

Abstract
Our understanding of the complex, transcriptional feedback loops in the circadian clock mechanism has depended upon quantitative, timeseries data from disparate sources. We measure clock gene RNA profiles in Arabidopsis thaliana seedlings, grown with or without exogenous sucrose, or in soil-grown plants and in wild-type and mutant backgrounds. The RNA profiles were strikingly robust across the experimental conditions, so current mathematical models are likely to be broadly applicable in leaf tissue. In addition to providing reference data, unexpected behaviours included co-expression of PRR9 and ELF4, and regulation of PRR5 by GI. Absolute RNA quantification revealed low levels of PRR9 transcripts (peak approx. 50 copies cell−1) compared with other clock genes, and threefold higher levels of LHY RNA (more than 1500 copies cell−1) than of its close relative CCA1. The data are disseminated from BioDare, an online repository for focused timeseries data, which is expected to benefit mechanistic modelling. One data subset successfully constrained clock gene expression in a complex model, using publicly available software on parallel computers, without expert tuning or programming. We outline the empirical and mathematical justification for data aggregation in understanding highly interconnected, dynamic networks such as the clock, and the observed design constraints on the resources required to make this approach widely accessible.
Keywords: circadian rhythms, plant biology, gene regulatory networks, biological clocks, model optimization, data management
1. Introduction
Circadian clocks are found widely among organisms from archaea to mammals [1,2]. These internal time-keepers generate approximately 24 h rhythms in the expression of 10–30% of genes, even without environmental cues. In natural conditions, circadian rhythms are entrained by light and temperature cycles. Their function is to coordinate internal processes with the external day/night cycle [3,4] and also, through photoperiodism, relative to the seasonal cycle [5]. The circadian system of each organism includes a phylum-specific gene regulatory network that is required for most rhythmicity [6], as well as non-transcriptional oscillator(s) that are less well characterized in eukaryotes [7].
In plants, the clock gene network includes highly connected, negative regulators forming a complicated circuit. This has been best studied in Arabidopsis thaliana. One simplification (figure 1a) visualizes the circuit as a three-loop structure of morning and evening loops coupled around a repressilator [10,11]. The morning loop includes the MYB-related transcription factors LHY and CCA1, which activate expression of the pseudo-response regulators PRR9 and PRR7 [12,13], but inhibit expression of later-expressed genes including PRR5 and TOC1 (PRR1). PRR9, PRR7, PRR5 and TOC1 bind to and inhibit LHY and CCA1 expression, as predicted by modelling [10,14] and demonstrated by experiments [15–18]. LHY and CCA1 also inhibit expression of ELF3, ELF4 and LUX (PCL1), whose protein products interact to form another repressor, the evening complex (EC) [19–22]. The EC is thought to inhibit the expression of at least ELF4 and LUX, forming a negative feedback loop, whose continued function might explain the damped oscillation of clock gene expression observed in lhy cca1 double mutant plants [10]. GI, a large plant-specific protein, is also rhythmically expressed but functions at a post-translational level through, for example, stabilization of the TOC1-degradation factor ZTL [23]. Light signalling controls multiple processes (electronic supplementary material, figure S1) that entrain the clock circuit to the day–night cycle. A growing number of identified processes and components remain to be fully integrated into the circuit, though even the components described are challenging to analyse.
Figure 1.

The clock gene network and experimental protocols. (a) The clock gene network summarized in the activity-flow language of SBGN v. 1.0 [8], with the principal connections in the P2012 model [9]. The repressilator is denoted by green lines; morning loop components are filled yellow; LHY/CCA1, red; evening loop components, blue. Light inputs are shown in electronic supplementary material, figure S1 and all modelled connections of P2011 [10] in electronic supplementary material, figure S2. (b) Peak-normalized RNA profiles of genes depicted in (a), in plants of the Col-0 accession under a 12 h light : 12 h dark cycle (LD 12 : 12; experiment 2b of panel (c)). (c) Graphical representation of the growth conditions. Experiments 1, 4, 5, 6 and 7 used seedlings grown in LD 12 : 12 for the number of days indicated; experiments 2 and 3 used plants grown on soil in LD 12 : 12 for the number of days indicated. Sucrose concentrations, growth temperatures and genotypes tested are shown for each experiment. Open box, light interval; black box, dark interval; light grey box, predicted darkness in constant light; dark grey box, predicted light in constant darkness; red box, red light. Sampling time in ZT (h), relative to lights-on of the first day of sampling or the last dawn before experimental treatment (ZT0). Ros, rosette; sd, seedling.
Formal, mathematical models have been helpful in understanding the plant clock, because its components are highly interconnected by nonlinear regulation (electronic supplementary material, figure S2; reviewed in [24]). Model development was necessarily based upon timeseries data, where the system was manipulated using mutations and by varying light or temperature inputs. More detailed models demanded greater precision and breadth in the data, which raised two major issues. First, data collation was laborious, because the numerical data underlying published timeseries graphs were rarely accessible [25]. Although the potential benefits of data sharing are recognized [26,27], in practice, useful sharing requires cyber infrastructure, which is currently best-developed for omics data rather than the many focused studies in the clock literature [28]. Second, the published data on Arabidopsis clocks used several genetic backgrounds and growth conditions, introducing ill-defined variation to the results.
To provide directly comparable data, we conducted large-scale qRT-PCR assays for the RNA levels of multiple clock genes. Overlapping studies in four laboratories using different growth stages and conditions highlighted the robustness of most expression profiles and the few instances where they varied. Visualizing the data as phase plane plots suggested new dynamic interactions and their genetic regulators. Absolute RNA quantification revealed the low expression levels of ELF3 and PRR9. To facilitate similar projects, we introduce data aggregation in the online BioDare resource, and illustrate the utility of our datasets by reoptimizing the P2011 clock model [10] with the open-source application Systems Biology Software Infrastructure (SBSI) [29], highlighting key areas for future experiments.
2. Results
2.1. Large-scale measurement of clock gene RNA profiles
This study was motivated by two projects that integrated circadian regulation into research on other plant physiological systems, which were incompatible with the growth conditions used in earlier circadian research. The Regulation of Biological Signalling by Temperature (ROBuST) project studied the interactions of ambient temperature with circadian and light signalling circuits; exogenous sucrose inhibits light signalling [30,31] and was therefore excluded. The Timing of Metabolism (TiMet) project studied circadian regulation of the starch pathway, among others, which is best characterized in rosette plants grown on soil. To measure the rhythmic expression in a set of clock-related genes (figure 1b), we used automated systems in Golm and Edinburgh to quantify mRNA levels for components of the clock circuit every 2 h, in multiple conditions and mutant backgrounds [32,33] (figure 1c). The ROBuST dataset tested 13-day-old, wild-type (WT) and mutant seedlings grown at 17°C on agar medium without additional sucrose. Datasets from the TiMet project tested 21-day-old rosette plants grown at 20°C on soil (TiMet ros) and 13-day-old seedlings on soil (TiMet sd1). The TiMet rosette data were collected from WT and clock mutant Arabidopsis thaliana plants grown under light : dark (LD) cycles in two experiments, followed by constant light (LL) or constant dark (DD) in one study. Three further studies were compared, from seedlings grown on sterile agar media without sucrose (TiMet sd2, using the same medium as the ROBuST data), or with exogenous sucrose under white (McWatters, this paper; and Edwards et al. [34]) or red light (Southern, this paper; and [21,35]).
2.2. Data presentation
Time is expressed as zeitgeber time (ZT) in hours since the last dark–light transition, by convention; the first dark–light transition within the sampling interval is 0 h on our plots. TiMet data are presented as absolute values [33], obtained by calibrating RNA extraction efficiency with heterologous control RNAs (electronic supplementary matetial, table S1) to calculate the number of copies of each RNA per gram fresh weight (gFW). Estimated cell numbers per gFW (see electronic supplementary material) were used to calculate RNA copies per cell. The other datasets are normalized relative to a control transcript (ACTIN7 for ROBuST; ACTIN2 for Edwards and Southern; βTUBULIN4 for McWatters). ACTIN2 and GAPDH controls were also assayed with two amplicons each in the TiMet assays, for comparison among datasets. Data were replicated in biological duplicate or triplicate samples and in equivalent sampling on successive days (0–12 h and 24–36 h in the TiMet and Edwards datasets). Data are presented on linear scales to reflect the potential for protein synthesis and hence regulatory effects on downstream targets (in keeping with most of the literature; figures 2 and 3; electronic supplementary material, figure S5) and on logarithmic scales to reveal the full dynamic range of RNA expression, and hence the influence of multiple upstream regulators (figures 4–6; electronic supplementary material, figure S3 and S4). Further technical comparison among the studies is presented in the electronic supplementary material.
Figure 2.

Clock gene expression in wild-type plants under LD cycles. Transcript levels in Col-0 and Ws-2 WT under LD 12 : 12 were measured by qRT-PCR, in experiment 2 (TiMet ros) including eight external RNA standards to allow absolute quantification in Col-0 and Ws-2 (a,c,e) and in experiment 1 (ROBuST) normalized to the ACTIN7 control in Col-4 and Ws-2 (b,d,f). Data represent transcripts of (a,b) LHY and CCA1, (c,d) PRR9, and (e,f) TOC1 and GI. Error bars show SD, for two to three biological replicates. Electronic supplementary material, figure S3 shows the data on logarithmic plots.
Figure 3.
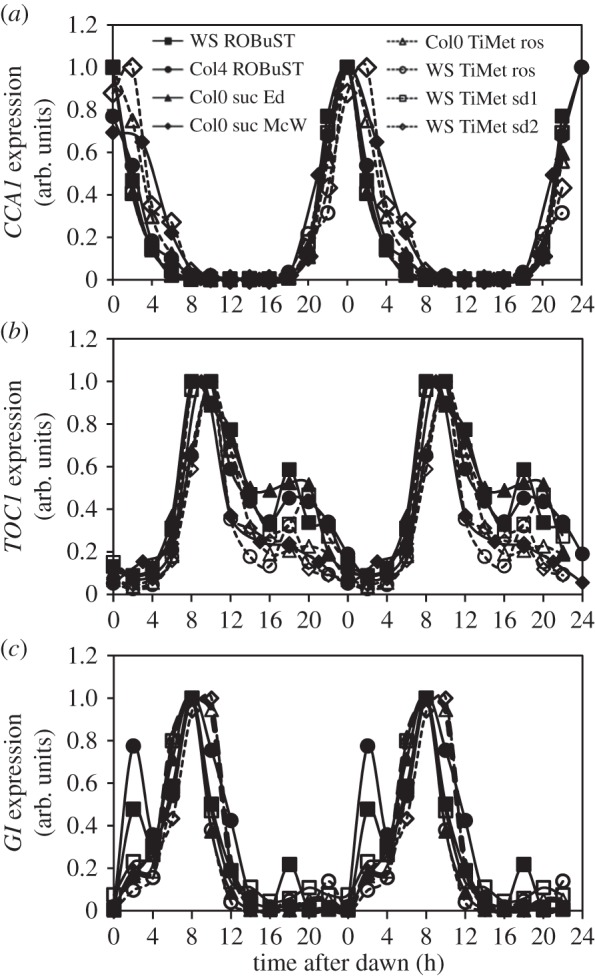
Waveforms of clock gene expression across experiments at different plant age and in the absence and presence of exogenous sucrose. This plot compares transcript abundance of CCA1, TOC1 and GI in 12 h photoperiods in three WTs grown in different experimental conditions in different laboratories. The data are taken from the following experiments (figure 1): WS ROBuST (1, seedlings), Col4 ROBuST (1, seedlings), Col0 suc Ed (6, seedlings provided with 3% exogenous sucrose), Col0 suc McW (5, seedlings provided with 3% sucrose), Col0 TiMet ros (2B, 21 day-old rosettes), WS TiMet ros (2, 21 day-old rosettes), WS TiMet sd1 (3, 10 day-old seedlings), WS TiMet sd2 (4, 13-day-old seedlings). All plants were entrained in LD 12 : 12 (figure 1). Values for each transcript are normalized to the peak. The results are the mean of duplicate or triplicate samples, double-plotted; error bars are not shown for clarity.
Figure 4.

Range of transcript abundance for clock genes in clock mutants. The bars show the highest and lowest mean values for the absolute abundance of transcripts for clock genes in a given genotype. The genotypes are, from left to right, Col-0 wild-type, gi-201, prr9 prr7 double mutant, toc1, WS WT, lhy cca1 double mutant (from experiments 2 and 2B of figure 1c, 21-day-old rosettes) and WS (designated WS_2) and elf3 from experiment 3 (13-day-old seedlings), (a) LHY, (b) CCA1, (c) PRR9, (d) PRR7, (e), PRR5, (f), TOC1, (g) LUX, (h) GI, (i) ELF3, (j) ELF4. The underlying data are as in figures 5 and 6.
Figure 6.

Clock gene expression in wild-type plants and elf3 mutants in LD. Ws-2 WT (solid lines) and elf3–4 mutant plants (dashed lines) were grown in a 12 h photoperiod for 12 days and harvested through one LD cycle (TiMet sd, dataset 3 of figure 1c). Transcript levels for clock genes were measured by qRT-PCR, including eight external RNA standards to allow absolute quantification. (a) LHY, (b) CCA1, (c) PRR9, (d) PRR7, (e), PRR5, (f), TOC1, (g) LUX, (h) GI, (i) ELF3, (j) ELF4. The results are the mean of duplicate samples. Error bars show SD.
2.3. Similarity and specific variations of wild-type RNA profiles across datasets
Clock gene RNA expression profiles in WT plants of two accessions (Col and Ws-2) grown in LD are presented in figure 2; profiles were similar across the TiMet and ROBuST datasets, despite major differences in growth conditions. The morning clock components, CCA1 and LHY, peaked as expected at dawn (figure 2a,b), followed by PRR7 (ZT6; figure 2c,d), PRR5 and GI (ZT8; figure 2e–h). Expression of the evening components, LUX, ELF4 and TOC1, peaked at ZT8–12 (figure 2e–j); peak expression of LUX was delayed by about 2 h in Col plants relative to Ws-2 in both datasets (figure 2g,h; replicated in LL data). ELF3 had a low-amplitude profile in both datasets, with lowest expression around ZT4.
The TiMet and ROBuST datasets differed at particular timepoints for PRR9, GI and TOC1. PRR9 expression was highest at ZT2–6 in both cases, with a clear peak at ZT2 in the ROBuST seedling data (consistent with many other reports from seedlings) but a broader profile in the TiMet data (figure 2c,d). After its major peak at ZT8–12, TOC1 expression is consistently observed (since [36]) to increase around ZT18, but the level of this night-time peak varied (figure 2e,f). The ROBuST data for seedlings showed a peak of GI expression at ZT2 (figure 2f); little induction is evident at ZT2 in the TiMet rosette data on a linear scale (figure 2e) though the logarithmic scale reveals the response (electronic supplementary material, figure S3e). The morning peak in GI is likely to be an acute response to lights-on. Rapid sampling in the Southern data [35] and in a follow-up microarray study [10] suggested that induction is rapid but transient, and therefore sensitive to sampling time. Nonetheless, the data suggest that either the magnitude or kinetics of light responsiveness vary across the conditions tested. The difference in PRR9 profiles could reflect slower activation of PRR9 in the TiMet data, consistent with lower light responsiveness in rosettes than in seedlings or with faster repression of PRR9 in seedlings. The level of GI transcripts at ZT12 also varied from 4% to 40% of the peak level, with the lowest level in rosettes of Ws-2 (figures 2e,f and 3c). GI expression is light sensitive at this phase [37], so our results are consistent with variation in light responsiveness.
Sucrose modestly increases expression of the evening clock components TOC1 and GI [38], particularly in darkness [39], and can repress PRR7 with subsequent effects on CCA1 under low light [40], along with transcriptome-wide effects under LD cycles [41,42]. We therefore compared the expression profiles for CCA1, TOC1 and GI in plants grown without (ROBuST and TiMet data) or with exogenous sucrose (McWatters, Edwards and Southern datasets; figure 3). To facilitate comparison, TiMet data were normalized to control transcripts (two amplicons each in GAPDH and ACTIN2), as for the other studies. Each profile was normalized to its maximum. Expression profiles of CCA1 across the different timeseries matched closely despite the differences in accession and experimental protocols (figure 3a). The times of peak, mid-rising and mid-falling phases differed by at most 2 h (one sampling interval) among datasets. In the falling phase at ZT4, the profiles in McWatters, TiMet ros and TiMet sd2 data were delayed relative to the other data. The night-time expression of TOC1 at ZT18 varied from 20% to 60% of the main peak level (figure 3b), with high expression in ROBuST, Edwards and TiMet sd2 datasets. The expression of GI at ZT2 in the TiMet and Edwards seedling data was about 20% of the main peak level (figure 3c, also in Southern data [35]), intermediate between the levels in ROBuST and TiMet rosette data (discussed above). These features of the expression profiles showed no clear relationship with growth medium or developmental stage.
2.4. Absolute quantification of clock gene transcripts
The absolute quantification in the TiMet ros data, which is based ultimately upon the certified amounts of synthetic commercial standards [33], revealed wide variation in peak RNA levels among clock genes in WT plants (figure 4). Highest RNA levels were detected for LHY at 1000–2100 copies per cell, similar to the control genes GAPDH and ACT2. PRR9 was least abundant at the peak, with 40–70 copies per cell; LUX and ELF3 peaked at 105–130 copies per cell; PRR7, PRR5, GI and TOC1 at 120–270 copies per cell; ELF4 and CCA1 at 250–600 copies per cell. RNA copy number of LHY was threefold greater than that of CCA1 (figure 4a,b).
Peak levels for the evening-expressed genes (figure 4f–j) were slightly higher in Ws-2 than Col-0 plants, by 1.2-fold (LUX) to 2.0-fold (ELF4), average 1.6-fold. Several clock gene RNAs fell to low copy numbers per cell at the trough. Consequently, rhythmic amplitudes (defined here as peak divided by trough levels) also varied greatly among clock genes. The TOC1 and ELF3 profiles showed only eight- to 20-fold amplitude in Col-0, and generally smaller amplitudes in other, mutant genotypes than the other clock genes (figure 4f,i), whereas LHY, CCA1, GI, ELF4 and PRR5 RNAs showed over 100-fold amplitude. This distinction was consistent in other datasets [21,34]. Amplitude estimates can be significantly affected by variation in the very low trough levels, which were higher in the TiMet sd1 dataset relative to the TiMet rosette data for LHY and all the evening-expressed genes in the Ws-2 accession, for example (figure 4). Transcripts with high-amplitude profiles might be expected to control circadian timing more effectively than the low-amplitude profiles of TOC1 and ELF3.
2.5. Regulation of clock genes under environmental and genetic manipulation
The TiMet project measured clock gene expression in LL and DD following LD entrainment, in seedlings of two WT and four clock mutant backgrounds (figure 5), revealing novel aspects of clock gene regulation as well as replicating regulation observed in many earlier, smaller studies. The results are discussed below with respect to the upstream regulators of each gene, rather than the effect on the gene's downstream targets. The RNA data are therefore presented in semi-logarithmic plots that show regulator activity even at low RNA levels.
Figure 5.


Clock gene expression in wild-type plants and clock mutants in LD, and after transition to constant light (LL) or darkness (DD). Col-0 and Ws-2 WT, the lhy-21 cca1-11 and prr7-3 prr9-1 double mutants, and the toc1-101 and gi-201 single mutants were grown in a 12 h photoperiod for 20 days, harvested through a LD cycle and then transferred to LL (a–j) or DD (k–t; TiMet ros, dataset 2 of figure 1c). Transcript levels for clock genes were measured by qRT-PCR, including eight external RNA standards to allow absolute quantification. (a,k) LHY, (b,l) CCA1, (c,m) PRR9, (d,n) PRR7, (e,o), PRR5, (f,p), TOC1, (g,q) LUX, (h,r) GI, (i,s) ELF3, (j,t) ELF4. The results are the mean of duplicate samples, error bars show SD. Open box, light interval; black box, dark interval; light grey box, predicted darkness in LL; dark grey box, predicted light in DD.
Comparing the three environmental conditions, peak RNA expression levels tended to fall in LL, consistent with the loss of dark-dependent regulation. The acute gene induction at the dark–light transition, faster degradation of PRR repressors in darkness and of the EC in the light are all expected to enhance rhythmic amplitude in LD. Expression levels of the clock RNAs were maintained in the first cycle in DD, except for the strongly light-regulated ELF4 [43,44]. Comparing the six genotypes, mutations that removed the repressors revealed the key connections in the clock circuit (figure 1a). The gi mutation, in contrast, had small or negligible effects on the timing and levels of expression except for PRR5, as noted below.
2.5.1. LHY and CCA1
Our results are consistent with PRR repressors controlling both the rising and falling phases of LHY and CCA1 expression at the transcriptional level [14,16–18,45]; several observations suggest that this activity is light-dependent. Both transcripts retain strikingly higher expression in the prr7;prr9 double mutant than in the WT, at ZT6–12 in LD and LL (figure 5a,b; p < 0.05; 20- to 30-fold higher at ZT8), consistent with the absence of repression from PRR9 and PRR7 proteins. By the second day in LL, the trough of LHY and CCA1 expression at ZT44 (68 h in figure 5) was also 20-fold higher than the WT trough level at ZT36–38 (60–62 h). Comparing LD and LL data with DD conditions revealed broader peaks of LHY and CCA1 RNA in DD (figure 5k,l), consistent with slower degradation of these transcripts in darkness [34,46]. In darkness, however, LHY and CCA1 levels in the prr7;prr9 mutant behaved very similarly to the WT, both during the falling phase in DD (ZT28–38; figure 5k,l) and during the rising phase in LD (ZT16–22; figure 5a,b). By dawn in LD, both transcripts peaked at the WT level, consistent with previous reports [12,13]. Thus, the misregulation of LHY and CCA1 in the light in the prr7;prr9 double mutant was abolished during the dark in LD.
Removing TOC1, the last of the PRR repressors to be expressed, would be expected to allow an earlier rise in expression of LHY and CCA1 during the night in the toc1 mutant under LD. This effect was relatively small (two- to 2.5-fold higher at ZT18, p = 0.02). LHY and CCA1 levels in toc1 mutants differed less than fourfold from WT at any point in LD. The mutant phenotype was not enhanced in the first DD cycle (figure 5k,l). In LL, however, LHY and CCA1 expression in the toc1 mutant peaked at ZT22 (46 h in figure 5) rather than at ZT26 (50 h) in Col, reached only 30–50% of WT peak level consistent with earlier data [47], and fell much earlier than the WT (19- to 27-fold lower at ZT30, time 54 h in figure 5a,b). Thus, the molecular phenotypes of both prr7;prr9 and toc1 mutants were light-dependent.
The elf3 mutant reduced peak expression of both LHY and CCA1 by five- to 10-fold (figure 6a,b; electronic supplementary material, figure S5g), with greatest effects at ZT20–24. This effect is thought to be indirect, as the EC (comprising ELF4, ELF3 and LUX) is proposed to repress the PRRs (figure 1a), as well as LUX and ELF4 [10,19,20,22]. De-repression of PRR expression in mutants of the EC should therefore explain the effects of elf3 on LHY and CCA1.
2.5.2. PRR9 and PRR7
PRR7 was the most severely affected gene in the elf3 mutant under LD, maintaining 25–85% of the WT peak level at all times (figure 6d), consistent with de-repression of the PRR7 promoter [21]. The resulting, 30- to 50-fold overexpression of PRR7 in elf3 at ZT20–24 is consistent with reduced expression of LHY and CCA1 at this time. PRR9 transcript levels retained a 100-fold rhythmic amplitude under LD in the elf3 mutant, indicative of ELF3-independent regulation (see Discussion). Nonetheless, PRR9 expression was also de-repressed from ZT10 in elf3 (p = 0.05), rising 2–4 h before dawn (figure 6c), and presumably also contributing to reduce LHY and CCA1 expression.
The early-expressed PRRs are thought to be repressed by the later-expressed PRR5 and TOC1 (figure 1a). The toc1 mutation had modest effects on PRR9 or PRR7 profiles under LD cycles (figure 5c,d), though the changes observed (such as an early rise in PRR7 at ZT20–24) were not consistently significant in the TiMet and ROBuST datasets, or in DD in the TiMet data (figure 5n). toc1 also had little effect on LHY and CCA1 levels in these conditions (figure 5a,b). In LL, however, removing TOC1 prevented full repression of the PRRs. The trough of PRR7 expression was at a 10-fold higher level than in the WT (p < 0.05) and 8 h earlier (ZT12 rather than ZT20, 36 h rather than 44 h in figure 5d). Higher expression of the repressor PRR7 at 38–52 h (figure 5d) is consistent with the lower peak expression of CCA1 and LHY in toc1 under LL (figure 1a [9]). Taken together, these results suggested that TOC1 repressor function was most effective under constant light conditions, where the toc1 mutant was originally identified [48].
Light-dependent regulation was also evident in WT plants. Peak PRR9 expression levels fell less than twofold in the first cycle of DD (p > 0.16; figure 5m). Peak PRR7 expression tended to increase (threefold or less) in all genotypes in DD (figure 5n; electronic supplementary material, figure S4c). The gi mutant was an exception, which slowed the rise of all the transcripts in DD except ELF3 and ELF4 (figure 5k–t; electronic supplementary material, figure S5d). Peak expression for some genes was reduced in gi below WT levels, including PRR7 (p = 0.02–0.03 at ZT26–28 h). Trough RNA levels in the WT plants rose more dramatically in DD, for PRR7 and other clock genes (except for LHY): the lowest expression of PRR7 in Col was 1.5 ± 0.4 copies per cell at ZT20 but 65 ± 6.8 copies per cell at ZT40 (electronic supplementary material, figure S4c). The Edwards dataset showed similar de-repression of CCA1 and GI trough levels in DD (electronic supplementary material, figure S4a,b [34]). Lastly, we tested the effect of CCA1 and LHY on the PRR transcripts, using the lhy;cca1 double mutant. In the WT, the repression of the evening-expressed genes by LHY and CCA1 in the early day delays the expression of these and other target genes until the evening. The double mutation advanced the peak phase of all the other clock genes to ZT2–4, as expected, except for ELF3 (see below). Despite the de-repression, peak levels were not consistently increased relative to the Ws-2 control. Peak expression of PRR9, PRR7 and PRR5 (figure 5c–e) was slightly reduced (up to twofold) in the lhy;cca1 background under LD, consistent with earlier results [12]. By ZT8 (or ZT4 for PRR9), all the clock genes were expressed at lower levels in lhy;cca1 than in the WT (p < 0.01–0.04), consistent with expression of all the PRR repressors. In the lhy;cca1 double mutant in DD, however, the PRR genes had broad peaks that rose earlier than in the WT (ZT22–30) but did not fall earlier (ZT34–40; figure 5m–o; electronic supplementary material, figure S4d). The absence of early repression in DD again suggests that inter-regulation of the PRRs is light-dependent.
2.5.3. PRR5 and TOC1
The later-expressed PRRs are repressed by LHY and CCA1, so longer expression of LHY and CCA1 in the prr7;prr9 double mutants delayed their expression in LD and LL conditions (figure 5e,f), as expected. In contrast, under DD conditions, PRR5 expression in prr7;prr9 rose indistinguishably from the WT at ZT26–34 h and peaked slightly (twofold) above the WT level (figure 5o). The lhy;cca1 double mutant caused the phase advance noted above, as the loss of LHY and CCA1 repressors increased TOC1 levels in the early day. Peak TOC1 RNA levels in the lhy;cca1 mutant did not change consistently from WT levels in the TiMet data under LD (figure 5f), and were lower than the WT in the ROBuST dataset (p < 0.01; electronic supplementary material, figure S5c).
Our detailed datasets also allowed us to compare expression waveforms. For example, PRR5 rises and falls 10-fold within 5 h in both TiMet and ROBuST data (figure 5e,o; electronic supplementary material, figures S3g,h, S5a). This narrow peak indicates highly nonlinear control, consistent with negative autoregulation and/or inhibition by TOC1 [15,49]. Moreover, our results indicate that this PRR5 waveform depends upon GI function. The gi-201 mutant had limited effects overall but slowed the fall in PRR5 mRNA in LD and LL (figure 5e), creating an asymmetric profile in PRR5 RNA that was also observed in the gi-11 tested in the ROBuST data (electronic supplementary material, figure S5a,b). Repression by the EC might also contribute to the falling phase of the PRR5 profile. Removing this repression in the elf3 mutant resulted in moderate de-repression of PRR5 and TOC1 in the late night (p < 0.01, ZT0/24; p = 0.01 for PRR5 ZT22) and potentially in the early morning (p = 0.06–0.08; ZT2–4; figure 6e,f). In contrast, de-repression of the early PRRs in elf3 was greatest in the early night (see above), indicating that the profile of regulators varies among the PRR family members (see Discussion).
2.5.4. GI
The main peak of GI expression in the late day behaves similarly to PRR5, with delayed expression in the prr7;prr9 double mutant owing to longer expression of LHY and CCA1 under LD and LL but not DD, and an advanced phase in the lhy;cca1 double mutant (figure 5h,p). In contrast to PRR5 but similarly to PRR9 and PRR7, GI was de-repressed from ZT10 in the elf3 mutant (p < 0.01), consistent with [21] and the Southern dataset (electronic supplementary material, figure S5f). The Southern dataset showed that the expression of GI was similar in elf3 and elf4 mutants, but there was much less effect on CCA1 in elf4 than elf3 (electronic supplementary material, figure S5g), indicating that the effects of the EC components can be distinct.
2.5.5. ELF3
The ELF3 rhythmic profile has low amplitude, as noted above, with a trough at ZT2–4 and peaks at both ZT8 and ZT18–20 in WT plants under LD in the TiMet and ROBuST datasets (figure 5i,s; electronic supplementary material, figures S3i–j, S5d). The trough of ELF3 expression is de-repressed at ZT4 in the lhy;cca1 double mutant (p < 0.01), though there is no peak at this time, in contrast to all the other clock genes. The rise in ELF3 expression is delayed in the prr7;prr9 double mutant (p < 0.01–0.05, at ZT6–10), consistent with repression by increased levels of LHY and CCA1 (figure 5i). The elf3–4 allele contains a small deletion in the coding region [50] and accumulates the mutant RNA. The mutant expression profile suggests de-repression at ZT2 (p = 0.06; figure 6i), consistent with lower expression of LHY and CCA1 in elf3 (noted above).
2.5.6. ELF4 and LUX
The two remaining EC components tested, ELF4 and LUX, share the evening expression peak determined by LHY/CCA1-mediated repression, with a phase advance in lhy;cca1 and a delay in prr7;prr9 in LD and LL conditions (figure 5g,j). Strikingly, however, the phase separation among the clock genes was lost in the lhy;cca1 double mutant under LL, such that PRR9 and ELF4 peaked together at 50 and 66 h (discussed below). Thus, LHY and CCA1 contribute to the 4 h separation of peak times between PRR9 (54 h) and ELF4 (58 h) in the Ws-2 control under LL. In DD, peak expression of ELF4 was the most reduced of all the genes, to less than 10% of the LD peak level (p < 0.01 in Col and Ws; figure 5t), consistent with the loss of light activation [44] and/or sugar signalling. ELF4 was also de-repressed earlier in the toc1 mutant under DD than the other genes (ZT28–36 h; figure 5j), rising as early as in the lhy;cca1 double mutant. Under LD conditions, the toc1 mutant de-repressed ELF4 at ZT2–6, earlier than WT. Peak expression of LUX did not fall significantly in DD (figure 5q).
LUX was broadly de-repressed in the elf3 mutant, remaining at the WT peak level at ZT6–22 h (figure 6g), in a similar pattern to PRR7. This result is consistent with LUX binding to its cognate promoter [20] resulting in negative autoregulation (figure 1a [10]). ELF4 expression in the elf3 mutant, in contrast, showed a pattern more similar to TOC1 and PRR5 (see above), with de-repression only from ZT22–ZT6 h (figure 6j).
2.6. Alternative visualization gives new insights into co-regulation of clock genes
Data visualization is critical in analysing the complex interactions within the clock gene circuit, in order to generate new hypotheses. Timeseries plots do not show these interactions directly. They can be revealed in phase plane diagrams that plot the levels of two components against each other (figure 7), though this format is less familiar (see electronic supplementary material). First, phase plane plots emphasize the relative timing of clock components, rather than control by the light : dark cycle. For example, GI rose without (before) TOC1, especially in Col plants of the TiMet and ROBuST datasets that were grown without exogenous sucrose. High TOC1 levels extended later than high GI, particularly in Ws-2 plants of the TiMet datasets (figure 7a). Second, this visualization can reveal interactions among the components plotted. For example, figure 7b shows TOC1 RNA levels in younger plants were maintained at 35–55% of the peak level at ZT20–22, when CCA1 expression rose above 50% of its peak level. TOC1 levels were lower for the same CCA1 level in rosette plants. The logarithmic scale shows this more clearly (figure 7c). This suggests that CCA1 protein is not yet an effective repressor of TOC1 at this phase, especially in younger tissues.
Figure 7.

Phase plane diagrams reveal pairwise gene interactions. (a–c) Normalized RNA profiles of figure 3 are represented as phase plane diagrams, plotting (a) GI and TOC1, and TOC1 and CCA1 on (b) linear and (c) logarithmic scales. Larger markers indicate ZT0 datapoint, arrows indicate the direction of time. (d–f) RNA profiles of figure 5 are represented as phase plane diagrams on logarithmic scales, plotting data for ELF4 and PRR9 (d) in wild-type Col plants under LD and LL (0–22 h in figure 5, dashed line; 24–70 h, solid line), and (e) in Col plants under LD and lhy cca1 double mutants under LD and LL (solid blue line), with (f) a rescaled view of a subset of the data from the lhy cca1 double mutants. Larger markers indicate 0 (ZT0) and 12 h (ZT12) datapoints in the cycle labelled LD. These timepoints are equivalent to 24 (ZT0) and 36 h (ZT12) in the cycle labelled LL. Arrows indicate the direction of time. (d) Red dashed line marks falling ELF4 levels during the night-time trough of PRR9 in LD. (f) Red dashed line marks correlated PRR9 and ELF4 levels; arrowheads mark an earlier peak on each cycle in PRR9. Timepoints 48 (ZT24) to 70 h (ZT46) under LL are plotted in brown to emphasize the similar profiles on successive days.
Finally, the phase plane diagrams can show how the interaction of two genes depends upon a third regulator. Expression peaks of PRR9 and ELF4 were far out of phase in the WT (figure 7d), for example. Data from LL (filled symbols) suggest a negative correlation in the subjective night, when ELF4 falls as PRR9 rises. However, the two genes peak then fall together in the lhy cca1 double mutant under LL, at ZT26 and ZT42 (figure 7e; equivalent to timepoints 50 and 66 h in figure 5), creating a diagonal with a positive gradient (red dashed line, figure 7f). PRR9 also had an earlier peak that was not shared by ELF4 (ZT22 and ZT38, or 46 and 62 h in figure 5; black arrowheads in figure 7f). Both features were reproduced on two successive cycles, though PRR9 expression was less than 1% of the WT peak level. Thus LHY, CCA1 and the LD cycle all differentiate PRR9 expression from ELF4, but in their absence, PRR9 and ELF4 expression profiles are similar for much of the circadian cycle (six of eight timepoints in the short, 16 h cycle of the mutant), presumably controlled by the other PRRs and/or the EC. Likewise, phase plane diagrams for the prr7;prr9 double mutant (electronic supplementary material, figure S6) suggested that not only CCA1 and LHY, but also the PRRs repress ELF4 in the WT. In addition to visualization, many other aspects of data management benefit significantly from online data infrastructure.
2.7. Online infrastructure for data sharing
Our open-source BioDare (Biological Data repository) [51] supports data from many small-scale experiments that collectively represent a significant resource (table 1). Empirical evidence indicates that these data are essential to understand complex biological regulation, and mathematical analysis shows why this is the case (see Discussion). In addition to six rhythm-analysis algorithms [52] and protocols for analysis, statistical summary and visualization [53], BioDare facilitates data sharing and public dissemination by providing a stable identifier for each experiment. Detailed metadata (experimental description) ensure that the data can be reused appropriately. Results can be compared across studies and laboratories (‘data aggregation’) by searching the metadata for genotype, marker gene and other terms (figure 8). Increased expression of GI in the elf3 mutant, for example, is highlighted despite the greater technical variability of manual assay preparation in the Southern dataset compared with the later, robotized assays in the TiMet data (figure 6h; electronic supplementary material, figure S5f and Methods).
Table 1.
Usage statistics of BioDare (Feb 2015), from originating groups and selected external users. An experiment represents a dataset similar to one of the above-described studies, which includes multiple timeseries, from samples of multiple genotypes, assays or reporters and/or environmental conditions. Totals include minor users that are not listed individually; the total number of data points is over 41 million.
| research group | location | experiments | % total experiments | timeseries | % total timeseries |
|---|---|---|---|---|---|
| A. J. Millar | Edinburgh, UK | 332 | 14 | 41 890 | 18 |
| A. Hall | Liverpool, UK | 261 | 11 | 79 228 | 34 |
| D. Bell-Pedersen | Texas A&M, USA | 138 | 6 | 1428 | 1 |
| J. Agren | Uppsala, Sweden | 18 | 1 | 9370 | 4 |
| K .J. Halliday | Edinburgh, UK | 230 | 10 | 5043 | 2 |
| L. Larrondo | Santiago, Chile | 75 | 3 | 6429 | 3 |
| M. Jones | Essex, UK | 89 | 4 | 3148 | 1 |
| M. Hastings | MRC LMB, UK | 1071 | 45 | 58 770 | 25 |
| S. Harmer | UC Davis, USA | 37 | 2 | 11 353 | 5 |
| S. A. Kay | USC, USA | 38 | 2 | 12 972 | 6 |
| All BioDare | 2344 | 232 844 |
Figure 8.
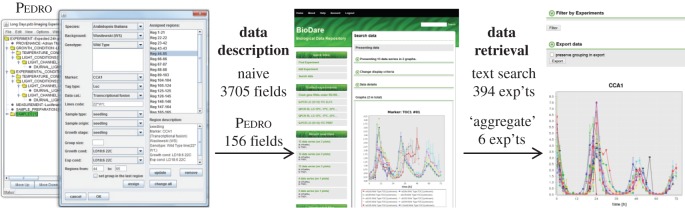
Computational infrastructure for systems chronobiology. Customized wizards in the Pedro XML editor capture detailed metadata (right panel, showing CCA1 : LUC in sample wizard). Rather than filling 3705 metadata fields for this experiment, as a naive spreadsheet would require, Pedro captures the information with only 156 entries. After uploading the metadata and numerical data to BioDare, results can be displayed in the web browser (centre panel) with powerful secondary processing functions. The left-hand sidebar in this screen has shortcuts to common tasks and recent activity. A naive text search for ‘CCA1’ returned 394 experiments (exp'ts), whereas BioDare's ‘aggregate’ function retrieved six specific results by searching the structured metadata, with secondary filters. The search shown (right panel) aggregated qPCR assays of CCA1 in wild-type plants (see main text) including datasets 1, 3, 4 and 6 of figure 1c. The export button above the graph downloads the data shown to a spreadsheet-compatible file.
2.8. Optimizing clock models with public resources
One goal of such comparisons is to determine how much of the available data is matched by a particular mathematical model: the ROBuST and TiMet experiments were designed to test models of the clock gene circuit under different growth conditions. However, testing complex models against large datasets requires skills that are rare among plant molecular researchers. We therefore tested whether our comprehensive data and better computational resources could make modelling more accessible. The open-source SBSI allows non-programmers to optimize model parameters in order to match diverse data, on large, parallel computers [29]. As a test case, we addressed a recognized limitation of the original P2011 model [10], termed P2011.1.1. The model was developed to understand circadian clock function under light–dark cycles and, separately, under constant light. Following a transition from LD to LL (as in figure 5a–j), the first peak in expression of the combined LHY/CCA1 component under constant light occurred at ZT28.4 h (52.4 h in figure 7a), about 2.5 h later than in the TiMet ros data (as noted [25,54]). The model's light–dark function was replaced with the input signal step function [55] to represent the LD–LL transition in the community model exchange format, SBML [56]. The resulting model P2011.1.2 was optimized in SBSI (see electronic supplementary material), testing model simulations with many alternative parameter sets against the TiMet ros RNA dataset, including the LD–LL transition (figure 5a–j), and against circadian period values for clock mutants and WT plants [29].
The optimized parameter set of model P2011.2.1 more closely matched the data, including an earlier peak of LHY/CCA1 in LL at ZT26.5 h (figure 9a) and a closer match to TOC1 and GI profiles in LD (ZT10–12 h; figure 9b,c), while retaining other qualitative behaviours. LHY/CCA1 expression rises in LL after the PRR repressor proteins are degraded. Consistent with this notion, removing TOC1, the last gene in the PRR repressor wave, advanced the phase of the entire clock mechanism in LL. Results for PRR7 are shown in figure 9d,e. PRR protein degradation rates were not strongly affected in P2011.2.1; rather, overall PRR levels were lower than in P2011.1.2 (not shown). In the simulated toc1 mutant, the peak of LHY/CCA1 was 1.4 h earlier than simulated WT in P2011.1.2, 2.5 h earlier in P2011.2.1, but 4 h earlier in the data (figure 5a,b). The simulations of PRR7 show the same improved timing of the new model for the WT (figure 9d) and the toc1 mutant in LD (figure 9e), but an earlier phase of the toc1 mutant data under LL. Regulatory interactions among the PRR genes will repay further analysis [9,10] in future models (see Discussion).
Figure 9.

Model re-optimization. Comparison of measured transcript levels from figure 5 (experimental data, symbols), with simulation of models P2011.1.2 (old model, dotted line) and P2011.2.1 (new model, solid line), which resulted from fitting to these data using SBSI. 0–24 h, LD; 24–72 h, LL. (a) LHY and CCA1 transcripts are combined in the model, so the average of LHY and CCA1 data is plotted. The peak of LHY/CCA1 under LL was delayed in the P2011.1.2 model (52.4 h) relative to the peak in the data (50 h), which was closely matched by the P2011.2.1 model (50.5 h). (b) GI transcript, (c) TOC1 transcript and (d) PRR7 transcript in Col-0 WT. (e) PRR7 transcript in the toc1 mutant shows a greater phase-advance in LL than either model. Chi-square cost value for match to TiMet ros Col-0 data in LD-LL was 20.2 for P2011.1.2, 7.6 for P2011.2.1. Chi-square cost for match to TiMet ros toc1 data in LD-LL was 39.7 for P2011.1.2, 13.1 for P2011.2.1.
The computation time required for P2011.2.1 was only approximately 30 core-hours, because the model parameters were varied within only a narrow range (two- to threefold change) from their starting values in P2011.1.2 [10]. The P2011.1.2 parameters had been manually determined to match a wide range of data and qualitative behaviours in the clock literature; many were derived from the parent model P2010 [14]. When the first model of a system is developed, in contrast, most or all parameter values may be unknown. We therefore tested our approach in such scenarios (table 2). Allowing parameter values to differ by up to 100-fold from the values in P2011.1.2 created a very large parameter space that was nonetheless centred on a known, viable region. In contrast, starting parameters from nominal values (0.1, 1, etc.) and testing each parameter over the same range (such as 0.001–10) removed that anchor. Viable parameter sets that gave cost values similar to the unmodified P2011.2.1 were identified in each test, with computation times up to four core years for P2011.6.1, using the UK national supercomputing resource HECToR. These parameter sets are not intended to replace P2011.1.2 but to demonstrate that similar results can be achieved by a more accessible approach using the TiMet data and SBSI, without new programming or laborious, manual model development. The P2011 model versions and the cognate graphical network diagram (electronic supplementary material, figure S2) are publicly accessible from the PlaSMo repository and elsewhere (see appendix A).
Table 2.
Optimization of model parameters from loose constraints. The starting P2011.1.2 model was optimized in SBSI to fit the TiMet ros dataset and additional period constraints (see electronic supplementary material, Methods). Model, version number of the resulting model. PlaSMo ID, model identifier in the PlaSMo resource. Job, computational job code. Start, the default parameters values from P2011.1.2 or nominal values (Nom). Range, the range of parameter values that were searched, either as fold change above and below the P2011.1.2 values or as a fixed range. Set-up trials, the number of randomly chosen parameter sets tested to initialize the optimization. Cost, the best cost value (closest fit to all constraints).
| model | PlaSMo ID | internal job ID | start | range | set-up trials | cost |
|---|---|---|---|---|---|---|
| P2011.1.2 | PLM_71 ver 1 | — | — | — | — | 171 |
| P2011.2.1 | PLM_71 ver 2 | .599 | P2011.1.2 | 2–3× | 5000 | 77 |
| P2011.3.1 | PLM_1041 ver 1 | t30 | P2011.1.2 | 100× | 2 097 152 | 175 |
| P2011.4.1 | PLM_1042 ver 1 | t34 | Nom | 0.001–10 | 67 108 864 | 270 |
| P2011.5.1 | PLM_1043 ver 1 | t37 | Nom | 0.001–10 | 67 108 864 | 190 |
| P2011.6.1 | PLM_1044 ver 1 | t40 | Nom | 0.0005–20 | 134 217 728 | 185 |
3. Discussion
3.1. Robust regulation of clock gene expression
Quantitative timeseries data are crucial to understand the dynamics of any moderately complex regulatory system. As understanding advances, more precise questions can be formulated that demand both consistent and comprehensive datasets. We provide such data for the RNA profiles of genes associated with the Arabidopsis circadian clock, with an online resource to facilitate comparisons within and across datasets. Our experiments were designed to test clock function under the distinct conditions required for separate studies, on light signalling (in the ROBuST project) and carbon metabolism (in the TiMet project), using different technical platforms. The results presumably include the variation previously observed among experiments designed to be replicated across laboratories [57]. We compared two Arabidopsis accessions. Significant differences in circadian timing have been demonstrated among Arabidopsis accessions, albeit using long-term, imaging assays that integrate the effects of small timing changes over many cycles [58–60]. Importantly, the rhythmic RNA profiles tested here were remarkably consistent (figure 3). Progress in understanding the clock gene network must, in part, be attributed to this robustness of circadian regulation.
Several clock genes are regulated with high daily amplitude, more than 100-fold for LHY, CCA1, GI, ELF4 and PRR5 under LD (figure 4; electronic supplementary material, figures S3 and S4 [21,34]), falling to low RNA copy numbers per cell. Our data necessarily reflect the mean expression across cells in the rosette, greater than 80% of which are in the leaf mesophyll [61]. Nonetheless, the absolute calibration of our RNA assays provides one approach for future estimation of the average copy number for the cognate proteins.
The most striking variations of RNA profiles among WT plants involved the acutely light-responsive genes GI and PRR9. The ROBuST dataset showed the highest levels of GI and strong induction of PRR9 at ZT2 (figures 2 and 3). This is consistent with strong light induction, which might be mediated by direct photoreceptor signalling and/or by indirect sugar signalling. The absence of exogenous sucrose in the ROBuST conditions was not the sole cause, as the TiMet sd2 data used the same, sucrose-free media but did not show such strong GI induction (figure 3c). The lower growth temperature in ROBuST conditions (17°C rather than 20–22°C in other datasets) might also increase light responsiveness. Consistent with this notion, both exogenous sucrose and higher ambient temperature limit other light responses [30,31].
3.2. Regulation of the PRR repressors
RNA profiles of the PRR gene family varied among datasets in the WT under LD, as well as among conditions and genotypes. The variable expression of TOC1 around ZT18 (figure 3b) awaits a mechanistic explanation, as do the de-repression of multiple genes in DD (for example, figure 5n) and of PRR5 in the gi mutant (figure 5e; electronic supplementary material, figure S5b). TOC1 is thought to be an active repressor at ZT18, so variable auto-repression is possible and might also explain variation in PRR5 expression at this phase (figure 2g). Alternatively, TOC1 expression might rise during a transition between one repressor in the early night (such as the EC) and another in the late night (such as LHY and CCA1).
The tight interconnections among the clock components complicate the analysis of these data, though the resulting combination of direct and indirect effects is now interpretable. For example, removing EC regulation in the elf3 mutant de-repressed the direct EC targets PRR9 and PRR7 in the early night, when the EC is active in WT plants. PRR5 and TOC1 were noted as potential targets based on mutant RNA profiles [10], but both genes were de-repressed around dawn in elf3, suggesting that an indirect mechanism owing to lower LHY and CCA1 levels is more significant than the loss of direct regulation by the EC in the mutant. PRR9 and PRR7 are both proposed EC targets (along with ELF4 and LUX), yet PRR9 (and ELF4) retains rhythmic regulation in the elf3 mutant under LD, whereas PRR7 (and LUX) is more severely affected (figure 6). To understand such differences in response, it will now be important to measure the affinity of regulators for their target genes, extending initial data [62]. Previous modelling results indicated that the different daily profiles of the PRR genes allow flexible responses to dawn and dusk [14], so the mechanisms that generate the PRR profiles will repay further analysis [10,11].
Several results suggested that regulation by the PRR genes is light-dependent. First, in the prr7;prr9 double mutant, LHY and CCA1 expression was de-repressed during the day but returned to match the WT profile at night in LD (figure 5). One explanation might be that PRR9 and PRR7 (directly or indirectly) antagonize the light activation of LHY and CCA1 during the day in the WT [14,63], and the absence of these PRR proteins in the double mutant has little effect in darkness. Consistent with this notion, the prr9 single mutant also showed a day-time de-repression of CCA1 in the ROBuST dataset (electronic supplementary material, figure S5e), albeit less than in the double mutant. However, the CCA1 profile in the prr7 single mutant was unaffected in the daytime, but de-repressed 2 h earlier in the night (electronic supplementary material, figure S5e). Thus, inter-regulation of the early PRR genes is important, in addition to regulation by TOC1 [10]. Second, in the lhy;cca1 double mutant, PRR gene expression is repressed to low levels at the end of the day in LD, consistent with simultaneous, early expression of all the PRR repressors in these mutant plants. In DD, however, the falling phase of PRR expression is the same in WT and double mutant plants (figure 5). The higher and earlier expression of the PRR RNAs in the double mutant in DD does not appear to be effective in suppressing PRR expression. The faster degradation of the PRR proteins in darkness presumably contributes to these effects; it will be interesting to determine whether the interaction of the photoreceptor PHYB with clock proteins (including TOC1 [64]) also mediates the light sensitivity of this process.
3.3. Effects of exogenous sucrose
Current models of the Arabidopsis circadian clock are necessarily based on disparate data, much of it derived from seedlings grown on media containing high levels of sucrose. The presence or absence of exogenous sucrose under the conditions tested here affected the clock RNA profiles less, or at least no more, than other experimental parameters, despite the widespread regulation of plant genes by sucrose [41,65]. Consistent with this, effects of exogenous sucrose on clock gene expression in WT plants have previously been reported under very low light fluence rates or in the presence of photosynthetic inhibitors [40], in DD, CO2-free air or the starchless pgm mutant [39,66,67]. PRR7 was induced in sugar-starved conditions (extended DD and at night in pgm) and was repressed by resupply of 3% exogenous sucrose (electronic supplementary material, figure S7a). Only the TiMet rosette study tested PRR7 in DD, finding increased PRR7 levels (figure 5n), especially in the trough of the profile (electronic supplementary material, figure S4c). Trough levels of CCA1 and GI were also raised in DD in the TiMet data, and in the Edwards experiment that included 3% exogenous sucrose (electronic supplementary material, figure S3 [34]). De-repression of the trough levels in DD is neither specific to PRR7 nor to sugar limitation. Transcript levels of the TOC1- and PRR5-degrading F-box protein ZTL, and its homologues LKP2 and FKF1, also rose slightly in sugar-starved conditions (electronic supplementary material, figure S7b [67]), suggesting one possible mechanism for de-repression of PRR7 via faster degradation of PRR repressors.
3.4. Open resources for small-scale results
Our results will be useful to generate and test many hypotheses beyond those reported here. The potential for such future value might, in principle, justify the additional effort in curating and disseminating our data. In practice, future value motivated little data sharing, compared with present value. We therefore outline the mathematical understanding of and empirical evidence for such present value, together with practical steps that increased both present and future value relative to the effort involved in sharing data.
No suitable community repository existed for our results. One reason was the relatively large effort required to describe accurately many small data files, which deters researchers and resource developers from sharing such data [68]. The largest-scale omics and sequencing studies have different data structures, motivations, stakeholders and economics, which can facilitate data sharing [69] including exemplary resources in the circadian field [70–72]. However, mathematical analysis explains why the results of small-scale experiments are often particularly valuable in understanding biological systems. Gutenkunst et al. [73] showed that parameters were ‘sloppy’ in dynamic models of a range of biological regulatory systems, meaning that a wide range of parameter values could generate the simple behaviours that they tested. Rand et al. [74–76] tested how many parameter changes could affect the dynamic behaviour of such systems. All possible behaviours were tested and only a handful of behaviours could be readily achieved by changing parameters (these behaviours have also been termed the ‘dynatype’ of the system, by analogy to the phenotype of an organism [77]). For circadian clocks, a change in period was the most accessible behaviour: many different parameter changes altered period under constant conditions [74]. The related, empirical result is that genetic screens seeking mutants with altered circadian period have not only identified clock components, but also many genes that affect the clock less directly [78,79]. Observing a change in period gives little evidence for the role of the mutated gene in the plant and does not strongly constrain any particular parameter in the model, but rather has a small constraining effect upon a large number of parameters, in agreement with Gutenkunst et al. [73]. A measured period value can therefore easily be accommodated without fundamentally changing the model. In contrast, manipulating the system to test less accessible behaviours provides strong constraints, albeit potentially on fewer parameters [76]. It is much more likely that such results would not be accommodated by any reasonable parameter values, falsifying the current model and leading to new understanding during the development of a better model. Thus, the number of manipulations tested is crucial; model analysis can prioritize the most informative manipulations [80,81].
One consequence for experimental design is that the number of manipulations is more important than the number of components tested. This concept is familiar from the statistical clustering of microarray timeseries. The behaviour of a single cluster mean can adequately represent hundreds of individual transcripts, even for genes with complex light and circadian regulation [82]. The individual transcript data are more valuable in identifying coregulated, downstream genes than in understanding the clock system upstream. Thus, targeted qRT-PCR or reporter gene assays have been more widely used in understanding the clock gene circuit, although they lacked a data-sharing resource. Despite the limited justification for costly omic assays, targeted data-sharing resources [67,70] have ensured that a subset of transcriptomics data have been reused effectively in clock studies.
Empirical evidence for the value of multiple manipulations comes from 10 years of modelling the plant clock gene circuit and output pathways. Constraining the models with timeseries data from many conditions was a critical tool [83], resulting in multiple, experimentally validated predictions. Gleaning the data from electronic supplementary material or by ‘scraping’ numerical values from published charts made this possible. In practice, aggregating the numerical data has often taken a major effort, after which the data were shared on author web sites [25,84] or on BioDare [82,85,86].
BioDare [51] was developed to share timeseries data from relatively small-scale experiments conducted within individual laboratories (such as the Edwards, Southern and McWatters datasets) or in collaborative projects with few partners (such as ROBuST and TiMet). A regular user might upload an experiment with several hundred timeseries each week [87,88]. However, the user must also provide experimental metadata that are sufficiently detailed to pinpoint the most relevant experiment among hundreds to thousands of similar studies (table 1). The resource must therefore streamline the process of writing the structured metadata to minimize the weekly effort involved, and then use the metadata to provide powerful search functions, for later users to discover relevant data that were previously unknown to them. Figure 8 illustrates metadata capture using ‘wizard’ forms in BioDare, data aggregation based upon the resulting metadata, and visualization of the data from a small set of relevant experiments, whereas a naive text search returned an impractically large number of results.
The potential future value of shared data resulted in fewer than a dozen datasets being shared in the early phases of our projects. To provide immediate value from depositing data, BioDare therefore offers data processing (detrending, averaging) and visualization along with specialized circadian data analysis [52,53]. Stable identifier URLs conveniently direct collaborators to specific datasets and can be cited in publications [88,89]. The citations will be tracked by the Thomson Reuters Data Citation Index, giving a metric analogous to publication citations to recognize data-sharing contributions [90]. BioDare is available as a community resource that could be linked to organism-specific databases [91]. BioDare complements our repository of plant systems models (PlaSMo) [92].
3.5. From visualization to modelling
Our analysis here was model-assisted but manual, so data visualization was important. For example, phase-plane diagrams can reveal conditional pairwise interactions including subtle effects at low RNA levels, such as the correlation of PRR9 and ELF4 expression in the lhy cca1 double mutant under LL (figure 6d–f). In contrast, PRR9 and ELF4 expression are uncorrelated or anticorrelated in the WT under LD. Such changes in dynamics are important in forming hypotheses during model development. Expert modelling has a subjective element. Objective machine-learning methods can also to contribute to hypothesis generation [93], though understanding such a conditionally connected network (electronic supplementary material, figure S2) is challenging by any approach [94].
Dense transcriptional regulatory interactions might be general for plant environmental response pathways [95], justifying investment in infrastructure to support their analysis. Mathematical models can powerfully express hypotheses about such circuits, so long as the starting model adequately recapitulates most data. Qualitatively, the variation among our datasets was smaller than the departure of the model simulations from the data (figures 3 and 9). The existing circadian clock models are therefore equally applicable to the several growth conditions tested, at least in leaf tissue.
The transition from LD to LL is one case where the model departed from the data, to which it had not previously been constrained (also noted in references [54,88]). The P2011.2.1 model's 2 h late phase in LL (figure 9) is caused by the slower degradation of PRR proteins in the light than in the dark [16,45]. Without a dark night to reduce PRR levels, their slow degradation delays the rise in LHY/CCA1 on the first cycle in LL in the model. PRR9, PRR7 and PRR5 RNA levels are reduced in the second cycle in LL in both model and data (figures 5c–e and 9e), restoring an approximately 24 h period in subsequent cycles. It is reassuring but not surprising that re-optimization of the model could better match this behaviour, but the models' detailed behaviour is non-trivial. Reducing the levels of PRR proteins in the new parameter set advanced the phase of the first peak in LL. Simplified models that included only the PRR protein changes also reduced the effect of the PRRs on the period of the clock in constant light (data not shown), contradicting the data. The re-optimization allowed multiple parameter changes to advance the phase of the P2011.2.1 model under LL while retaining the observed effects of PRRs on clock period, such as the short period of the toc1 mutant (figure 9e).
Most significantly, this result was obtained using tools designed to be accessible to biological researchers with no specialist computing or mathematical skills. Development of P2011.2.1 required no new programming, nor the hand-crafted cost functions that were used to optimize previous models [25,83–85], nor the laborious, expert parameter exploration used to construct its parent models [10,14,96]. Our intention was that the scarcity of these skills should no longer present an insuperable barrier, though of course they remain beneficial, not least to keep abreast of relevant method development [80]. To test whether this approach could assist new model development, as well as adjustment of an existing model, we repeated the parameter search within a wide range of values and/or after setting P2011.1.2 model parameters to nominal values. Greater computational power is required when there are fewer constraints on the model's parameter values; however, viable solutions were identified (table 2) and suitable computing resources are increasingly accessible [97]. The approach and infrastructure presented here allow a wider range of biologists to engage with complicated models, which will be essential tools to understand the mechanisms and physiological functions of complex biological networks.
Supplementary Material
Acknowledgements
We are grateful to Gavin Steel and Kelly Stewart for expert technical assistance, Martin Beaton and Richard Adams for support of BioDare and SBSI, Uriel Urquiza for preparing SBML files, David Rand for insightful discussion, and members of the A.J.M. and K.J.H. laboratories, who curated data for BioDare.
Appendix A
A.1. Experimental procedures
Experimental methods were similar or identical to published protocols [33,88,98], as detailed in the electronic supplementary material. Statistical significance of comparisons reported in the results is from two-tailed t-tests compared with the cognate WT plants at the same timepoint, unless otherwise stated. Homoscedasticity is assumed, because all comparisons reported are within individual datasets for the same PCR primers. Significance is not corrected for multiple comparisons (reducing significance), nor for support from neighbouring timepoints or replication across cycles or studies (which can increase significance).
A.2. Biodare and computational methods
The BioDare online resource (www.biodare.ed.ac.uk) uses a desktop application to prepare metadata (describe experiments). The XML editor Pedro [99] was customized for each experimental protocol to speed up metadata entry, as each experiment can comprise several hundred samples. Numerical data are uploaded in a spreadsheet-compatible format, with the XML metadata, and stored in a relational database. Password-protected access allows controlled data sharing or public dissemination. Searching the metadata (by genotype, marker, etc.) allows aggregation of similar data from multiple sources, followed by secondary processing (detrending, normalization, averaging), visualization (figure 7a) and download. Rhythm analysis in BioDare was recently described [52,53]. Model optimization used SBSIvisual v. 1.4.5 [29] and SBSInumerics v. 1.2 (see electronic supplementary material, Methods). Graphical network diagrams used SBGN-ED in VANTED [38].
A.3. Data, network diagram, model and code accessibility
The accessibility of resources used in the publication is summarized at the University of Edinburgh's institutional repository, at http://www.research.ed.ac.uk/portal/en/datasets/data-code-and-models-for-flis-et-al-rs-open-biology-2015(fd297498-7d0d-4d57-9040-769af9c65212).html
A.3.1. RNA expression profile data
The RNA datasets reported here are publicly available from BioDare with the permanent data identifiers listed below, using login name ‘public’ with password ‘public’. Numbers below match figure 1.
(1) Pinas-Fernandez and K.J. Halliday (2015) ROBuST RNA timeseries data at 17°C for clock model parameterisation. BioDare accessions:
ROBuST sd for CCA1, BioDare accession 12820611467827, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12820611467827
ROBuST sd for LHY, BioDare accession 3492, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=3492
ROBuST sd for PRR9, BioDare accession 12820610743262, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12820610743262
ROBuST sd for PRR7, BioDare accession 12820611319996, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12820611319996
ROBuST sd for PRR5, BioDare accession 12820611188065, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12820611188065
ROBuST sd for TOC1, BioDare accession 12820611587928, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12820611587928
ROBuST sd for GI, BioDare accession 12820606741450, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12820606741450
ROBuST sd for LUX, BioDare accession 12820610913763, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12820610913763
ROBuST sd for CAB2, BioDare accession 13228354371807, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=13228354371807
ROBuST sd for ELF4, BioDare accession 12962296599986, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12962296599986
ROBuST sd for ELF3, BioDare accession 12962294335805, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=12962294335805
(2) Flis, V. Mengin, R. Sulpice and M. Stitt (2015) TiMet RNA timeseries data from rosette plants for clock model parameterisation. TiMEt ros, BioDare accession 2841, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=2841
(3) Flis, V. Mengin, R. Sulpice and M. Stitt (2015) TiMet RNA timeseries data from elf3 mutant plants for clock model parameterisation. TiMet sd1, BioDare accession 2842, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=2842
(4) Flis, V. Mengin, R. Sulpice and M. Stitt (2015) TiMet RNA timeseries data from seedlings for clock model parameterisation. TiMet sd2, BioDare accession 2843, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=2843
(5) H. G. McWatters (2015) Clock RNA timeseries in light and temperature entrainment. McWatters sd, BioDare accession 3488, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=3488
(6) K.D. Edwards and A.J. Millar (2010). Clock RNA timeseries in multiple photoperiods. Edwards sd, BioDare accession 13227622661196, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=13227622661196
(7) M.M. Southern and A.J. Millar (2015). Clock RNA timeseries in wild-type and mutant plants under red light.
Independent biological replicates are presented for this experiment, with the same genotypes and markers tested in replicates (1 + 2) and (3 + 4):
Southern sd replicate 1, Biodare accession 13228298288040, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=13228298288040
Southern sd replicate 2, Biodare accession 13227619871305, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=13227619871305
Southern sd replicate 3, Biodare accession 13228357055348, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=13228357055348
Southern sd replicate 4, Biodare accession 13228357183121, https://www.biodare.ed.ac.uk/robust/ShowExperiment.action?experimentId=13228357183121
A.3.2. Code
BioDare and SBSI are open-source and available from Sourceforge (www.sourceforge.net).
BioDare: Sourceforge project, http://sourceforge.net/projects/biodare/. The online resource is available at www.biodare.ed.ac.uk.
SBSI: Sourceforge project, http://sourceforge.net/projects/sbsi/. Related materials, plugins and tutorials are available at www.sbsi.ed.ac.uk.
A.3.3. Graphical network diagram
The diagram of the Arabidopsis clock model (electronic supplementary material, figure S2) is available from the PlaSMo repository (www.plasmo.ed.ac.uk), which handles a variety of XML file formats.
D. D. Seaton and A. J. Millar (2015), Graphical network diagram of the Arabidopsis clock model P2011.1 in SBGN PD: PlaSMo accession 1045 version 1, http://www.plasmo.ed.ac.uk/plasmo/models/model.shtml?accession=PLM_1045&version=1
A.3.4. Models
A. Pokhilko et al. (2012), Arabidopsis clock model P2011.1.1 (published in Molecular Systems Biology, 2012): BioModels identifier BIOMD0000000412 [100].
The Arabidopsis clock models below are available from the PlaSMo repository (www.plasmo.ed.ac.uk) and will also be submitted to Biomodels when the present publication has a digital identifier. The model versioning convention is described in the electronic supplementary material.
A. J. Millar and A. Hume (2015) Arabidopsis clock model P2011.1.2: PlaSMo accession PLM_71 version 1, http://www.plasmo.ed.ac.uk/plasmo/models/model.shtml?accession=PLM_71&version=1
A. J. Millar and A. Hume (2015) Arabidopsis clock model P2011.2.1: PlaSMo accession PLM_71 version 2, http://www.plasmo.ed.ac.uk/plasmo/models/model.shtml?accession=PLM_71&version=2
K. Stratford, A. Hume and A. J. Millar (2015) Arabidopsis clock model P2011.3.1: PlaSMo accession PLM_1041 version 1, http://www.plasmo.ed.ac.uk/plasmo/models/model.shtml?accession=PLM_1041&version=1
K. Stratford, A. Hume and A. J. Millar (2015) Arabidopsis clock model P2011.4.1: PlaSMo accession PLM_1042 version 1, http://www.plasmo.ed.ac.uk/plasmo/models/model.shtml?accession=PLM_1042&version=1
K. Stratford, A. Hume and A. J. Millar (2015) Arabidopsis clock model P2011.5.1: PlaSMo accession PLM_1043 version 1, http://www.plasmo.ed.ac.uk/plasmo/models/model.shtml?accession=PLM_1043&version=1
K. Stratford, A. Hume and A. J. Millar (2015) Arabidopsis clock model P2011.6.1: PlaSMo accession PLM_1044 version 1, http://www.plasmo.ed.ac.uk/plasmo/models/model.shtml?accession=PLM_1044&version=1
Author contributions
Designed experiments: A.F., R.S., K.S., H.G.M., M.S., A.J.M., K.J.H. Performed experiments: A.F., A.P.F., V.M., M.M.S., H.G.M., K.S., A.J.M. Designed infrastructure: T.Z., A.J.M., K.J.H. Built infrastructure: T.Z., A.H.; Analysed data: D.D.S., A.P., M.M.S., H.G.M., A.J.M., M.S., K.J.H. A.F., H.G.M., M.S. and A.J.M. wrote the paper with input from all authors.
Competing interests
We declare we have no competing interests.
Funding
Supported by awards from UK BBSRC and EPSRC (ROBuST BB/F005237/1 and SynthSys BB/D019621/1) and from the European Commission (FP7 collaborative project TiMet, contract 245143). This work made use of the facilities of HECToR, the UK's national high-performance computing service, which was provided by UoE HPCx Ltd at the University of Edinburgh, Cray Inc. and NAG Ltd, and funded by the Office of Science and Technology through EPSRC's High End Computing Programme.
References
- 1.Zhang EE, Kay SA. 2010. Clocks not winding down: unravelling circadian networks. Nat. Rev. Mol. Cell Biol. 11, 764–776. (doi:10.1038/nrm2995) [DOI] [PubMed] [Google Scholar]
- 2.Dong G, Golden SS. 2008. How a cyanobacterium tells time. Curr. Opin. Microbiol. 11, 541–546. (doi:10.1016/j.mib.2008.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dodd AN, et al. 2008. Plant circadian clocks increase photosynthesis, growth, survival, and competitive advantage. Science 309, 630–633. (doi:10.1126/science.1115581) [DOI] [PubMed] [Google Scholar]
- 4.Ouyang Y, Andersson CR, Kondo T, Golden SS, Johnson CH. 1998. Resonating circadian clocks enhance fitness in cyanobacteria. Proc. Natl Acad. Sci. USA 95, 8660–8664. (doi:10.1073/pnas.95.15.8660) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinmonth-Schultz HA, Golembeski GS, Imaizumi T. 2013. Circadian clock-regulated physiological outputs: dynamic responses in nature. Semin. Cell Dev. Biol. 24, 407–413. (doi:10.1016/j.semcdb.2013.02.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young MW, Kay SA. 2001. Time zones: a comparative genetics of circadian clocks. Nat. Rev. Genet. 2, 702–715. (doi:10.1038/35088576) [DOI] [PubMed] [Google Scholar]
- 7.van Ooijen G, Millar AJ. 2012. Non-transcriptional oscillators in circadian timekeeping. Trends Biochem. Sci. 37, 484–492. (doi:10.1016/j.tibs.2012.07.006) [DOI] [PubMed] [Google Scholar]
- 8.Le Novere N, et al. 2009. The systems biology graphical notation. Nat. Biotechnol. 27, 735–741. (doi:10.1038/nbt.1558) [DOI] [PubMed] [Google Scholar]
- 9.Pokhilko A, Mas P, Millar AJ. 2013. Modelling the widespread effects of TOC1 signalling on the plant circadian clock and its outputs. BMC Syst. Biol. 7, 23 (doi:10.1186/1752-0509-7-23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pokhilko A, Fernandez AP, Edwards KD, Southern MM, Halliday KJ, Millar AJ. 2012. The clock gene circuit in Arabidopsis includes a repressilator with additional feedback loops. Mol. Syst. Biol. 8, 574 (doi:10.1038/msb.2012.6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamichi N. 2011. Molecular mechanisms underlying the Arabidopsis circadian clock. Plant Cell Physiol. 52, 1709–1718. (doi:10.1093/pcp/pcr118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farre EM, Harmer SL, Harmon FG, Yanovsky MJ, Kay SA. 2005. Overlapping and distinct roles of PRR7 and PRR9 in the Arabidopsis circadian clock. Curr. Biol. 15, 47–54. (doi:10.1016/j.cub.2004.12.067) [DOI] [PubMed] [Google Scholar]
- 13.Salome PA, McClung CR. 2005. PSEUDO-RESPONSE REGULATOR 7 and 9 are partially redundant genes essential for the temperature responsiveness of the Arabidopsis circadian clock. Plant Cell 17, 791–803. (doi:10.1105/tpc.104.029504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pokhilko A, Hodge SK, Stratford K, Knox K, Edwards KD, Thomson AW, Mizuno T, Millar AJ. 2010. Data assimilation constrains new connections and components in a complex, eukaryotic circadian clock model. Mol. Syst. Biol. 6, 416 (doi:10.1038/msb.2010.69) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamichi N, Kiba T, Kamioka M, Suzuki T, Yamashino T, Higashiyama T, Sakakibara H, Mizuno T. 2012. Transcriptional repressor PRR5 directly regulates clock-output pathways. Proc. Natl Acad. Sci. USA 109, 17 123–17 128. (doi:10.1073/pnas.1205156109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamichi N, Kiba T, Henriques R, Mizuno T, Chua NH, Sakakibara H. 2010. PSEUDO-RESPONSE REGULATORS 9, 7, and 5 are transcriptional repressors in the Arabidopsis circadian clock. Plant Cell 22, 594–605. (doi:10.1105/tpc.109.072892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang W, Perez-Garcia P, Pokhilko A, Millar AJ, Antoshechkin I, Riechmann JL, Mas P. 2012. Mapping the core of the Arabidopsis circadian clock defines the network structure of the oscillator. Science 336, 75–79. (doi:10.1126/science.1219075) [DOI] [PubMed] [Google Scholar]
- 18.Gendron JM, Pruneda-Paz JL, Doherty CJ, Gross AM, Kang SE, Kay SA. 2012. Arabidopsis circadian clock protein, TOC1, is a DNA-binding transcription factor. Proc. Natl Acad. Sci. USA 109, 3167–3172. (doi:10.1073/pnas.1200355109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nusinow DA, Helfer A, Hamilton EE, King JJ, Imaizumi T, Schultz TF, Farré EM, Kay SA. 2011. The ELF4-ELF3-LUX complex links the circadian clock to diurnal control of hypocotyl growth. Nature 475, 398–402. (doi:10.1038/nature10182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helfer A, Nusinow DA, Chow BY, Gehrke AR, Bulyk ML, Kay SA. 2011. LUX ARRHYTHMO encodes a nighttime repressor of circadian gene expression in the Arabidopsis core clock. Curr. Biol. 21, 126–133. (doi:10.1016/j.cub.2010.12.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon LE, Knox K, Kozma-Bognar L, Southern MM, Pokhilko A, Millar AJ. 2011. Temporal repression of core circadian genes is mediated through EARLY FLOWERING 3 in Arabidopsis. Curr. Biol. 21, 120–125. (doi:10.1016/j.cub.2010.12.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrero E, et al. 2012. EARLY FLOWERING4 recruitment of EARLY FLOWERING3 in the nucleus sustains the Arabidopsis circadian clock. Plant Cell 24, 428–443. (doi:10.1105/tpc.111.093807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim WY, et al. 2007. ZEITLUPE is a circadian photoreceptor stabilized by GIGANTEA in blue light. Nature 449, 356–360. (doi:10.1038/nature06132) [DOI] [PubMed] [Google Scholar]
- 24.Bujdoso N, Davis SJ. 2013. Mathematical modeling of an oscillating gene circuit to unravel the circadian clock network of Arabidopsis thaliana. Front. Plant Sci. 4, 3 (doi:10.3389/fpls.2013.00003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogelmark K, Troein C. 2014. Rethinking transcriptional activation in the Arabidopsis circadian clock. PLoS Comput. Biol. 10, e1003705 (doi:10.1371/journal.pcbi.1003705) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Royal Society. 2012. Science as an open enterprise 2012. 29 June 2012. See https://royalsociety.org/~/media/policy/projects/sape/2012-06-20-saoe.pdf.
- 27.Hey T, Tansley S, Tolle K (eds). 2009. The fourth paradigm: data-intensive scientific discovery. Redmond, WA: Microsoft Research.
- 28.Bastow R, Leonelli S. 2010. Sustainable digital infrastructure. Although databases and other online resources have become a central tool for biological research, their long-term support and maintenance is far from secure. EMBO Rep. 11, 730–734. (doi:10.1038/embor.2010.145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams R, et al. 2013. SBSI: an extensible distributed software infrastructure for parameter estimation in systems biology. Bioinformatics 29, 664–665. (doi:10.1093/bioinformatics/btt023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart JL, Maloof JN, Nemhauser JL. 2011. PIF genes mediate the effect of sucrose on seedling growth dynamics. PLoS ONE 6, e19894 (doi:10.1371/journal.pone.0019894) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franklin KA, Toledo-Ortiz G, Pyott DE, Halliday KJ. 2014. Interaction of light and temperature signalling. J. Exp. Bot. 65, 2859–2871. (doi:10.1093/jxb/eru059) [DOI] [PubMed] [Google Scholar]
- 32.Salvo-Chirnside E, Kane S, Kerr LE. 2011. Protocol: high throughput silica-based purification of RNA from Arabidopsis seedlings in a 96-well format. Plant Methods 7, 40 (doi:10.1186/1746-4811-7-40) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piques M, Schulze WX, Hohne M, Usadel B, Gibon Y, Rohwer J, Stitt M. 2009. Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 5, 314 (doi:10.1038/msb.2009.68) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards KD, et al. 2010. Quantitative analysis of regulatory flexibility under changing environmental conditions. Mol. Syst. Biol. 6, 424 (doi:10.1038/msb.2010.81) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Locke JC, Southern MM, Kozma-Bognar L, Hibberd V, Brown PE, Turner MS, Millar AJ. 2005. Extension of a genetic network model by iterative experimentation and mathematical analysis. Mol. Syst. Biol. 1, 20050013 (doi:10.1038/msb4100018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strayer C, et al. 2000. Cloning of the Arabidopsis clock gene TOC1, an autoregulatory response regulator homolog. Science 289, 768–771. (doi:10.1126/science.289.5480.768) [DOI] [PubMed] [Google Scholar]
- 37.Fowler S, et al. 1999. GIGANTEA: a circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. EMBO J. 18, 4679–4688. (doi:10.1093/emboj/18.17.4679) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Czauderna T, Klukas C, Schreiber F. 2010. Editing, validating and translating of SBGN maps. Bioinformatics 26, 2340–2341. (doi:10.1093/bioinformatics/btq407) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalchau N, et al. 2011. The circadian oscillator gene GIGANTEA mediates a long-term response of the Arabidopsis thaliana circadian clock to sucrose. Proc. Natl Acad. Sci. USA 108, 5104–5109. (doi:10.1073/pnas.1015452108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haydon MJ, Mielczarek O, Robertson FC, Hubbard KE, Webb AA. 2013. Photosynthetic entrainment of the Arabidopsis thaliana circadian clock. Nature 502, 689–692. (doi:10.1038/nature12603) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Usadel B, et al. 2005. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 138, 1195–1204. (doi:10.1104/pp.105.060459) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osuna D, et al. 2007. Temporal responses of transcripts, enzyme activities and metabolites after adding sucrose to carbon-deprived Arabidopsis seedlings. Plant J. 49, 463–491. (doi:10.1111/j.1365-313X.2006.02979.x) [DOI] [PubMed] [Google Scholar]
- 43.Doyle MR, Davis SJ, Bastow RM, McWatters HG, Kozma-Bognar L, Nagy F, Millar AJ, Amasino RM. 2002. The ELF4 gene controls circadian rhythms and flowering time in Arabidopsis thaliana. Nature 419, 74–77. (doi:10.1038/nature00954) [DOI] [PubMed] [Google Scholar]
- 44.Li G, et al. 2011. Coordinated transcriptional regulation underlying the circadian clock in Arabidopsis. Nat. Cell Biol. 13, 616–622. (doi:10.1038/ncb2219) [DOI] [PubMed] [Google Scholar]
- 45.Kiba T, Henriques R, Sakakibara H, Chua NH. 2007. Targeted degradation of PSEUDO-RESPONSE REGULATOR5 by an SCFZTL complex regulates clock function and photomorphogenesis in Arabidopsis thaliana. Plant Cell 19, 2516–2530. (doi:10.1105/tpc.107.053033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yakir E, Hilman D, Hassidim M, Green RM. 2007. CIRCADIAN CLOCK ASSOCIATED1 transcript stability and the entrainment of the circadian clock in Arabidopsis. Plant Physiol. 145, 925–932. (doi:10.1104/pp.107.103812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alabadi D, Oyama T, Yanovsky MJ, Harmon FG, Mas P, Kay SA. 2001. Reciprocal regulation between TOC1 and LHY/CCA1 within the Arabidopsis circadian clock. Science 293, 880–883. (doi:10.1126/science.1061320) [DOI] [PubMed] [Google Scholar]
- 48.Millar AJ, Carré IA, Strayer CA, Chua NH, Kay SA. 1995. Circadian clock mutants in Arabidopsis identified by luciferase imaging. Science 267, 1161–1163. (doi:10.1126/science.7855595) [DOI] [PubMed] [Google Scholar]
- 49.Carre I, Veflingstad SR. 2013. Emerging design principles in the Arabidopsis circadian clock. Semin. Cell Dev. Biol. 24, 393–398. (doi:10.1016/j.semcdb.2013.03.011) [DOI] [PubMed] [Google Scholar]
- 50.Hicks KA, Albertson TM, Wagner DR. 2001. EARLY FLOWERING3 encodes a novel protein that regulates circadian clock function and flowering in Arabidopsis. Plant Cell 13, 1281–1292. (doi:10.1105/tpc.13.6.1281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.BioDare 2011 The Biological Data Repository. See www.biodare.ed.ac.uk.
- 52.Zielinski T, Moore AM, Troup E, Halliday KJ, Millar AJ. 2014. Strengths and limitations of period estimation methods for circadian data. PLoS ONE 9, e96462 (doi:10.1371/journal.pone.0096462) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore A, Zielinski T, Millar AJ. 2014. Online period estimation and determination of rhythmicity in circadian data, using the BioDare data infrastructure. In Plant circadian networks (ed. Staiger D.), pp. 13–44. Clifton, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- 54.Dodd AN, Dalchau N, Gardner MJ, Baek SJ, Webb AA. 2014. The circadian clock has transient plasticity of period and is required for timing of nocturnal processes in Arabidopsis. New Phytol. 201, 168–179. (doi:10.1111/nph.12489) [DOI] [PubMed] [Google Scholar]
- 55.Adams RR, Tsorman N, Stratford K, Akman OE, Gilmore S, Juty N, Le Novere N, Millar AJ, Millar AJ. 2012. The Input Signal Step Function (ISSF), a standard method to encode input signals in SBML models with software support, applied to circadian clock models. J. Biol. Rhythms 27, 328–332. (doi:10.1177/0748730412451077) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hucka M, et al. 2003. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531. (doi:10.1093/bioinformatics/btg015) [DOI] [PubMed] [Google Scholar]
- 57.Massonnet C, et al. 2010. Probing the reproducibility of leaf growth and molecular phenotypes: a comparison of three Arabidopsis accessions cultivated in ten laboratories. Plant Physiol. 152, 2142–2157. (doi:10.1104/pp.109.148338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swarup K, Alonso-Blanco C, Lynn JR, Michaels SD, Amasino RM, Koornneef M, Millar AJ. 1999. Natural allelic variation identifies new genes in the Arabidopsis circadian system. Plant J. 20, 67–77. (doi:10.1046/j.1365-313X.1999.00577.x) [DOI] [PubMed] [Google Scholar]
- 59.Michael TP, et al. 2003. Enhanced fitness conferred by naturally occurring variation in the circadian clock. Science 302, 1049–1053. (doi:10.1126/science.1082971) [DOI] [PubMed] [Google Scholar]
- 60.Darrah C, Taylor BL, Edwards KD, Brown PE, Hall A, McWatters HG. 2006. Analysis of phase of LUCIFERASE expression reveals novel circadian quantitative trait loci in Arabidopsis. Plant Physiol. 140, 1464–1474. (doi:10.1104/pp.105.074518) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wuyts N, Palauqui JC, Conejero G, Verdeil JL, Granier C, Massonnet C. 2010. High-contrast three-dimensional imaging of the Arabidopsis leaf enables the analysis of cell dimensions in the epidermis and mesophyll. Plant Methods 6, 17 (doi:10.1186/1746-4811-6-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O'Neill JS, van Ooijen G, Le Bihan T, Millar AJ. 2011. Circadian clock parameter measurement: characterization of clock transcription factors using surface plasmon resonance. J. Biol. Rhythms 26, 91–98. (doi:10.1177/0748730410397465) [DOI] [PubMed] [Google Scholar]
- 63.Locke JC, Kozma-Bognar L, Gould PD, Feher B, Kevei E, Nagy F, Turner MS, Hall A, Millar AJ. 2006. Experimental validation of a predicted feedback loop in the multi-oscillator clock of Arabidopsis thaliana. Mol. Syst. Biol. 2, 59 (doi:10.1038/msb4100102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yeom M, Kim H, Lim J, Shin AY, Hong S, Kim JI, Nam HG. 2014. How do phytochromes transmit the light quality information to the circadian clock in Arabidopsis? Mol. Plant. 7, 1701–1704. (doi:10.1093/mp/ssu086) [DOI] [PubMed] [Google Scholar]
- 65.Price J, Laxmi A, St Martin SK, Jang JC. 2004. Global transcription profiling reveals multiple sugar signal transduction mechanisms in Arabidopsis. Plant Cell 16, 2128–2150. (doi:10.1105/tpc.104.022616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blasing OE, et al. 2005. Sugars and circadian regulation make major contributions to the global regulation of diurnal gene expression in Arabidopsis. Plant Cell 17, 3257–3281. (doi:10.1105/tpc.105.035261) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Usadel B, Blasing OE, Gibon Y, Retzlaff K, Hohne M, Gunther M, Stitt M. 2008. Global transcript levels respond to small changes of the carbon status during progressive exhaustion of carbohydrates in Arabidopsis rosettes. Plant Physiol. 146, 1834–1861. (doi:10.1104/pp.107.115592) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leonelli S, Smirnoff N, Moore J, Cook C, Bastow R. 2013. Making open data work for plant scientists. J. Exp. Bot. 64, 4109–4117. (doi:10.1093/jxb/ert273) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toronto International Data Release Workshop A, et al. 2009. Prepublication data sharing. Nature 461, 168–170. (doi:10.1038/461168a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mockler TC, Michael TP, Priest HD, Shen R, Sullivan CM, Givan SA, McEntee C, Kay SA, Chory J. 2007. The DIURNAL project: diurnal and circadian expression profiling, model-based pattern matching, and promoter analysis. Cold Spring Harb. Symp. Quant. Biol. 72, 353–363. (doi:10.1101/sqb.2007.72.006) [DOI] [PubMed] [Google Scholar]
- 71.Zhang EE, et al. 2009. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 139, 199–210. (doi:10.1016/j.cell.2009.08.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patel VR, Eckel-Mahan K, Sassone-Corsi P, Baldi P. 2012. CircadiOmics: integrating circadian genomics, transcriptomics, proteomics and metabolomics. Nat. Methods 9, 772–773. (doi:10.1038/nmeth.2111) [DOI] [PubMed] [Google Scholar]
- 73.Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP. 2007. Universally sloppy parameter sensitivities in systems biology models. PLoS Comput. Biol. 3, 1871–1878. (doi:10.1371/journal.pcbi.0030189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rand DA, Shulgin BV, Salazar D, Millar AJ. 2004. Design principles underlying circadian clocks. Interface 1, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rand DA, Shulgin BV, Salazar JD, Millar AJ. 2006. Uncovering the design principles of circadian clocks: mathematical analysis of flexibility and evolutionary goals. J. Theor. Biol. 238, 616–635. (doi:10.1016/j.jtbi.2005.06.026) [DOI] [PubMed] [Google Scholar]
- 76.Rand DA. 2008. Mapping global sensitivity of cellular network dynamics: sensitivity heat maps and a global summation law. J. R. Soc. Interface 5(Suppl 1), S59–S69. (doi:10.1098/rsif.2008.0084.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daniels BC, Chen YJ, Sethna JP, Gutenkunst RN, Myers CR. 2008. Sloppiness, robustness, and evolvability in systems biology. Curr. Opin. Biotechnol. 19, 389–395. (doi:10.1016/j.copbio.2008.06.008) [DOI] [PubMed] [Google Scholar]
- 78.Dunlap JC. 1993. Genetic analysis of circadian clocks. Annu. Rev. Physiol. 55, 683–728. [DOI] [PubMed] [Google Scholar]
- 79.McWatters HG, Devlin PF. 2011. Timing in plants: a rhythmic arrangement. FEBS Lett. 585, 1474–1484. (doi:10.1016/j.febslet.2011.03.051) [DOI] [PubMed] [Google Scholar]
- 80.Transtrum MK, Machta BB, Sethna JP. 2010. Why are nonlinear fits to data so challenging? Phys. Rev. Lett. 104, 060201 (doi:10.1103/PhysRevLett.104.060201) [DOI] [PubMed] [Google Scholar]
- 81.Domijan M, Rand DA. 2015. Using constraints and their value for optimization of large ODE systems. J. R. Soc. Interface 12, 20141303 (doi:10.1098/rsif.2014.1303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seaton DD, et al. 2015. Linked circadian outputs control elongation growth and flowering in response to photoperiod and temperature. Mol. Syst. Biol. 11, 776 (doi:10.15252/msb.20145766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Locke JC, Millar AJ, Turner MS. 2005. Modelling genetic networks with noisy and varied experimental data: the circadian clock in Arabidopsis thaliana. J. Theor. Biol. 234, 383–393. (doi:10.1016/j.jtbi.2004.11.038) [DOI] [PubMed] [Google Scholar]
- 84.Salazar JD, Saithong T, Brown PE, Foreman J, Locke JC, Halliday KJ, Carré IA, Rand DA, Millar AJ. 2009. Prediction of photoperiodic regulators from quantitative gene circuit models. Cell 139, 1170–1179. (doi:10.1016/j.cell.2009.11.029) [DOI] [PubMed] [Google Scholar]
- 85.Troein C, Corellou F, Dixon LE, van Ooijen G, O'Neill JS, Bouget FY, Millar AJ. 2011. Multiple light inputs to a simple clock circuit allow complex biological rhythms. Plant J. 66, 375–385. (doi:10.1111/j.1365-313X.2011.04489.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Song YH, Smith RW, To BJ, Millar AJ, Imaizumi T. 2012. FKF1 conveys timing information for CONSTANS stabilization in photoperiodic flowering. Science 336, 1045–1049. (doi:10.1126/science.1219644) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brancaccio M, Maywood ES, Chesham JE, Loudon AS, Hastings MH. 2013. A Gq-Ca2+ axis controls circuit-level encoding of circadian time in the suprachiasmatic nucleus. Neuron 78, 714–728. (doi:10.1016/j.neuron.2013.03.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gould PD, et al. 2013. Network balance via CRY signalling controls the Arabidopsis circadian clock over ambient temperatures. Mol. Syst. Biol. 9, 650 (doi:10.1038/msb.2013.7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dixon LE, Hodge SK, van Ooijen G, Troein C, Akman OE, Millar AJ. 2014. Light and circadian regulation of clock components aids flexible responses to environmental signals. New Phytol. 203, 568–577. (doi:10.1111/nph.12853) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Force MM, Robinson NJ. 2014. Encouraging data citation and discovery with the data citation index. J. Compt-aided Mol. Des. 28, 1043–1048. (doi:10.1007/s10822-014-9768-5) [DOI] [PubMed] [Google Scholar]
- 91.International Arabidopsis Informatics Consortium. 2012. Taking the next step: building an Arabidopsis information portal. The Plant Cell 24, 2248–2256. (doi:10.1105/tpc.112.100669) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.PlaSMo 2010 The Plant Systems Modelling portal See www.plasmo.ed.ac.uk.
- 93.Akman OE, Watterson S, Parton A, Binns N, Millar AJ, Ghazal P. 2012. Digital clocks: simple Boolean models can quantitatively describe circadian systems. J. R. Soc. Interface 9, 2365–2382. (doi:10.1098/rsif.2012.0080) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aderhold A, Husmeier D, Grzegorczyk M. 2014. Statistical inference of regulatory networks for circadian regulation. Stat. Appl. Genet. Mol. Biol. 13, 227–273. (doi:10.1515/sagmb-2013-0051) [DOI] [PubMed] [Google Scholar]
- 95.Carrera J, Rodrigo G, Jaramillo A, Elena SF. 2009. Reverse-engineering the Arabidopsis thaliana transcriptional network under changing environmental conditions. Genome Biol. 10, R96 (doi:10.1186/gb-2009-10-9-r96) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pokhilko A, Ramos JA, Holtan H, Maszle DR, Khanna R, Millar AJ. 2011. Ubiquitin ligase switch in plant photomorphogenesis: a hypothesis. J. Theor. Biol. 270, 31–41. (doi:10.1016/j.jtbi.2010.11.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goff SA, et al. 2011. The iPlant collaborative: cyberinfrastructure for plant biology. Front. Plant. Sci. 2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Knight H, Thomson AJ, McWatters HG. 2008. Sensitive to freezing integrates cellular and environmental inputs to the plant circadian clock. Plant Physiol. 148, 293–303. (doi:10.1104/pp.108.123901) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garwood KL, Taylor CF, Runte KJ, Brass A, Oliver SG, Paton NW. 2004. Pedro: a configurable data entry tool for XML. Bioinformatics 20, 2463–2465. (doi:10.1093/bioinformatics/bth251) [DOI] [PubMed] [Google Scholar]
- 100.Le Novere N, et al. 2006. BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 34, D689–D691. (doi:10.1093/nar/gkj092) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.