

Abstract
Background
Chlamydia pecorum is a globally recognised pathogen of livestock and koalas. To date, comparative genomics of C. pecorum strains from sheep, cattle and koalas has revealed that only single nucleotide polymorphisms (SNPs) and a limited number of pseudogenes appear to contribute to the genetic diversity of this pathogen. No chlamydial plasmid has been detected in these strains despite its ubiquitous presence in almost all other chlamydial species. Genomic analyses have not previously included C. pecorum from porcine hosts. We sequenced the genome of three C. pecorum isolates from pigs with differing pathologies in order to re-evaluate the genetic differences and to update the phylogenetic relationships between C. pecorum from each of the hosts.
Methods
Whole genome sequences for the three porcine C. pecorum isolates (L1, L17 and L71) were acquired using C. pecorum-specific sequence capture probes with culture-independent methods, and assembled in CLC Genomics Workbench. The pairwise comparative genomic analyses of 16 pig, sheep, cattle and koala C. pecorum genomes were performed using several bioinformatics platforms, while the phylogenetic analyses of the core C. pecorum genomes were performed with predicted recombination regions removed. Following the detection of a C. pecorum plasmid, a newly developed C. pecorum-specific plasmid PCR screening assay was used to evaluate the plasmid distribution in 227 C. pecorum samples from pig, sheep, cattle and koala hosts.
Results
Three porcine C. pecorum genomes were sequenced using C. pecorum-specific sequence capture probes with culture-independent methods. Comparative genomics of the newly sequenced porcine C. pecorum genomes revealed an increased average number of SNP differences (~11 500) between porcine and sheep, cattle, and koala C. pecorum strains, compared to previous C. pecorum genome analyses. We also identified a third copy of the chlamydial cytotoxin gene, found only in porcine C. pecorum isolates. Phylogenetic analyses clustered porcine isolates into a distinct clade, highlighting the polyphyletic origin of C. pecorum in livestock.
Most surprising, we also discovered a plasmid in the porcine C. pecorum genome. Using this novel C. pecorum plasmid (pCpec) sequence, a) we developed a pCpec screening assay to evaluate the plasmid distribution in C. pecorum from different hosts; and b) to characterise the pCpec sequences from available previously sequenced C. pecorum genome data. pCpec screening showed that the pCpec is common in all hosts of C. pecorum, however not all C. pecorum strains carry pCpec.
Conclusions
This study provides further insight into the complexity of C. pecorum epidemiology and novel genomic regions that may be linked to host specificity. C. pecorum plasmid characterisation may aid in improving our understanding of C. pecorum pathogenesis across the variety of host species this animal pathogen infects.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2053-8) contains supplementary material, which is available to authorized users.
Keywords: C. pecorum plasmid, Porcine hosts, C. pecorum comparative genomics, Cytotoxin gene, Phylogenetic relationships
Background
Chlamydia pecorum is a globally distributed animal pathogen of domesticated and wild ruminants, and the iconic native Australian marsupial, the koala. Expansion of the number of available whole genome sequences of C. pecorum from sheep, cattle, and koala hosts has provided insights into its lifestyle, associated virulence factors, and evolution [1–3]. The genetic differences that may influence host specificity and/or pathogenicity in C. pecorum remain largely unknown, however, with only (i) single nucleotide polymorphisms (SNPs); (ii) a limited number (four to five) of pseudogenes; and (iii) variable numbers of tandem repeats in certain C. pecorum genes (eg. ORF663 and IncA genes), differentiating strains from different hosts or associated with different diseases [1–5].
In other chlamydial species, the almost ubiquitous non-integrative chlamydial plasmid [6, 7] has been recognised as a major chlamydial virulence factor [8, 9]. This plasmid also has value as a target for molecular typing [10] and as an immunogenic vaccine candidate both for humans and animals such as ducks, pigeons, cats and pigs [11, 12]. Two studies have previously detected a C. pecorum plasmid in isolates from three koalas and one bovid [13, 14], but further evidence for a C. pecorum plasmid has remained absent despite the growing number of genomics studies for this species [1–3, 15].
Although C. pecorum infects a wide range of hosts, including some other Australian marsupials, European wild ruminants, and pigs, C. pecorum comparative genomics studies thus far have focused only on sheep, cattle, and koala strains [16–18]. Porcine C. pecorum infections are underestimated as a source of on-farm economic loss for producers, manifesting as a variety of diseases such as pneumonia, pleuritis, polyserositis, polyarthritis, conjunctivitis, and enteritis. As with other hosts, C. pecorum subclinical infections in pigs are also common [18–22]. In an effort to broaden our knowledge of the genetic diversity of C. pecorum in other hosts, in the present study, we sequenced three porcine C. pecorum genomes and compared them to the available sheep, cattle, and koala C. pecorum genomes. In doing so, we also provide the first detailed description of the genetic structure and distribution of the C. pecorum plasmid, a genetic feature associated with virulence in other chlamydial species.
Methods
Descriptions of pig C. pecorum isolates, clinical samples and other C. pecorum strains used in this study
The three C. pecorum strains L1, L17 and L71 isolated from pigs presenting with pneumonia (L1 and L17) and polyarthritis (L71) were utilised for whole genome sequencing and plasmid analyses in the present study. These strains were one of the first pig chlamydial strains, isolated together with many others in 1969 in Austria, from pigs presenting with chlamydiosis during a mass chlamydial outbreak [20, 21].
Plasmid screening was performed on a large collection of (i) previously tested C. pecorum PCR positive clinical samples from sheep, cattle and koalas (Additional file 6: Table S4); (ii) and a small collection of cultured C. pecorum isolates from a variety of hosts (Additional file 4: Table S3); (iii) additional previously untested but C. pecorum PCR positive sheep and koala samples identified in this study (Additional file 6: Table S4). This latter testing, combined with our previous screening for C. pecorum DNA in clinical samples, resulted in (i) 89 C. pecorum PCR positive ocular and urogenital swab samples from 53 koalas from different populations across Australia; (ii) 83 ocular, joint, vaginal and rectal swabs collected from 41 sheep from nine different flocks from the Central NSW region in Australia [23, 24]; and (iii) ocular, nose, internal organs, and rectal samples (n = 9) from four cattle from two herds, one previously described from Western Australia (WA) [25] and a second from Central NSW (Additional file 6: Table S4). Cultured C. pecorum isolates from pigs, sheep, cattle, and koalas screened are listed in Additional file 4: Table S3.
Genomic DNA extraction and C. pecorum-specific qPCR screen
New sheep, cattle and koala clinical swab samples used in this study were processed as previously described [23]. The cultures of three C. pecorum porcine isolates L1, L17 and L71 were propagated in chicken embryos and purified, as previously described [22], while the cultures of 14 koala isolates were propagated in Hep-2 and/or McCoy cells and semi-purified using a probe sonication and density gradient centrifugation, as previously described [1]. DNA extraction for both samples and cultures was performed using the QIAmp DNA mini kit (Qiagen), as per the manufacturer’s instructions. C. pecorum infectious load and/or presence were determined using a C. pecorum species-specific qPCR assay targeting 202 bp of 16S rDNA [5]. Samples with < 50 copies C. pecorum 16S rDNA were considered negative. DNA concentration for each sample was also measured in duplicates on a Nanodrop. Additionally, 1 μl of extracted, screened and quantified gDNA of three porcine (L1, L17 L71) C. pecorum samples were run on a 1 % TBE agarose gel to confirm high molecular weight DNA prior to whole genome sequencing.
Sequence capture, whole genome sequencing and assembly
Sequence capture was performed on total DNA extracted from C. pecorum L1, L17 and L71 cultures with the C. pecorum custom-designed probes (made by Agilent Technologies) [2], using a DNA hybridisation capture and amplification process based on the methods described elsewhere [26]. L1, L17 and L71 genomes were sequenced at the Institute for Genome Sciences (IGS), Baltimore, USA, using the Illumina HiSeq 2500 platform producing paired-end 101 base-pair reads. Read quality, de novo assembly and read-mapping of paired-end reads from L1, L17 and L71 was performed using CLC Genomics Workbench (CLC bio, Qiagen), after filtering and trimming for size and quality. The read mapping was visualised with BLAST ring image generator (BRIG) software [27]. After de novo assembly, L1, L17 and L71 contigs were blasted to confirm identity using BLASTn (Basic Local Alignment Search Tool from http://blast.ncbi.nlm.nih.gov/Blast.cgi) and ordered against complete C. pecorum E58 genome (accession number: CP002608) with progressive Mauve [28] to produce single genome scaffold. Genome annotations were done using RAST [29] and the IGS prokaryotic annotation pipeline, assisted by mapping onto the C. pecorum E58 type strain sequence, as previously described [15]. Number and distribution of SNPs was determined using the Probabilistic variant detection plug-in with default settings with L1, L17 and L71 reads mapped to a reference genome, as implemented in CLC genomics workbench. General descriptions and accession numbers of L1, L17 and L71 genomes were outlined in Table 1.
Table 1.
General description of pig C. pecorum L1, L17 and L71 samples and their genomes
| L1 | L17 | L71 | |
|---|---|---|---|
| Host disease | Pneumonia | Pneumonia | Polyarthritis |
| Tissue isolated from | Lung | Lung | Joint |
| Year of isolation | 1969 | 1969 | 1969 |
| Country | Austria | Austria | Austria |
| C. pecorum load and amount of DNA | 2.15 X 106 copies/μl; 1000 ng | 2 X 106 copies/μl; 1000 ng | 1.5 X 106 copies/μl; 1000 ng |
| Total No. of filtered reads | 48 485 057 | 41 185 214 | 42 490 870 |
| Av. filtered read length | 95.1 | 95.4 | 95.1 |
| Total No. of filtered reads for de novo assembly | 38 807 222 (80 %) | 31 115 875 (76 %) | 32 594 606 (77 %) |
| Contigs | 5 (9.7 – 623 Kbp) | 3 (10–790 Kbp) | 5 (11.9 –780 Kbp) |
| N50 | 623 872 | 293 736 | 780 721 |
| Av. read depth | 2569X | 1800X | 3020X |
| Genome size (bp) | 1 106 140 | 1 106 210 | 1 115 012 |
| No. of predicted CDS | 1007 | 1002 | 1002 |
| % GC | 41.1 % | 42 % | 41.1 % |
| Plasmid | Yes (7.5 Kbp) | No | No |
| Tryptophan operon | Present (TrpABFCDR, kynU, prsA) | Present (TrpABFCDR, kynU, prsA) | Present (TrpABFCDR, kynU, prsA) |
| Biotin and pyrimidine genes | Present (bioBFDA; pyrEHG, ndk) | Present (bioBFDA; pyrEHG, ndk) | Present (bioBFDA; pyrEHG, ndk) |
| Accession numbers | LFRH01000000 | LFRK01000000 | LFRL01000000 |
Macroscopic comparative genomics and phylogenetic analyses using new porcine genomes
Alongside the newly sequenced three porcine C. pecorum genomes, we also used publicly available genome sequences of C. pecorum from: cattle encephalomyelitis E58 [15] (accession number: CP002608); sheep arthritis P787 (accession number CP004035.1), sheep fecal W73 (accession number CP004034.1), cattle metritis PV3056/3 (accession number CP004033.1) [3]; koala cystitis Mc/Marsbar (accession number CM002310.10) and DBDeUG (accession number CM002308.1), koala conjunctivitis IpTaLE (accession number CM002309.1), sheep polyarthritis IPA (VR629) (accession number CM002311.1) [1]; cattle encephalomyelitis NSW/Bov/SBE (accession number SRR1693788), sheep polyarthritis Mer/Ovi1/Jnt (accession number SRR1693791), sheep conjunctivitis Nar/S22/Rec (accession number SRR1693794), koalas with cystitis Gun/koa1/Ure (accession number SRR1693763) and SA/K2/UGT (accession number SRR1693792) [2] for whole genome comparative analyses.
Pairwise comparative genomic analyses were performed in ACT, the Artemis Comparison Tool [30] and Geneious 7.1.4 [31] using alignments generated with progressive Mauve [28]. Polymorphic loci and/or regions of interest in the genomes were extracted and further aligned with ClustalX [32]. DnaSP 5.0 [33] was used to analyse the level of sequences polymorphisms by: determining the ratio of non-synonymous to synonymous substitutions (dn/ds) (Jukes-Cantor corrected); calculating the number of polymorphic (segregating) sites and haplotype diversity. Further, if a polymorphism resulted in a non-synonymous amino acid change or premature STOP codon in the loci of interest in each of the analysed C. pecorum genome sequence, the translated sequence was further analysed in BLAST for comparison (http://blast.ncbi.nlm.nih.gov/Blast.cgi),UniProt (Universal Protein Resource from http://www.uniprot.org/) and Conserved Domains Database (CDD from http://www.ncbi.nlm.nih.gov/cdd) to assess protein functionality. Figures of the genomic regions of interest, such as the PZ was generated using Easyfig [34], based on tblastx comparison. Construction of phylogenetic relationships between the loci was performed using MrBayes [35] as implemented in Geneious 7.1.4.
In addition to the subset of 16 C. pecorum genomes used for comparative analyses, for phylogenetic analyses we have included: (i) sheep Nar/S22/RE (accession number SRR1693793); (ii) as well as the two secondary koala C. pecorum strains (Gun/koa1/Ure-ß, SA/K2/UGT-ß) identified in the same study [2]. Conserved regions were identified in 19 C. pecorum genomes using all-versus-all BLAST search. Syntenic regions were individually aligned using Muscle [36] and concatenated to produce a ~ 280 kbp core genome alignment. Poorly aligned regions were removed from the alignment using GBLOCKs with the minimum length of a block set to 5 and no gap positions were allowed. Recombination regions were predicted using Gubbins [37] and subsequently filtered from the alignment. A mid-point rooted phylogenetic tree was constructed from the genome alignment with PhyML 3.1 using the GTR model. Bootstrap values were calculated using 500 replicates.
PCR amplification and phylogenetic analyses of toxC
During pairwise comparative genomic analyses, the presence of a third copy of the cytotoxin gene (toxC) in porcine L71 genome was identified. Primers (toxC For (5’ TCA GAG AGA AGC AGC TTA G 3’) and toxC Rev (5’ TTC TTG AGA AGT AAC ACT ACG 3’)) were designed to amplify a 644 bp fragment of the toxC gene in order to confirm the presence of this sequence. Using purified L71 genomic DNA as a template (and positive control), and purified C. pecorum koala MC/Marsbar and IpTaLe, cattle E58, porcine L1, L17 and sheep IPA genomic DNA as negative controls, conventional PCR assay was performed to amplify the 644 bp toxC fragment. PCR assays for 644 bp toxC fragment were prepared to a total reaction volume of 50 μl including 1X Amplitaq Gold® 360 Master Mix (Life Technologies, Victoria, Australia), 0.3 μM of forward and reverse primer each (Integrated DNA Technologies New South Wales, Australia) and 3 μl DNA template. The cycling conditions included an initial denaturation (10 min, 95 °C) followed by 35 cycles of denaturation (30s, 95 °C), annealing (30s, 53.5 °C) and extension (1 min, 72 °C), followed by a final extension (7 min, 72 °C). The amplicon was then purified and dideoxy sequenced (described in more detail in the following section below). After successful amplification and confirmation of the toxC fragment in L71 only, we tested the remaining eight porcine, seven cattle, two sheep (W73 and JP-1-751) and five koala C. pecorum isolates (Additional file 4: Table S3) for the presence of the toxC 644 bp fragment sequence.
Identification of the full length L71 toxC sequence was checked in BLAST and the midpoint rooted Bayesian phylogeny was constructed using alignment of all three cytotoxin (tox A, B and C) genes from a subset of 12 C. pecorum analysed in this study. The phylogenetic tree was constructed with MrBayes as implemented in Geneious 7.1.4, using the HKY + I model with four Markov Chain Monte Carlo (MCMC) chains with a 150 000 generations, sampled every 100 generations and with the first 10 000 trees were discarded as burn-in. C. muridarum Nigg (accession number: AE002160) three cytotoxin gene sequences were also included in the alignment for phylogenetic analyses, with the C. muridarum Nigg tox 3 used as an out-group.
C. pecorum plasmid contig identification
The L1 7.5kbp plasmid contig was annotated with RAST [29], and further curated in Geneious 7.1.4. Additionally, pCpec L1 sequence was also blasted against raw reads of C. pecorum porcine L17 and L71, sheep IPA, cattle NSW/Bov/SBE, and koala Mc/Marsbar, DBDeUG, IpTaLE, SA/K2/UGT and Gun/k1/Ure. pCpec 7.5kbp sequence was only identified in the three koala C. pecorum Mc/Marsbar, DBDeUG and IpTaLE raw reads and also used in this study.
pCpec conv. PCR confirmatory assay design
Newly identified pCpec sequences from a porcine L1 and three koala C. pecorum genomes were aligned using ClustalX. Primer pair (P frag For 5’ GTT CAC ACT CTG CCT CAT C 3’ and P frag Rev 5’ CCT ATT TAT TGG CGT CTA GG 3’) was designed to amplify a 522 bp plasmid fragment which includes a conserved intergenic region and a part of the plasmid gene CDS8. Primer sequences were tested in silico for secondary structures and dimerisations at Integrated DNA Technologies, Inc OligoAnalyzer 3.1 online tool (https://sg.idtdna.com/calc/analyzer). Additionally, plasmid isolation was performed from fresh available koala C. pecorum MC/Mars, DBDeUG, and IpTaLE cultures. Plasmid isolation and purification was performed using GeneJET Plasmid Midiprep 25 reactions, ThermoFisher Scientific, Victoria, Australia, as per manufacturer’s instructions. Freshly extracted plasmids were digested with NEB SacI (Genesearch Pty Ltd, Queensland, Australia) and visualised on the Ethidium Bromide stained 1.2 % gel, using NEB 2-Log DNA Ladder(0.1 – 10kbp) (Genesearch Pty Ltd, Queensland, Australia).
Using purified porcine L1 genomic DNA and extracted plasmid DNA from koala C. pecorum MC/Mars, DBDeUG, and IpTaLE as templates and positive controls (as plasmid contigs were identified in their genome sequences), conventional PCR to amplify 522 bp plasmid fragment was performed. In the same assay we used purified pig L17 and L71 genomic DNA as negative controls (as plasmid contigs were not identified in their genome sequences). The plasmid primers were additionally tested against genomic avian C. psittaci DNA extracted from a cultured isolate in order to test for unspecified primer binding. After successful amplification of the 522 bp plasmid fragments from templates only, the presence of the amplicon was confirmed on 1.5 % TBE gel, purified and sent off for confirmatory dideoxy sequencing.
We tested the detection limit of our pCpec screening assay, based on a conventional PCR using: i) serially diluted pCpec from 1010 to 101 copies/μl as a template in triplicate; and ii) C. pecorum positive clinical sample Bella UGT, with 106C. pecorum genome copy number/μl diluted to 101C. pecorum genome copy number/μl, as a template in duplicate. The detection limit for pCpec assay used in the present study was from 1010 to 102 pCpec fragment copies/μl, as determined by amplicon visualisation on 1.5 % TBE gel.
PCR-based pCpec screening
After confirming the 522 bp amplicon sequence, we applied the above described assay to screen for plasmid presence in koala and livestock C. pecorum samples. The full lists of screened samples are outlined in Additional file 4: Table S3 and Additional file 6: Table S4. The testing of these swab samples, collected as a part of routine diagnostic investigations of koalas and livestock, has been considered by the University of Sunshine Coast (USC) Animal Ethics Committee and the requirement for ethics approval was waived (AN/E/14/01 and AN/E/14/02). PCR assays for screening 522 bp pCpec fragment were prepared to a total reaction volume of 50 μl including 1X Amplitaq Gold® 360 Master Mix (Life Technologies, Victoria, Australia), 0.3 μM of forward and reverse primer each (Integrated DNA Technologies New South Wales, Australia) and 3 μl DNA template. Negative (dH20 and L17 and/or L71 DNA) and positive (MC/Marsbar plasmid DNA and L1 gDNA) controls were included in each amplification assay. The cycling conditions included an initial denaturation (10 min, 95 °C) followed by 35 cycles of denaturation (30s, 95 °C), annealing (30s, 57.5 °C) and extension (1 min, 72 °C), followed by a final extension (7 min, 72 °C). Upon amplification, PCR products were detected on a 2 % Ethidium bromide agarose gel and visualised under an UV transilluminator and purified, using a High Pure PCR product purification kit (Roche, New South Wales, Australia). A subset of 15 PCR products was directly sequenced using a BigDye Terminator v3.1 Cycle Sequencing kit (Life Technologies, Victoria, Australia) and subsequently purified according to the manufacturer’s instructions. Sequencing was performed at the Institute for Future Environments (IFE), Queensland University of Technology (QUT), Brisbane, Australia using the Applied Biosystems ABI3500 Gene analyser.
Results and discussion
Porcine C. pecorum L1, L17 and L71 genome assemblies
Using C. pecorum-specific sequence capture probes [2], we acquired whole genome sequences for three porcine C. pecorum isolates. C. pecorum strains L1 and L17 were isolated from the lungs of two different pigs presenting with pneumonia, while the L71 strain was a joint isolate from a case of polyarthritis [20, 21]. Consistent with the high average read depth (~2400X), de novo assembly resolved the genomes of these C. pecorum isolates into 3 – 5 contigs for each isolate (Table 1). Read-mapping to a reference C. pecorum E58 genome [15] confirmed almost whole chromosome coverage (~100 %) for all three genomes. Similar to the previously described C. pecorum genomes [1–3, 15], our annotation pipelines predicted ~ 1000 coding DNA sequences (CDS’s), including the three rRNA and 38 tRNA genes. The draft genome sizes of ~1.106 Mbp are almost identical to all other available C. pecorum genomes, with the exception of L71 which has a slightly larger draft genome size of 1.115 Mbp (Table 1).
Interestingly, we also identified a distinct 7.5kbp contig (with 153X coverage), assembled only from C. pecorum L1 strain reads. BLAST searches revealed that this contig shared 70 % sequence identity to other related chlamydial species plasmids and 99 % sequence identity to a 710 bp sequence previously identified from a suspected C. pecorum plasmid (accession number M32752.1) [13], based on blastn and discontiguous megablast BLAST searches (Additional file 1: Table S1). The annotation of this L1 7.5kbp contig resulted in eight CDSs specific for chlamydial plasmids.
Sequence capture methodology continues to be an effective tool for acquiring high quality whole genome sequence data [2, 38, 39], especially when cultured isolate gDNA of high concentration and genome copy number is used as a template, as in the present study. High read depths, complete genome coverage, and sequence homogeneity for our three pig strains was not surprising, as they were all pure cultured isolates, contrasting the sequence heterogeneity previously seen in C. pecorum-positive clinical swab studies [2]. Interestingly, design of the C. pecorum species-specific probes [2] also allowed for identification of distinct and/or novel sequences, such as the full length plasmid contig detected in the L1 draft genome. Now that whole genome sequences for porcine C. pecorum are available, the use of C. pecorum-specific RNA probes for sequence capture of C. pecorum DNA can be further improved in order to reveal features not previously observed or investigated, and to improve coverage of the polymorphic genomic regions in this species.
Re-evaluating the genetic differences between C. pecorum strains
The previously described C. pecorum gene order and synteny [3, 15] remained conserved in the porcine C. pecorum genomes with nearly all variation, once again, limited to SNPs (Fig. 1). Comparison of the porcine L1, L17 and L71 genome sequences against those from other hosts revealed the most SNPs between our C. pecorum porcine strains and the genomes of C. pecorum strains previously sequenced from sheep (~11 800 SNPs; Table 2). The closest C. pecorum strains from other hosts, on the other hand, were from a cow (PV3056/3) and a koala (SA/K2/UGT) with approximately 5800 and 6600 SNPs, respectively. Interestingly, the majority of koala C. pecorum strains differed from porcine strains by ~ 9500 SNPs, less than sheep and cattle strains (Table 2). Pairwise comparison of the genome sequences revealed that the majority of SNPs were found in two major clusters, including (i) the plasticity zone (PZ); and (ii) the major polymorphic membrane (pmp) gene cluster (Fig. 1).
Fig. 1.
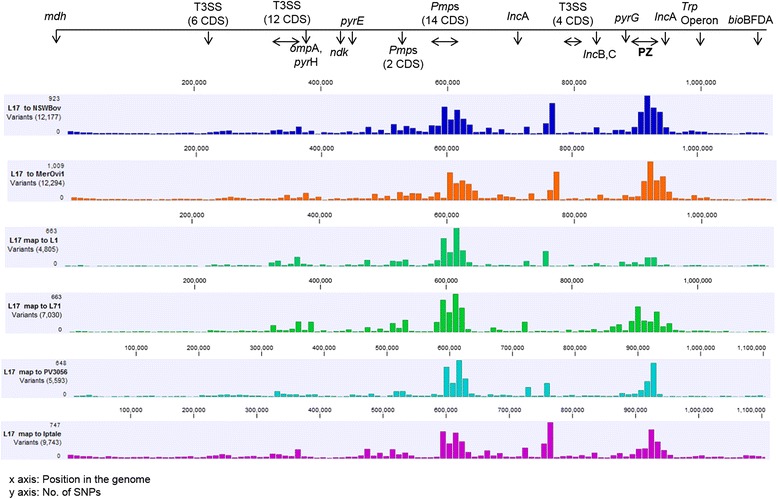
SNP distribution and identification of genomic elements across the porcine L17 genome in comparison to cattle NSW/Bov/SBE and PV3056, sheep Mer/Ovi1/Jnt, porcine L1 and L71, and koala IpTaLe genomes. All genomes start from malate dehydrogenase (mdh) gene. Genomic location of Type 3 Secretion System gene clusters (T3SS), major outer membrane protein (ompA), pyrimidine genes (pyrEHG, ndk), polymorhic membrane protein (pmp) gene clusters, inclusion proteins genes (IncABC), tryptophan (TrpABFCDR, kynU, prsA) and biotin operon (bioBFDA) genes are denoted on the horizontal line above the histograms. Variants, represented as histograms of a 10kbp size, were determined in CLC Genomics (CLCBio, Qiagen) platform
Table 2.
The number of nucleotide differences between porcine C. pecorum genomes when mapped to a reference porcine, sheep, cattle, and koala C. pecorum genome
| Mapped to Reference | Porcine L1 | Porcine L17 | Porcine L71 |
|---|---|---|---|
| Porcine L1 | – | 4 376 | 6 665 |
| Porcine L17 | 4 805 | – | 7 030 |
| Porcine L71 | 6926 | 6276 | – |
| Cattle PV3056/3 | 4 924 | 5 593 | 7 199 |
| Koala Sa/K2/UGT | 6321 | 6533 | 7215 |
| Sheep W73 | 11 514 | 12 682 | 11 957 |
| Sheep IPA | 11 386 | 12 695 | 11 939 |
| Sheep Mer/Ovi1/Jnt | 10 850 | 12 294 | 11 502 |
| Sheep Nar/S22/Rec | 11 986 | 12 533 | 11 347 |
| Cattle NSW/Bov/SBE | 10 798 | 12 177 | 11 457 |
| Koala Mc/Marsbar | 8 235 | 9 482 | 9 916 |
| Koala IpTaLE | 8 565 | 9 743 | 10 171 |
Not surprisingly, averaging at ~600 SNPs/10Kbp, the pmp gene regions were a major contributor of single nucleotide differences (Fig. 1, Additional file 2: Table S2). Overall sequence similarity of different C. pecorum pmp gene families from the porcine, sheep, cattle, and koala strains ranged from 80 – 100 % (Additional file 2: Table S2). The Bayesian phylogenetic analyses resolved the pmp A, B, E, E’, D, and H gene families in clades respective to their designated gene family, while the pmpG family was the most divergent, resolving each of the eight pmpG subtypes into its own diverse clade (Additional file 3: Figure S1, A-B). Porcine C. pecorum L1, L17, and L71 strains had an identical genomic organisation, subtype and number of predicted pmp genes (n = 15) as the previously characterised sheep, cattle, and koala strains [1, 3] (Additional file 3: Figure S1, A-B; Additional file 2: Table S2). Sequence analyses of each of the 15 pmp genes from the porcine C. pecorum strains, indicated that only pmpG1 and pmpG9 type appear to be under positive selection pressure, with observed dn/ds ratios of 1.49, and 1.63, respectively (Additional file 2: Table S2). Subsequent sequence analyses of the pmpG1, and pmpG9 from all 16 C. pecorum strains analysed in this study, also returned dn/ds ratios > 1. Interestingly, pmp G1 and G9 phylogenies were similar to the phylogenetic relationships constructed from the core genomes of the 16 C. pecorum strains analysed in this study (described below), as well as the previous study [2] (Additional file 3: Figure S1,C-D).
In the recent sheep, cattle and koala C. pecorum comparative genomic study [1], the pmpG region was also identified as the most diverse genomic region, however, most of the genetic variation observed resulted in synonymous substitutions. In our study, variation in the pmp genes also mainly resulted in synonymous SNPs, with the exception of C. pecorum pmp genes G1 and G9, which appeared to be under positive selection. Predicted to have a major role in chlamydial infection due to their adhesive function in the interaction with host [40, 41], pmp genes are predicted to maintain some hypervariability in order to evade immune defences [42, 43]. Rapid diversification and evolution of the pmpG genes family has been previously observed in the genomic studies of the related ovine pathogen C. abortus [44], and avian pathogen C. psittaci, where it has been suggested that these genes may play a role in adaptation to different hosts and environments [45, 46]. The observed positive selection on these genes in the C. pecorum genomes may support a role in host adaptation for this pathogen, an aspect that may warrant extended investigation.
While the remaining genetic variation was evenly distributed across the porcine C. pecorum chromosomes, we identified additional eight genes under positive selection in the polymorphic regions. As outlined in Additional file 4: Table S3, cpL1_0338 and cpL1_0441, were both identified as effectors of the chlamydial Type 3 Secretion System (T3SS), a system of structural, chaperone and secreted effector proteins with its main function in host cell manipulation and subversion of the host cellular processes [47, 48]. The cpL1_0338 was predicted to be orthologous to a C. psittaci secreted T3SS protein (SINC) [49], while the cpL1_0441 was a well-studied chlamydial T3SS translocated actin-recruiting protein (Tarp) homologue [50]. In the porcine C. pecorum genomes, although polymorphic, the remaining T3SS genes were under negative selection. Positive selection on the C. pecorum Tarp homologue is perhaps not surprising, as the chlamydial Tarp was previously recognised as an important antigenic protein [51, 52]. Further, genomic studies of the C. psittaci and C. trachomatis revealed that the variation in the T3SS effector genes (such as Tarp) could also contribute to the differences in virulence, and host and/or tissue tropism associated with these pathogens [46, 53]. Presently the exact role of T3SS effectors in C. pecorum infections remains largely unknown, and will require more comprehensive in silico as well as cell biology analyses comprising of isolates from healthy, as well as diseased hosts to fully dissect T3SS effector roles. The remaining genes under positive selection were: (i) cpL1_0291, identified as a homologue of a previously described C. pecorum surface binding protein gene (SrpA), also under positive selection in koala strains [1]; (ii) cpL1_0675, a homologue of a macro domain (ADP-ribose binding) protein; and (iii) four genes encoded conserved chlamydial hypothetical proteins of unknown function (Additional file 4: Table S3).
Variation in the C. pecorum PZ
In addition to the analysed pmp genes, the PZ, spanning from inosine-5'-monophosphate dehydrogenase (impd) to acetyl-CoA carboxylase (accB), also displayed significant variability (Fig. 1). The PZ is a region of focus in chlamydial comparative genomic studies due to the presence and/or absence of a range of established chlamydial virulence factors [54–56]. In our study, like other C. pecorum strains [1–3], the porcine C. pecorum PZs were found to contain: (i) three purine biosynthesis genes, (ii) a MAC/Perforin, (iii) a variable number of phospholipase D (PLD) genes, (iv) two copies of cytotoxin genes, (v) and two acetyl-CoA-carboxylase genes (Fig. 2). Some notable genetic variation, however, was also observed in these porcine C. pecorum strains including (i) major differences in the number of PLD genes and; (ii) the presence of a third copy of the cytotoxin gene in the L71 strain, as summarised in Fig. 2. In terms of PLD genes, five were observed in L17, a comparable number to that identified in the genome sequences of C. pecorum strains from sheep (IPA, W73 and P787), koalas (MC/Marsbar, IpTaLe, DBDeUG) and cattle (E58, NSW/Bov/SBE). The previously sequenced bovine PV3056/3 isolate also had four PLDs like L71, while the porcine L1 isolate is presently the only C. pecorum strain with six PLDs (Fig. 2). The variable number of PLDs is not unique to C. pecorum [3], as related species such as C. trachomatis and C. muridarum have also been shown to have a variable number of PLD genes among strains [55]. It has been suggested that the number of PLD genes could influence virulence [56], but whether this is the case for C. pecorum is still unclear, as the majority of the sequenced C. pecorum were sampled from hosts with evidence of chlamydial disease.
Fig. 2.

Graphical representation of the C. pecorum cattle NSW/Bov/SBE and PV3056, sheep Mer/Ovi1/Jnt, pigs L1, L17 and L71, and koala SA/K2/Ugt plasticity zones (PZs) tblastX comparisons. The coloured arrows represent PZ genes according to their function, as outlined in the legend. The intensity of grey shaded areas corresponds to the sequence identity. The image was generated with Easyfig [34]
In terms of cytotoxin genes, almost all of the analysed strains had two copies of the cytotoxin gene, with observed toxA truncation in C. pecorum cattle NSW/Bov/SBE and koala SA/K2/UGT strains (Fig. 2). The porcine L71 strain however, had three copies of the cytotoxin gene (toxA: cpL71_0929, toxB: cpL71_0933, toxC: cpL71_0931) (Fig. 2). Among the genus Chlamydia, only C. muridarum has three copies of the cytotoxin gene [55]. Sequences from this latter species were included as an out-group in the tox phylogenetic tree to investigate whether the third tox copy identified in the L71 is similar to the C. muridarum third tox copy (Additional file 5: Figure S2). Analyses of the C. pecorum cytotoxin genes revealed that L71 toxC is more similar to C. pecorum toxA (69 % sequence similarity) than toxB (50.5 % sequence similarity). A mid-point rooted Bayesian phylogenetic tree of 11 C. pecorum toxA and toxB sequences with tox sequences from L71 and C. muridarum further revealed that the L71 toxC may be a duplication of the C. pecorum toxA, as evidenced by the clustering of this gene with C. muridarum tox1 and other C. pecorum toxA genes.
To see if toxC could be found in strains from other hosts, we designed a toxC-specific PCR (amplifying a 644 bp toxC specific fragment) to screen C. pecorum strains from pigs, sheep, cattle, and koalas (Additional file 4: Table S3). This toxC fragment was detected only in three more isolates: porcine pneumonia L39, L40 and HsLuRz (Additional file 4: Table S3). These strains were isolated from the same chlamydial outbreak in 1969 among Austrian pigs as the L1, L17 and L71 strains and the remaining five porcine strains (outlined in Additional file 4: Table S3) [20, 21]. Thus, based on our genomic and preliminary toxC analyses, it appears that the 1969 C. pecorum outbreak among Austrian pigs was polyclonal, with strains involved having two or three cytotoxin genes. Variability in the number of tox genes and our previously noted truncation of certain tox genes in some C. pecorum strains [1], raises the question over the function and impact of this gene in C. pecorum virulence.
Expansion on C. pecorum phylogenetic relationships
Using a C. pecorum Multi Locus Sequence Typing (MLST) scheme [23], we recently showed that porcine C. pecorum strains clustered in a clade separate to that of C. pecorum strains from a diverse range of hosts including sheep, koalas, and deer [16]. In the current study, core genome alignment, including all 19 available C. pecorum sequences, resolved six clades in the maximum likelihood tree (Fig. 3). The three porcine C. pecorum strains clustered with cattle PV3056 and a koala SA/K2/UGT strains, forming the first well supported clade (Fig. 3). Four primary koala strains resolved into their own exclusive second clade, while the third clade consisted of the European sheep C. pecorum isolates W73 and P787. USA polyarthritis C. pecorum IPA isolate formed its own fourth clade, as did the Australian sheep rectal Nar/S22/Rec strain. The final clade mainly consisted of Australian cattle and sheep strains, along with two koala C. pecorum strains (SA/K2/UGT-ß and Gun/Koa1/Ure-ß) detected as mixed infections with genetically distinct C. pecorum strains [2], and the USA cattle encephalomyelitis E58 isolate (Fig. 3).
Fig. 3.

A mid-point rooted phylogenetic tree, constructed from the 280kbp core genome alignment with PhyML 3.1 using the GTR model, using 19 C. pecorum pig, cattle, sheep and koala strains. Bootstrap values were calculated using 500 replicates. Bootstrap values > 75 are displayed on the tree nodes. Hosts are indicated by the colouring in the legend
Sequence-based detection of the C. pecorum plasmid
No plasmids were reported in any of the C. pecorum genomes published prior to this study [1–3]. The unexpected finding of a C. pecorum 7.5 kbp plasmid (pCpec) in the L1 genome prompted us to re-investigate the presence of the plasmid in other sequenced livestock and koala C. pecorum, using the raw reads available in our collection [1, 2]. Interestingly, full length plasmid sequences were detected only in the genomic data of C. pecorum Mc/Marsbar, DBDeUG and IpTaLE strains from koala hosts [1]. These additional three plasmid contigs were confirmed based on BLAST homology search and read mapping to the originally identified L1 plasmid. All identified plasmids shared an identical annotation with eight CDSs and four 22 bp tandem repeats (Fig. 4), as previously described in related chlamydial plasmids [6]. Briefly, CDSs 1 (pGP8), 2 (pGP8), 3 (pGP1), and 7 (parA) were denoted as putative integrase, helicase, and a partioning plasmid proteins, while the CDSs 4 (pGP2), 5 (pGP3), 6 (pGP4), and 8 (pGP6) were denoted as putative chlamydia-specific plasmid virulence proteins. The 7.5 kbp pCpec sequences were 99.1 % similar to each other and shared an overall sequence similarity of 67 – 70 % to other chlamydial plasmids [6, 7] (Additional file 1: Table S1). The exact function of the pCpec genes and/or their predicted products will require further in vitro investigations.
Fig. 4.

Sequence alignment of the annotated characterised pCpecs from a pig and three koala C. pecorum strains. SNPs are highlighted in black. On the identity heat map above the alignment, green denotes 100 % sequence similarity while yellow denotes between 30 % to less than 100 % sequence similarity
Distribution of the C. pecorum plasmid
To investigate the plasmid distribution in C. pecorum strains, we developed a specific PCR assay that targets a 522 bp pCpec fragment to screen: (i) 114 C. pecorum PCR positive samples collected from 67 livestock (Table 3), and (ii) 113 C. pecorum PCR positive samples from 73 koalas from different populations (Table 4). As summarised in Table 3, 38.6 % of the livestock C. pecorum strains sampled had a plasmid, with the highest detection of 63.6 % in the porcine isolates (Additional file 4: Table S3 and Additional file 6: Table S4). pCpec was present in 38.4 % of the sheep C. pecorum strains sampled, with plasmids detected in C. pecorum strains detected at a range of anatomical sites (Table 3, Additional file 6: Table S4). In sheep strains, we were further able to observe that (i) genetically diverse strains (as determined by previous C. pecorum MLST typing [24]) infecting different sites in the same host (e.g. Nar/S45/LE and Rec; Hey/S129/Eye and Rec (Additional file 6: Table S4)) can carry pCpec; and (ii) both pCpec-positive and pCpec-negative genetically diverse strains can circulate in a single flock (e.g. Nar/S45/Eye and Rec; Nar/S25/Vag and Rec; Nar /S40/Eye, Vag and Rec; Nar/S84 Eye and Rec (Additional file 6: Table S4)).
Table 3.
Plasmid distribution in C. pecorum positive clinical samples and cultured isolates from pigs, sheep and cattle
| No. of hosts | No. of samples | Plasmid detection |
|---|---|---|
| Sheep (N = 44) | 86 | 33/86 (38.4 %) |
| Cattle (N = 12) | 17 | 4/17 (23.5 %) |
| Pigs (N = 11) | 11 | 7/11 (63.6 %) |
| Total: | ||
| 114 | 44/114 (38.6 %) |
Table 4.
Plasmid distribution in C. pecorum positive clinical samples and cultured isolates from different koala populations across Australia
| No. of hosts | No. of samples | Plasmid detection |
|---|---|---|
| QLD koalas (N = 30) | 44 | 32/44 (72.7 %) |
| NSW koalas (N = 15) | 19 | 16/19 (84.2 %) |
| Vic koalas (N = 9) | 14 | 11/14 (78.5 %) |
| SA koalas (N = 19) | 36 | 4/36 (11.1 %) |
| Total: | ||
| 113 | 63/113 (55.8 %) |
In cattle, pCpec was detected in ocular, brain, joint and faecal C. pecorum isolates (Table 3, Additional file 4: Table S3). pCpec was less common in the limited number of clinical samples analysed (Table 3, Additional file 6: Table S4). A subset of these C. pecorum positive clinical samples, collected from two calves (WA/B65 and WA/B31), with sporadic bovine encephalomyelitis (SBE) were described previously in a case study [25] where C. pecorum MLST [23] indicated that brain, liver and lung C. pecorum WA/B65 and WA/B31 strains were of the same C. pecorum genotype (denoted ST23) as other SBE isolates such as NSW/Bov/SBE and E58. In the present study we observed that all of these samples were pCpec negative, but that a genetically distinct C. pecorum positive ileal sample was pCpec positive. pCpec was similarly absent in several other C. pecorum SBE isolates (Additional file 4: Table S3). The absence of pCpec from SBE-associated C. pecorum strains based on this limited pCpec screening may imply that the pCpec is not a virulence factor that affects SBE pathogenesis.
The plasmid was more commonly detected in the koala C. pecorum samples studied, with 72.7 %, 84.2 % and 78.5 % distribution in the samples collected from Queensland (QLD), New South Wales (NSW) and Victoria (Vic), respectively (Table 4). In contrast, pCpec was less common in the South Australian (SA) koala samples analysed (11.1 %) (Table 4).
Together, this data indicates that the C. pecorum plasmid is not ubiquitous, in contrast to C. trachomatis, where nearly all naturally occurring strains contain the plasmid [57]. In terms of confidence in our detection, we believe that the sensitivity of our pCpec assay was sufficient to detect plasmid even in low C. pecorum (<1x102C. pecorum genome copies) positive samples as the chlamydial plasmid is thought to have between two and 10 copies/per chlamydial genome, based on other studies [57]. As outlined in Additional file 4: Table S3 and Additional file 6: Table S4, the number of C. pecorum genomes for each screened sample ranged from a 5.0x101 to 9.8x106C. pecorum genome copies/μl of the extracted DNA (as determined by the C. pecorum-specific qPCR screen [5]), while the tested pCpec fragment lower detection limit was 1x102 plasmid copies/μl. Indeed, in the present study, there was no apparent relationship between ability to detect plasmid and the number of genome copies in a sample, with plasmid detected in koala A3, R15, R1-17 UGT and sheep Cur/L236/Vag samples with only ~1x102C. pecorum genome copies (Additional file 6: Table S4). Nevertheless, we do also acknowledge that it is possible that we may have missed detection of pCpec in samples with < 1x102C. pecorum genome copies if each C. pecorum genome were to be associated with only single pCpec copy.
The reliance on archived samples in the present study precluded observation of trends in disease, tissue and/or host specificity for plasmid distributions; most PCR positive samples or isolates were from diseased animals, sample sizes were small, and sampling was spatially clustered (Additional file 4: Table S3 and Additional file 6: Table S4). As prevalence among such samples may not accurately reflect prevalence in wider populations and small sample sizes would confer very wide confidence intervals on prevalence estimates, it was not possible to compare prevalence across geographic locations, hosts, or disease states with any certainty. However, the markedly lower apparent prevalence of pCpec in South Australian koala samples (11 %) (Table 4, Additional file 6: Table S4) relative to koala samples from QLD, Vic, and NSW (73 – 84 %) appears worthy of further investigation. Anecdotal evidence and preliminary PCR-based screening studies [58] (Speight K.N., Polkinghorne A., Penn R., Boardman W., Timms P., Fraser T., Johnson K., Faull R., Bate S., Woolford L., 2015, unpublished observations), suggest that C. pecorum infections in SA may be less common and pathogenic than those described in eastern and northern Australian states (QLD and NSW) [17]. Similarly low prevalence of chlamydial disease has been proposed for Victorian koalas. In other chlamydial species, there is strong evidence linking the chlamydial plasmid to pathogenic potential. For example, in the mouse model, it was demonstrated that plasmid deficient C. trachomatis isolates are less infective and less virulent [9]. Similarly, C. muridarum studies demonstrated critical role of the plasmid in the development and severity of intrauterine infections [8, 59]. Given the multifactorial nature of disease pathogenesis, however, the picture is unlikely to be simple, particularly in the koala. As such, further studies are required to examine the impact of the presence or absence of this plasmid on C. pecorum infection, growth and pathogenicity.
As an interesting sidenote, in C. trachomatis, it was also observed that the chlamydial plasmids are correlated with the accumulation of glycogen granules inside the inclusion (used as an energy source) and that some plasmid loci (like pgp4) may regulate transcription of the corresponding chromosomal genes involved in glycogen pathways [7, 60]. In their 1992 study [22], Kaltenboeck and Storz examined the biological properties of the same pig strains that we used for our genomic and plasmid analyses. Inclusions of L1, R106, 1710S, and 1920Brz pig strains were observed to be aberrant, coarse and patchy, with pleomorphic reticulate bodies, in contrast to other pig strains (such as L71 and L40) with regular inclusions. In our study we detected plasmid in the same strains (Additional file 4: Table S3), with plasmid fully characterised from L1, while the regular inclusion strains L71 and L40 were plasmid negative. Given the differing distribution of the C. pecorum plasmid and the otherwise conserved coding sequences of this plasmid relative to the plasmid in other chlamydial species, this study raises important questions over the function of the C. pecorum plasmid. These questions can only be answered with additional in vitro and in vivo investigations.
Conclusion
In the present study, we sequenced three C. pecorum genomes from pigs presenting with pneumonia (L1 and L17 strains) and polyarthritis (L71), and compared them to genomes of other C. pecorum strains from sheep, cattle and koala. The main genetic differences were limited to the highly variable pmp region and the C. pecorum PZ. Besides the variable number of phospholipase D genes, we also observed a third full copy of the cytotoxin gene (toxC) in the PZ of the porcine L71 genome. Preliminary screening for a toxC fragment in other C. pecorum strains revealed that this additional tox gene could only be found in other porcine strains (L39, L40 and HsLuRz).
The most significant and novel genomic feature described in this study was the identification of the chlamydial plasmid in C. pecorum. Based on our PCR-based screening approach, this plasmid does not appear to be ubiquitous, raising questions over its function and impact on chlamydial pathogenesis in light of the important role it plays in other chlamydial species such as C. muridarum [8, 59]. It appears possible that the plasmid is less common in strains infecting koalas in South Australia, relative to those in other states. Although no association was observed between the presence or absence of the plasmid in association with tissue, host or disease in the present study, differences in the distribution of this plasmid among koala populations and individuals with different levels of chlamydial disease appears to warrant further investigations. Whether the C. pecorum plasmid is a virulence factor or just a “harmless accessory” otherwise, remains to be elucidated.
Availability of supporting data
All of the available supporting data from this manuscript is provided as Additional Files.
Acknowledgments
This work was funded by an ARC Discovery Project grant (DP130102066) and a Queensland Government Department of Environment and Heritage Protection Koala Research Grant (KRG18) awarded to AP and PT. We thank (i) Alyce Taylor-Brown and Marion Desclozeaux for helpful discussions and technical assistance, (ii) Mike Humphreys, Institute of Genome Sciences, University of Maryland for assistance with C. pecorum sequence capture, (iii) Tamieka Fraser for assisting with koala swab processing and screening, (iv) Evelyn Walker for providing sheep C. pecorum positive DNA samples used in our analyses; and (v) Eileen Roulis for her advice on bioinformatics analyses. We acknowledge the Queensland Department of Transport and Main Roads, specifically the Moreton Bay Rail Link project team, and the dedicated staff at Endeavour Veterinary Ecology for collecting samples from Queensland koalas.
Abbreviations
- Bp
Base pairs
- CDS
Coding DNA sequence
- kbp
Kilo base pairs
- Mbp
Mega base pairs
- MLST
Multi Locus Sequence Typing
- PCR
Polymerase chain reaction
- pmp
Polymorphic membrane protein
- PZ
Plasticity zone
- qPCR
Quantitative polymerase chain reaction
- rRNA
Ribosomal RNA
- SNP
Single nucleotide polymorphism
- tRNA
Transfer RNA
Additional files
C. pecorum L1 plasmid sequence identity comparisons based on discontiguous megaBLAST hits. (PDF 338 kb)
Sequence analyses of the porcine L1, L17, and L71 C. pecorum pmp gene families. (PDF 357 kb)
{kind=link}
Bayesian phylogenetic analyses of the C. pecorum pmp gene subtypes. Posterior probabilities > 0.70 are displayed on the tree nodes. A: C. pecorum pmps A,B, D, E, E, H gene family phylogenies from the 16 porcine, sheep, cattle, and koala C. pecorum strains analysed in this study; B: C. pecorum pmpG gene family phylogeny from the 16 porcine, sheep, cattle, and koala C. pecorum strains analysed in this study; C: pmpG1 phylogenetic tree constructed from the 2406 bp alignment of 16 porcine, sheep, cattle, and koala C. pecorum strains analysed in this study; and D: pmpG9 phylogenetic tree constructed from the 2841 bp alignment of 16 porcine, sheep, cattle, and koala C. pecorum strains analysed in this study. Hosts are indicated by the colouring in the legend. (PNG 300 kb)
Characteristics of the polymorphic chromosomal genes under positive selection analysed in the present study from three porcine C. pecorum L1, L17, and L71 strains. (PDF 267 kb)
{kind=link}
Bayesian phylogenetic analyses of the cytotoxin genes from the subset of 12 porcine, sheep, cattle, and koala C. pecorum strains. Posterior probabilities > 0.70 are displayed on the tree nodes, while C. muridarum Nigg tox 3 is used as an out-group. (PNG 219 kb)
C. pecorum isolates and clinical samples Additional file 4: Table S3 and Additional file 6: Table S4. (XLSX 23 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MJ performed the experimental studies, comparative genomics analyses and prepared the manuscript. NB assisted with bioinformatics and phylogenetic analyses. BK and DPH maintained the chlamydial strains used in this study. CW performed samples processing and PCR screening. LW, KNS, AG and CF collected koala samples used in the present study. GSM assisted with C. pecorum probe design and performed culture-independent genome sequencing. PT and AP conceived and designed the study and helped write the paper. All authors have read and approved the final manuscript.
References
- 1.Bachmann N, Fraser T, Bertelli C, Jelocnik M, Gillett A, Funnell O, et al. Comparative genomics of koala, cattle and sheep strains of Chlamydia pecorum. BMC Genomics. 2014;15(1):667. doi: 10.1186/1471-2164-15-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachmann NL, Sullivan MJ, Jelocnik M, Myers GSA, Timms P, Polkinghorne A. Culture-independent genome sequencing of clinical samples reveals an unexpected heterogeneity of infections by Chlamydia pecorum. J Clin Microbiol. 2015;53(5):1573–1581. doi: 10.1128/JCM.03534-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sait M, Livingstone M, Clark EM, Wheelhouse N, Spalding L, Markey B, et al. Genome sequencing and comparative analysis of three Chlamydia pecorum strains associated with different pathogenic outcomes. BMC Genomics. 2014;15(1):23. doi: 10.1186/1471-2164-15-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohamad KY, Kaltenboeck B, Rahman Kh S, Magnino S, Sachse K, Rodolakis A. Host adaptation of Chlamydia pecorum towards low virulence evident in co-evolution of the ompA, incA, and ORF663 loci. PLoS ONE. 2014;9(8):e103615. doi: 10.1371/journal.pone.0103615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marsh J, Kollipara A, Timms P, Polkinghorne A. Novel molecular markers of Chlamydia pecorum genetic diversity in the koala (Phascolarctos cinereus) BMC Microbiol. 2011;11(1):77. doi: 10.1186/1471-2180-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas NS, Lusher M, Storey CC, Clarke IN. Plasmid diversity in Chlamydia. Microbiology. 1997;143(6):1847–1854. doi: 10.1099/00221287-143-6-1847. [DOI] [PubMed] [Google Scholar]
- 7.Rockey DD. Unraveling the basic biology and clinical significance of the chlamydial plasmid. J Exp Med. 2011;208(11):2159–2162. doi: 10.1084/jem.20112088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Huang Y, Yang Z, Sun Y, Gong S, Hou S, et al. Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect Immun. 2014;82(12):5327–5335. doi: 10.1128/IAI.02576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sigar IM, Schripsema JH, Wang Y, Clarke IN, Cutcliffe LT, Seth-Smith HM, et al. Plasmid deficiency in urogenital isolates of Chlamydia trachomatis reduces infectivity and virulence in a mouse model. Pathog Dis. 2014;70(1):61–69. doi: 10.1111/2049-632X.12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seth-Smith HM, Harris SR, Persson K, Marsh P, Barron A, Bignell A, et al. Co-evolution of genomes and plasmids within Chlamydia trachomatis and the emergence in Sweden of a new variant strain. BMC Genomics. 2009;10:239. doi: 10.1186/1471-2164-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z, Chen D, Zhong Y, Wang S, Zhong G. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun. 2008;76(8):3415–3428. doi: 10.1128/IAI.01377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donati M, Laroucau K, Storni E, Mazzeo C, Magnino S, Di Francesco A, et al. Serological response to pgp3 protein in animal and human chlamydial infections. Vet Microbiol. 2009;135(1–2):181–185. doi: 10.1016/j.vetmic.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 13.Hugall A, Timms P, Girjes AA, Lavin MF. Conserved DNA sequences in Chlamydial plasmids. Plasmid. 1989;22(2):91–98. doi: 10.1016/0147-619X(89)90018-8. [DOI] [PubMed] [Google Scholar]
- 14.Girjes AA, Hugall AF, Timms P, Lavin MF. Two distinct forms of Chlamydia psittaci associated with disease and infertility in Phascolarctos cinereus (koala) Infect Immun. 1988;56(8):1897–1900. doi: 10.1128/iai.56.8.1897-1900.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mojica S, Huot Creasy H, Daugherty S, Read TD, Kim T, Kaltenboeck B, et al. Genome sequence of the obligate intracellular animal pathogen Chlamydia pecorum E58. J Bacteriol. 2011;193(14):3690. doi: 10.1128/JB.00454-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jelocnik M, Self R, Timms P, Borel N, Polkinghorne A. Novel sequence types of Chlamydia pecorum infect free-ranging Alpine ibex (Capra ibex) and red deer (Cervus elaphus) in Switzerland. J Wildl Dis. 2015;51(2):479–483. doi: 10.7589/2014-08-220. [DOI] [PubMed] [Google Scholar]
- 17.Polkinghorne A, Hanger J, Timms P. Recent advances in understanding the biology, epidemiology and control of chlamydial infections in koalas. Vet Microbiol. 2013;165(3–4):214–223. doi: 10.1016/j.vetmic.2013.02.026. [DOI] [PubMed] [Google Scholar]
- 18.Yousef Mohamad K, Rodolakis A. Recent advances in the understanding of Chlamydophila pecorum infections, sixteen years after it was named as the fourth species of the Chlamydiaceae family. Vet Res. 2010;41(3):27. doi: 10.1051/vetres/2009075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schautteet K, Vanrompay D. Chlamydiaceae infections in pig. Vet Res. 2011;42(1):29. doi: 10.1186/1297-9716-42-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koelbl O. Untersuchungen ueber das vorkommen von Miyagawanellen beim schwein. Wien Tierarztl Mschr. 1969;56:332–335. [Google Scholar]
- 21.Koelbl O, Burtscher H, Hebensreit J. Polyarthritis bei schlachtschweinen. Mikrobiologische, histologishe und fleishhygienische aspekte. Wien Tierarztl Mschr. 1970;57:355–361. [Google Scholar]
- 22.Kaltenboeck B, Storz J. Biological properties and genetic analysis of the ompA locus in chlamydiae isolated from swine. Am J Vet Res. 1992;53(9):1482–1487. [PubMed] [Google Scholar]
- 23.Jelocnik M, Frentiu FD, Timms P, Polkinghorne A. Multi-locus sequence analysis provides insights into the molecular epidemiology of Chlamydia pecorum infections in Australian sheep, cattle and koalas. J Clin Microbiol. 2013;51(8):2625–2632. doi: 10.1128/JCM.00992-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jelocnik M, Walker E, Pannekoek Y, Ellem J, Timms P, Polkinghorne A. Evaluation of the relationship between Chlamydia pecorum sequence types and disease using a species-specific multi-locus sequence typing scheme (MLST) Vet Microbiol. 2014;174(1–2):214–222. doi: 10.1016/j.vetmic.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 25.Jelocnik M, Forshaw D, Cotter J, Roberts D, Timms P, Polkinghorne A. Molecular and pathological insights into Chlamydia pecorum-associated sporadic bovine encephalomyelitis (SBE) in Western Australia. BMC Vet Res. 2014;10:121. doi: 10.1186/1746-6148-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christiansen MT, Brown AC, Kundu S, Tutill HJ, Williams R, Brown JR, et al. Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect Dis. 2014;14:591. doi: 10.1186/s12879-014-0591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24(23):2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 33.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 34.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 36.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43(3):e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seth-Smith HMB, Harris SR, Skilton RJ, Radebe FM, Golparian D, Shipitsyna E, et al. Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 2013;23(5):855–866. doi: 10.1101/gr.150037.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Putman TE, Suchland RJ, Ivanovitch JD, Rockey DD. Culture-independent sequence analysis of Chlamydia trachomatis in urogenital specimens identifies regions of recombination and in-patient sequence mutations. Microbiology. 2013;159(10):2109–2117. doi: 10.1099/mic.0.070029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becker E, Hegemann JH. All subtypes of the Pmp adhesin family are implicated in chlamydial virulence and show species-specific function. MicrobiologyOpen. 2014;3(4):544–556. doi: 10.1002/mbo3.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molleken K, Schmidt E, Hegemann JH. Members of the Pmp protein family of Chlamydia pneumoniae mediate adhesion to human cells via short repetitive peptide motifs. Mol Microbiol. 2010;78(4):1004–1017. doi: 10.1111/j.1365-2958.2010.07386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harley R, Catanese B, Helps C. Polymorphic membrane proteins 1 and 7 from Chlamydophila felis are significant immunodominant proteins. Vet Microbiol. 2010;144(3–4):415–421. doi: 10.1016/j.vetmic.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 43.Gomes JP, Nunes A, Bruno WJ, Borrego MJ, Florindo C, Dean D. Polymorphisms in the nine polymorphic membrane proteins of Chlamydia trachomatis across all serovars: evidence for serovar Da recombination and correlation with tissue tropism. J Bacteriol. 2006;188(1):275–286. doi: 10.1128/JB.188.1.275-286.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomson NR, Yeats C, Bell K, Holden MTG, Bentley SD, Livingstone M, et al. The Chlamydophila abortus genome sequence reveals an array of variable proteins that contribute to interspecies variation. Genome Res. 2005;15(5):629–640. doi: 10.1101/gr.3684805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voigt A, Schöfl G, Saluz HP. The Chlamydia psittaci Genome: A Comparative analysis of intracellular pathogens. PLoS ONE. 2012;7(4):e35097. doi: 10.1371/journal.pone.0035097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolff BJ, Morrison SS, Pesti D, Ganakammal SR, Srinivasamoorthy G, Changayil S, et al. Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and Type III Secretion System genes. Microbiology. 2015;161(7):1378–91. doi: 10.1099/mic.0.000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters J, Wilson DP, Myers G, Timms P, Bavoil PM. Type III secretion a la Chlamydia. Trends Microbiol. 2007;15(6):241–251. doi: 10.1016/j.tim.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 48.Hsia RC, Pannekoek Y, Ingerowski E, Bavoil PM. Type III secretion genes identify a putative virulence locus of Chlamydia. Mol Microbiol. 1997;25(2):351–359. doi: 10.1046/j.1365-2958.1997.4701834.x. [DOI] [PubMed] [Google Scholar]
- 49.Mojica SA, Hovis KM, Frieman MB, Tran B, Hsia RC, Ravel J, et al. SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol Biol Cell. 2015;26(10):1918–1934. doi: 10.1091/mbc.E14-11-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci U S A. 2006;103(42):15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rahman KS, Chowdhury EU, Poudel A, Ruettger A, Sachse K, Kaltenboeck B. Defining species-specific immunodominant B cell epitopes for molecular serology of Chlamydia species. Clin Vaccine Immunol. 2015;22(5):539–552. doi: 10.1128/CVI.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Forsbach-Birk V, Foddis C, Simnacher U, Wilkat M, Longbottom D, Walder G, et al. Profiling antibody responses to infections by Chlamydia abortus enables identification of potential virulence factors and candidates for serodiagnosis. PLoS ONE. 2013;8(11):e80310. doi: 10.1371/journal.pone.0080310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borges V, Gomes JP. Deep comparative genomics among Chlamydia trachomatis lymphogranuloma venereum isolates highlights genes potentially involved in pathoadaptation. Infect Genet Evol. 2015;32:74–88. doi: 10.1016/j.meegid.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 54.Nunes A, Gomes JP. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol. 2014;23:49–64. doi: 10.1016/j.meegid.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 55.Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28(6):1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomson NR, Holden MTG, Carder C, Lennard N, Lockey SJ, Marsh P, et al. Chlamydia trachomatis: Genome sequence analysis of lymphogranuloma venereum isolates. Genome Res. 2008;18(1):161–171. doi: 10.1101/gr.7020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pickett MA, Everson JS, Pead PJ, Clarke IN. The plasmids of Chlamydia trachomatis and Chlamydophila pneumoniae (N16): accurate determination of copy number and the paradoxical effect of plasmid-curing agents. Microbiology. 2005;151(3):893–903. doi: 10.1099/mic.0.27625-0. [DOI] [PubMed] [Google Scholar]
- 58.Funnell O, Johnson L, Woolford L, Boardman W, Polkinghorne A, McLelland D. Conjunctivitis associated with Chlamydia pecorum in three koalas (Phascolarctos cinereus) in the Mount Lofty Ranges, South Australia. J Wildl Dis. 2013;49(4):1066–1069. doi: 10.7589/2013-03-066. [DOI] [PubMed] [Google Scholar]
- 59.Chen J, Yang Z, Sun X, Tang L, Ding Y, Xue M, et al. Intrauterine infection with plasmid-free Chlamydia muridarum reveals a critical role of the plasmid in chlamydial ascension and establishes a model for evaluating plasmid-independent pathogenicity. Infect Immun. 2015;83(6):2583–2592. doi: 10.1128/IAI.00353-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, et al. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect Immun. 2013;81(3):636–644. doi: 10.1128/IAI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]