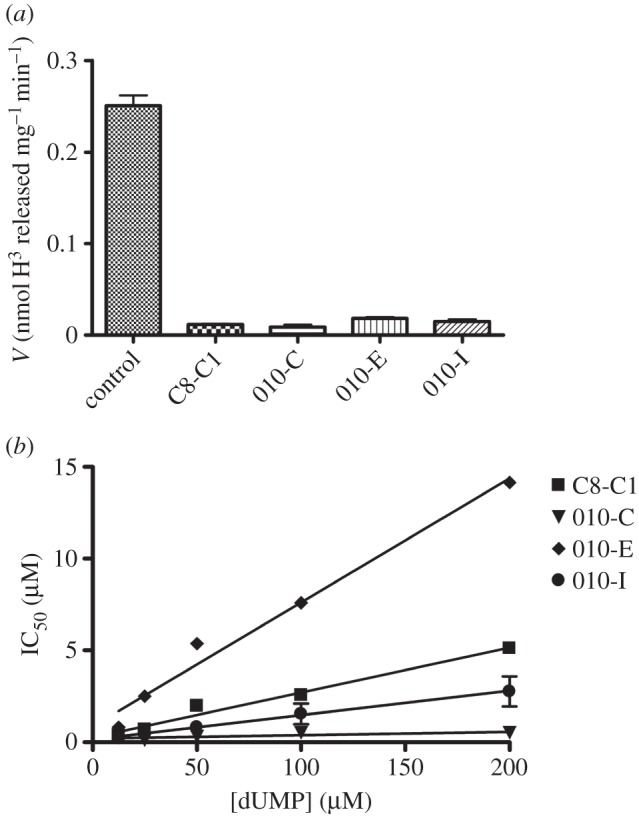
HpThyX inhibition in vitro. (a) Deprotonation assays using 10 µM of enzyme and 100 µM of different drugs. (b) Determination of IC50 values for different compounds as a function of the dUMP concentration. NADPH oxidation assays were used for activity measurements.
Figure 5.

Three-dimensional modelling of HpThyX–inhibitor complexes using Autodock Vina. FAD and HpThyX inhibitor are depicted in stick representations with carbon, oxygen, nitrogen and fluorine atoms coloured in yellow, red, blue and green, respectively. The HpThyX surface is shown with carbon, oxygen, nitrogen and sulfur atoms coloured grey, red, blue and yellow, respectively. Some amino acids were removed for clarity. Two orientations, separated by a 90° rotation along the y-axis, are shown. (a) 007-A (C8-C1). (b) 010-C. (c) 010-E. (d) 010-I.
3.5. Mouse infection and treatment
The in vivo efficacy of the molecules 010-C, 010-E and 010-I was assessed by testing their effect on mouse colonization by H. pylori (figure 6). The molecules were dissolved in β-cyclodextrin at 2% adjusted to pH 7.5, a drug delivery vehicle known to be harmless towards mice and H. pylori. These non-mitotoxic molecules were chosen for animal experiments not only because of their appropriate logS and logP values for oral absorption, but also for their markedly lower Ki-values when compared with the ‘parent’ molecule C8-C1. A previously established mouse model for following H. pylori infections was used for these experiments [40]. After an initial colonization period of one week, three groups of mice were treated orogastrically three times a day with 500 µl of compounds 010-C, 010-E or 010-I at 0.25 mg ml−1 (0.375 mg mouse day−1) during one additional week. The dosage of these treatments was 17 mg kg−1 of body weight, which is the same order of magnitude as antibiotics used for anti-H. pylori treatments. As a control, a group of mice was orogastrically treated with the same amount of 2% β-cyclodextrin. None of the aforementioned treatments affected either the body or stomach weight of the mice (data not shown). After treatment, viable H. pylori, colonizing the stomach of the different groups of mice, were enumerated by culturing serial dilutions of homogenized tissue (figure 6). We observed a statistically significant (Mann–Whitney test, one-tail, p = 0.0003) decrease of 1.22 log (17-fold) in the colonization loads (geometric means) of mice that were treated with the molecule 010-I as compared with the mice treated with the vehicle alone.
Figure 6.
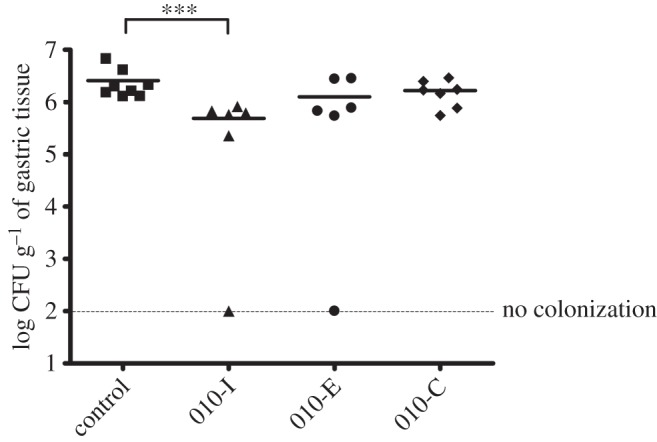
Gastric colonization of NMRI mice infected with H. pylori strain SS1 after treatment with three 2-OH-1,4-NQ compounds: 010-C, 010-E and 010-I. Mice were sacrificed two weeks after inoculation, including one week of orogastric treatment with the compounds. Each symbol corresponds to the H. pylori colonization load in the gastric mucosa of a single mouse. Squares correspond to infected mice treated with 2% β-cyclodextrin (vehicle control), triangles, circles and diamonds refer to infected mice treated with compounds 010-I, 010-E and 010-C, respectively. The horizontal bars correspond to the geometric mean calculated for each group of mice. The dashed horizontal bar represents the detection limit of colonization. Differences in the bacterial loads were statistically analysed by the Mann–Whitney test (GraphPad, Prism), ***p < 0.01 (one-tail).
4. Discussion
Helicobacter pylori infections are common, and are currently treated by either a classical triple therapy consisting of a combination of proton pump inhibitor (PPI) and two antibiotics, among them clarythromycin, amoxicillin and metronidazole or, alternatively, by the simultaneous administration of PPI, bismuth, tetracycline and metronidazole. Resistance to all of these antibiotics except amoxicillin is frequent among clinical strains of H. pylori. For instance, in Europe, the resistance rates for adults reach 17.5% for clarithromycin and 35% for metronidazole, thus justifying the search for new anti-microbial compounds with activity against H. pylori cells [41].
The TS from H. pylori and humans are mechanistically and structurally very distinct, thus prompting for the development of new anti-microbial compounds specifically targeting HpThyX. Towards this goal, we designed a set of eighteen 2-OH-1,4-NQ derivatives that are closely related to atovaquone, a clinically used anti-malarial compound (figure 1). Despite considerable variations in predicted solubility, hydrophobicity and size (electronic supplementary material, table S2), all 18 molecules acted as ThyX inhibitors in vitro under semi-quantitative assay conditions (table 1). Molecules 007-A (=C8-C1), 010-C, 010-E and 010-I were then selected for a more quantitative study (figure 4). Overall, our experiments resulted in identification of HpThyX inhibitors with nanomolar Ki-values and provided additional insight into further optimization. In all the structural models obtained (figure 5), the binding modes of the inhibitor and dUMP are similar, thus providing a plausible explanation for the simultaneous inhibition of NADPH oxidation and deprotonation activities. Thus, this mechanism of inhibition is not the result of the redox or chemical reactivity of NQs, but rather a direct effect of blocking a binding of a catalytic activator dUMP. Thus, binding of the inhibitor in the vicinity of the FAD cofactor first prevents activation of the NADPH oxidation/FAD reduction and, second, the binding of the substrate that receives a carbon from CH2H4folate is blocked [15]. The docked configuration for the molecules C8-C1 and 010-C is very similar to that observed in crystal structures of the PBCV-1 enzyme [11].
We moreover demonstrated that these compounds have a potent and concentration-dependent anti-microbial activity against two different strains of H. pylori grown in liquid or solid medium (MIC-values in the range 0.625–20 μg ml−1; table 1). In addition, this anti-microbial activity is bactericidal, as the number of viable cells drastically diminished when H. pylori cultures were continuously exposed to the molecule C8-C1 (figure 2). To date, our attempts to identify mutants that are resistant to these compounds were unsuccessful when using compound concentrations that inhibit growth on solid media, suggesting that these molecules may have several targets in bacterial cells. To exclude that the biological activities of our molecules simply resulted from non-specific redox activity or chemical reactivity, we performed cytotoxicity and mitotoxicity tests. These studies identified several new 2-hydroxy-1,4-naphthoquinones (2-OH-1,4-NQs) that were substantially less cytotoxic than atovaquone and an order of magnitude more potent HpThyX inhibitors than the founding molecule C8-C1. We also showed that cytotoxic 2-OH-1,4-NQs (including atovaquone) targeted mitochondria in our assays (electronic supplementary material, figure S3). Interestingly, we found that the compounds 010-C, 010-E, 010-I and C8-C1 activated the mitochondrial metabolism at low concentrations (figure 3b), suggesting that under these conditions these non-cytotoxic compounds probably mediate electron transfer from NAD(P)H to resazurin and/or might have antioxidant activity similar to that of idebenone, a synthetic analogue of coenzyme Q10 [42].
The three compounds 010-C, 010-E and 010-I were chosen for animal experimentation because of their lack of cytotoxicity and high affinity against the target enzyme. To obtain insight into the in vivo activity of these three HpThyX inhibitors, we investigated their effect in an H. pylori mouse model of infection. This model has been used in previous studies to validate the in vivo efficacy of H. pylori inhibitors. Two studies used the same mouse model set-up and the same H. pylori SS1 strain, one showing the in vivo efficacy of a metronidazole treatment to eradicate H. pylori [43], and a second one reporting the use of isopentenyloxycinnamyl derivatives to reduce the H. pylori colonization loads [44]. Despite widely assumed toxicity of NQs, we found that the three ThyX inhibitors tested were tolerated in mice. For the molecule 010-I, with relatively high predicted solubility, a statistically significant effect against whole cells of H. pylori was identified using the aforementioned animal model (figure 6). Although the observed in vivo effect remains modest (17-fold), we consider our observations a promising starting point for further small molecule optimization to improve, for example, solubility and bioavailability of this class of compounds.
In summary, we have characterized a series of new HpThyX inhibitors, allowing identification of non-mitotoxic NQs with high efficiency against the target enzyme. Our results provide proof-of-concept that targeting ThyX enzymes is a highly feasible strategy for the development of therapies against H. pylori and a high number of other ThyX-dependent pathogenic bacteria. Our results further underline that the widely assumed chemical reactivity of NQs does not necessarily prevent their exploitation as anti-microbial compounds, particularly when mitotoxicity screening is used to prioritize these compounds for further experimentation.
Supplementary Material
Acknowledgement
We thank Dr H. Becker, Dr S. Barth and Dr S. Lagrange for helpful discussions.
Ethics
Experiments in mice were carried out in strict accordance with the recommendations in the Specific Guide for the Care and the Use of Laboratory Animals of the Pasteur Institute, according to the European Directive (2010/63/UE) and the corresponding French law on animal experimentation (Arrêtés 19 April 1988). The protocol has been approved by the Committee of Central Animal Facility Board of the Pasteur Institute. The project was submitted and approved by the CETEA, Comité d’éthique en Expérimentation Animale of the Institut Pasteur (no. 2013-0051) and was submitted for final approval to the Ministère de l'Enseignement Supérieur et de la Recherche (no. 00317).
Authors' contributions
S.S., H.d.R. and H.M. designed and supervised experiments. S.S., K.D., I.L., K.A. and J.B. provided reagents and performed experiments. J.-C.L. performed computational analyses. S.S., U.L., H.d.R. and H.M. wrote the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the ANR project AMTHYX (ANR-10-EMMA-044-01) to H.M. and H.d.R.
References
- 1.Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. 2014. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 11, 282–298. (doi:10.1038/nrclinonc.2014.51) [DOI] [PubMed] [Google Scholar]
- 2.Gangjee A, Kurup S, Namjoshi O. 2007. Dihydrofolate reductase as a target for chemotherapy in parasites. Curr. Pharm. Des. 13, 609–639. (doi:10.2174/138161207780162827) [DOI] [PubMed] [Google Scholar]
- 3.Hawser S, Lociuro S, Islam K. 2006. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 71, 941–948. (doi:10.1016/j.bcp.2005.10.052) [DOI] [PubMed] [Google Scholar]
- 4.Carreras CW, Santi DV. 1995. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 64, 721–762. (doi:10.1146/annurev.bi.64.070195.003445) [DOI] [PubMed] [Google Scholar]
- 5.Gattis SG, Palfey BA. 2005. Direct observation of the participation of flavin in product formation by thyX-encoded thymidylate synthase. J. Am. Chem. Soc. 127, 832–833. (doi:10.1021/ja0432214) [DOI] [PubMed] [Google Scholar]
- 6.Graziani S, et al. 2006. Catalytic mechanism and structure of viral flavin-dependent thymidylate synthase ThyX. J. Biol. Chem. 281, 24 048–24 057. (doi:10.1074/jbc.M600745200) [DOI] [PubMed] [Google Scholar]
- 7.Koehn EM, Fleischmann T, Conrad JA, Palfey BA, Lesley SA, Mathews II, Kohen A. 2009. An unusual mechanism of thymidylate biosynthesis in organisms containing the thyX gene. Nature 458, 919–923 (doi:10.1038/Nature07973) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mishanina TV, Koehn EM, Conrad JA, Palfey BA, Lesley SA, Kohen A. 2012. Trapping of an intermediate in the reaction catalyzed by flavin-dependent thymidylate synthase. J. Am. Chem. Soc. 134, 4442–4448. (doi:10.1021/Ja2120822) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Myllykallio H, Lipowski G, Leduc D, Filee J, Forterre P, Liebl U. 2002. An alternative flavin-dependent mechanism for thymidylate synthesis. Science 297, 105–107. (doi:10.1126/science.1072113) [DOI] [PubMed] [Google Scholar]
- 10.Myllykallio H, Leduc D, Filee J, Liebl U. 2003. Life without dihydrofolate reductase FolA. Trends Microbiol. 11, 220–223. (doi:10.1016/S0966-842X(03)00101-X) [DOI] [PubMed] [Google Scholar]
- 11.Basta T, et al. 2012. Mechanistic and structural basis for inhibition of thymidylate synthase ThyX. Open Biol. 2, 120120 (doi:10.1098/rsob.120120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kogler M, Busson R, De Jonghe S, Rozenski J, Van Belle K, Louat T, Munier-Lehmann H, Herdewijn P. 2012. Synthesis and evaluation of 6-Aza-2’-deoxyuridine monophosphate analogs as inhibitors of thymidylate synthases, and as substrates or inhibitors of thymidine monophosphate kinase in Mycobacterium tuberculosis. Chem. Biodiv. 9, 536–556. (doi:10.1002/cbdv.201100285) [DOI] [PubMed] [Google Scholar]
- 13.Kogler M, et al. 2011. Synthesis and evaluation of 5-substituted 2‘-deoxyuridine monophosphate analogues as inhibitors of flavin-dependent thymidylate synthase in Mycobacterium tuberculosis. J. Med. Chem. 54, 4847–4862. (doi:10.1021/Jm2004688) [DOI] [PubMed] [Google Scholar]
- 14.Mathews II, Deacon AM, Canaves JM, McMullan D, Lesley SA, Agarwalla S, Kuhn P. 2003. Functional analysis of substrate and cofactor complex structures of a thymidylate synthase-complementing protein. Structure 11, 677–690. (doi:10.1016/S0969-2126(03)00097-2) [DOI] [PubMed] [Google Scholar]
- 15.Becker HF, Djaout K, Lamarre I, Ulmer JE, Schaming D, Balland V, Liebl U, Myllykallio H, Vos MH. 2014. Substrate interaction dynamics and oxygen control in the active site of thymidylate synthase ThyX. Biochem. J. 459, 37–45. (doi:10.1042/Bj20131567) [DOI] [PubMed] [Google Scholar]
- 16.Esra Onen F, Boum Y, Jacquement C, Spanedda MV, Jaber N, Scherman D, Myllykallio H, Herscovici J. 2008. Design, synthesis and evaluation of potent thymidylate synthase X inhibitors. Bioorg. Med. Chem. Lett. 18, 3628–3631. (doi:10.1016/j.bmcl.2008.04.080) [DOI] [PubMed] [Google Scholar]
- 17.Singh V, et al. 2014. The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem. Biol. 22, 63–75. (doi:10.1016/j.chembiol.2014.11.006) [DOI] [PubMed] [Google Scholar]
- 18.Dandawate P, et al. 2012. Synthesis, characterization, molecular docking and cytotoxic activity of novel plumbagin hydrazones against breast cancer cells. Bioorg. Med. Chem. Lett. 22, 3104–3108. (doi:10.1016/j.bmcl.2012.03.060) [DOI] [PubMed] [Google Scholar]
- 19.Dandawate P, Ahmad A, Deshpande J, Swamy KV, Khan EM, Khetmalas M, Padhye S, Sarkar F. 2014. Anticancer phytochemical analogs 37: synthesis, characterization, molecular docking and cytotoxicity of novel plumbagin hydrazones against breast cancer cells. Bioorg. Med. Chem. Lett. 24, 2900–2904. (doi:10.1016/j.bmcl.2014.04.100) [DOI] [PubMed] [Google Scholar]
- 20.Borges-Argaez R, Canche-Chay CI, Pena-Rodriguez LM, Said-Fernandez S, Molina-Salinas GM. 2007. Antimicrobial activity of Diospyros anisandra. Fitoterapia 78, 370–372. (doi:10.1016/j.fitote.2007.03.004) [DOI] [PubMed] [Google Scholar]
- 21.Kuete V, Tangmouo JG, Meyer JJ, Lall N. 2009. Diospyrone, crassiflorone and plumbagin: three antimycobacterial and antigonorrhoeal naphthoquinones from two Diospyros spp. Int. J. Antimicrob. Agents 34, 322–325. (doi:10.1016/j.ijantimicag.2009.04.008) [DOI] [PubMed] [Google Scholar]
- 22.Lall N, Das Sarma M, Hazra B, Meyer JJ. 2003. Antimycobacterial activity of diospyrin derivatives and a structural analogue of diospyrin against Mycobacterium tuberculosis in vitro. J. Antimicrob. Chemother. 51, 435–438. (doi:10.1093/jac/dkg068) [DOI] [PubMed] [Google Scholar]
- 23.Baggish AL, Hill DR. 2002. Antiparasitic agent atovaquone. Antimicrob. Agents Chemother. 46, 1163–1173. (doi:10.1128/AAC.46.5.1163-1173.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karkare S, Chung TT, Collin F, Mitchenall LA, McKay AR, Greive SJ, Meyer JJ, Lall N, Maxwell A. 2013. The naphthoquinone diospyrin is an inhibitor of DNA gyrase with a novel mechanism of action. J. Biol. Chem. 288, 5149–5156. (doi:10.1074/jbc.M112.419069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knecht W, Henseling J, Loffler M. 2000. Kinetics of inhibition of human and rat dihydroorotate dehydrogenase by atovaquone, lawsone derivatives, brequinar sodium and polyporic acid. Chem. Biol. Interact. 124, 61–76. (doi:10.1016/S0009-2797(99)00144-1) [DOI] [PubMed] [Google Scholar]
- 26.Guo J, Song W, Ding F, Zhang J, Sun Z. 2012. Study on cytotoxicity and structure-activity relationship of HL-7702 cell exposed to naphthoquinones. Environ. Toxicol. Pharmacol. 33, 408–413. (doi:10.1016/j.etap.2012.01.005) [DOI] [PubMed] [Google Scholar]
- 27.Pinho BR, Santos MM, Fonseca-Silva A, Valentao P, Andrade PB, Oliveira JM. 2013. How mitochondrial dysfunction affects zebrafish development and cardiovascular function: an in vivo model for testing mitochondria-targeted drugs. Br. J. Pharmacol. 169, 1072–1090. (doi:10.1111/bph.12186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birth D, Kao WC, Hunte C. 2014. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 5, 4029 (doi:10.1038/ncomms5029) [DOI] [PubMed] [Google Scholar]
- 29.Salama NR, Hartung ML, Muller A. 2013. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 11, 385–399. (doi:10.1038/nrmicro3016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomb JF, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547. (doi:10.1038/41483) [DOI] [PubMed] [Google Scholar]
- 31.Lee A, O'Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112, 1386–1397. (doi:10.1016/S0016-5085(97)70155-0) [DOI] [PubMed] [Google Scholar]
- 32.Leduc D, Graziani S, Lipowski G, Marchand C, Le Marechal P, Liebl U, Myllykallio H. 2004. Functional evidence for active site location of tetrameric thymidylate synthase X at the interphase of three monomers. Proc. Natl Acad. Sci. USA 101, 7252–7257. (doi:10.1073/pnas.0401365101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang XL, Zhang JY, Mao XH, Zou QM, Hu YL, Wang DC. 2010. Crystallization and preliminary crystallographic studies of a flavin-dependent thymidylate synthase from Helicobacter pylori. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 513–515. (doi:10.1107/S174430911000864x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang XL, Zhang JY, Guo G, Mao XH, Hu YL, Zou QM. 2012. Crystal structure of a flavin-dependent thymidylate synthase from Helicobacter pylori strain 26695. Prot. Pept. Lett. 19, 1225–1230. (doi:10.2174/092986612803217105) [DOI] [PubMed] [Google Scholar]
- 35.Schrödinger LLC. 2010. The PyMOL molecular graphics system, v. 1.3r1. New York, NY: Schrödinger LLC. [Google Scholar]
- 36.Seeliger D, de Groot BL. 2010. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 24, 417–422. (doi:10.1007/s10822-010-9352-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. (doi:10.1002/jcc.21334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bury-Mone S, Skouloubris S, Labigne A, De Reuse H. 2001. UreI: a Helicobacter pylori protein essential for resistance to acidity and for the early steps of murine gastric mucosa infection. Gastroenterol. Clin. Biol. 25, 659–663. [PubMed] [Google Scholar]
- 39.Bonini M, Rossi S, Karlsson G, Almgren M, Lo Nostro P, Baglioni P. 2006. Self-assembly of beta-cyclodextrin in water. Part 1: cryo-TEM and dynamic and static light scattering. Langmuir 22, 1478–1484. (doi:10.1021/la052878f) [DOI] [PubMed] [Google Scholar]
- 40.Skouloubris S, Thiberge JM, Labigne A, De Reuse H. 1998. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immunol. 66, 4517–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Megraud F, Coenen S, Versporten A, Kist M, Lopez-Brea M, Hirschl AM, Andersen LP, Goossens H, Glupczynski Y. 2013. Helicobacter pylori resistance to antibiotics in Europe and its relationship to antibiotic consumption. Gut 62, 34–42. (doi:10.1136/gutjnl-2012-302254) [DOI] [PubMed] [Google Scholar]
- 42.Mordente A, Martorana GE, Minotti G, Giardina B. 1998. Antioxidant properties of 2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1,4-benzoquinone (idebenone). Chem. Res. Toxicol. 11, 54–63. (doi:10.1021/tx970136j) [DOI] [PubMed] [Google Scholar]
- 43.Jenks PJ, Ferrero RL, Tankovic J, Thiberge JM, Labigne A. 2000. Evaluation of nitrofurantoin combination therapy of metronidazole-sensitive and -resistant Helicobacter pylori infections in mice. Antimicrob. Agents Chemother. 44, 2623–2629. (doi:10.1128/AAC.44.10.2623-2629.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.da Silva CR, Michel V, Genovese S, Prevost MC, Epifano F, Touati E. 2012. Anti-Helicobacter pylori activities of natural isopentenyloxycinnamyl derivatives from Boronia pinnata. Nat. Prod. Commun. 7, 1347–1350. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.