

Short abstract
We assess five years of usage of the major genome-wide collections of mutants from Saccharomyces cerevisiae.
Abstract
We assess five years of usage of the major genome-wide collections of mutants from Saccharomyces cerevisiae: single deletion mutants, double mutants conferring 'synthetic' lethality and the 'TRIPLES' collection of mutants obtained by random transposon insertion. Over 100 experimental conditions have been tested and more than 5,000 novel phenotypic traits have been assigned to yeast genes using these collections.
In April 1996, the completely annotated genome sequence of the yeast Saccharomyces cerevisiae was made publicly available [1,2], the first eukaryotic genome sequence to be completed. Eight years later, thanks to the united efforts of the large yeast research community and to the unique genetic and physiological properties of yeast, this humble servant of mankind provides by far the best annotated eukaryotic genome [3]. The completeness of the yeast genome sequence has allowed the development of many novel tools for analyzing all molecular components of the cell and their interactions. These tools include three high-throughput collections of mutants that were first produced in 1999 and that have been analyzed in the five years since then. Here, we review the uses of these collections and their contribution to the identification of the components of basic physiological and developmental pathways of S. cerevisiae.
The yeast deletion mutant collection
A set of over 20,000 knockout strains was created by a consortium of European and North American laboratories [4,5]. The collection currently contains homozygous and heterozygous diploid strains corresponding to deletions of each of 5,916 genes (including 1,159 essential genes) and one haploid strain of each mating type for every non-essential gene (4,757 genes). Each knockout strain is marked by two unique 20-nucleotide 'bar codes', allowing quantitative and qualitative identification by DNA microarray hybridization of each strain in the pools used to assess the strains under different growth conditions (see Figure 1). The original article [4] describing this collection has been cited more then 560 times in the five years since its publication, according to the ISI Web of Science [6]. The complete collection of strains can be obtained at low cost from Euroscarf [7], ATCC [8] and Invitrogen [9].
Figure 1.
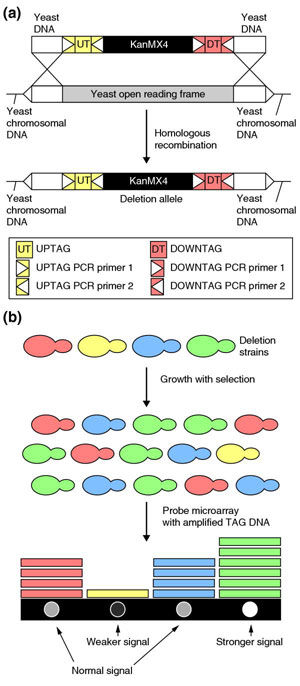
Construction and screening of the yeast deletion strain collection. (a) The cassette used consists of a kanamycin-resistance gene (KanMX4) flanked by two tags (also called barcodes), the UPTAG and the DOWNTAG, which are unique to each gene. The yeast DNA 5' and 3' to the barcodes is homologous to yeast DNA flanking the gene to be deleted. After homologous recombination, the gene is replaced by the cassette sequences, including the barcodes. (b) Screening the deletion strains for differences in fitness under selective conditions. Selection leads to an increase in the proportion of some strains in the culture and a decrease of others; these changes can be detected by probing a microarray containing the sequences complementary to the barcodes. A stronger signal, indicating a higher level of a barcode in the RNA extracted from the culture, shows strains that have increased in frequency after selection. Adapted with permission from [71].
The deletion collection has been used in dozens of novel exhaustive screens for phenotypes that occur under a variety of physiological conditions; these include growth in minimal medium, in high salt and low salt, in galactose or sorbitol, at pH 8, after heat or cold shock, under stress by hydrogen peroxide (all in [10]); growth on non-fermentable carbon substrates [11], in saline conditions [12] or after treatment by ionizing radiation or DNA-damaging agents [13-17]; and the collection has also been screened for defects in meiosis, sporulation and germination [18,19]. This approach has uncovered numerous new putative components of well-known pathways; for instance, the number of genes known to have sporulation or germination phenotypes when deleted has been doubled by these analyses [18].
More sophisticated screens, for example for suppressors of the accumulation of mutations [20], have been developed more recently, as well as screens involving transformation of the deletion strains in order to identify genes needed for non-homologous DNA end-joining [21]. Novel protocols requiring individual transformations of each mutant have allowed the identification of host factors that influence the fate of the Ty family of long-terminal-repeat retrotransposable elements [22] and of genes involved in the unfolded protein response induced by heterologous introduction of mutant human Huntingtin protein or fragments of α-synuclein, both of which form disease-associated aggregates [23]. Similarly, proteins that interfere with the assembly of endoplasmic-reticulum structures termed karmellae, which are induced by elevated levels of HMG-CoA reductase under specific growth and genetic conditions, have been identified using the collection [24]. Several morphological screens have been developed, for example for defects in the selection of the bipolar bud site [25], in cell-size distribution [26,27], in cell morphology [28] and in meiotic chromosomal segregation [29]. Another approach is the screening of individual colonies. For instance, mis-sorting and secretion of vacuolar carboxypeptidase Y were detected by colony immunoblotting [30]. A second example of colony screening is the transformation of each single-deletion strain to express viral replicase proteins and an RNA replication template in which the capsid gene was replaced by a luciferase reporter gene, which was used to monitor viral expression in yeast colonies [31]. Finally, to identify the genes affecting glycogen storage, the deletion mutant colonies were blotted and stained by iodine vapor; the intensity of coloration allowed assessment of glycogen accumulation [32].
Use of the yeast deletion collection in screens for synthetic lethal mutants and to study drug targets
Synthetic lethality is the phenomenon that occurs when two mutations that are each viable are combined and the double mutant is lethal. A method has been developed for the systematic construction of double mutants, which is called synthetic genetic array analysis (SGA) [33,34]. Haploid strains with mutations in non-essential genes were crossed to an array of the whole haploid deletion collection; the resulting diploid cells were made to sporulate and the lethal combinations identified, indicating the existence of essential interactions between gene products. In a first SGA screen using eight query genes, 291 interactions among 204 genes were identified [33]. Three years later [34], the search was expanded to 132 query genes, and 4,000 interactions were identified among 1,000 genes with roles in cytoskeletal organization, cell-wall biosynthesis, microtubule-based chromosome segregation and DNA metabolism.
A more recent development of this approach is the use of DNA-DNA hybridization protocols to assess lethality, termed 'synthetic lethality analysis by microarray' (SLAM). In this method, a pool of haploid deletion strains is transformed with a cassette that replaces the gene of interest with either a deletion construct or the wild-type form. Transformants are pooled and genomic DNA is isolated; the barcodes are amplified by PCR and labeled with either Cy3 (green) or Cy5 (red) fluorescent dyes; and hybridization to an array containing all the deletion tags allows identification of the synthetic-lethal combinations (which are missing). The SLAM method has been validated by identifying members of the DNA helicase interaction network [35]. Synthetic genetic arrays have also been used for high-resolution genetic mapping of suppressor mutations [36]; this method, termed SGA mapping (SGAM), is in principle also applicable to the analysis of multigenic traits.
The yeast deletion collection has also been used to identify members of the pathways modified by more than 25 different chemical ligands. This approach has identified the L-carnitine transporter Agp2p as a novel transporter of bleomycin in yeast, implicating membrane transport as a key determinant of resistance to this widely used anticancer agent [37]. In another study, the transcription factor Rpn4p was shown to compensate for proteasome inhibition by PS-341, a drug that is being studied as a treatment for cancer [38]. Other ligands tested include the phosphatidyl kinase inhibitor wortmannin [39]; anticancer agents [10,37,40,41]; antifungal agents including nystatin and calcofluor [10,41]; antibiotics including hygromycin B [42] and rapamycin [43]; statins (a class of drugs that lower cholesterol), the smooth muscle relaxant alverine citrate, the local anaesthetic dyclonine, yeast K1 killer toxin [44], 6-azauracil (a drug that depresses cellular nucleoside triphosphate levels [45]) and even the detergent sodium dodecyl sulfate [44].
Major scientific contributions arising from the use of the yeast deletion collection
In a total of 33 publications that have used the deletion collection, in which over 100 different conditions were explored, more then 5,000 genes were attributed a phenotype (see Table 1 for an overview). Many of these genes were not previously known to contribute to the pathways scrutinized, even though in many cases - such as in studies of sporulation, the cell cycle, radiation damage and growth in defined media - there had previously been apparently exhaustive classical mutagenesis or DNA microarray screens. Altogether, these analyses have shown 71 genes previously thought to be non-essential to have a nonviable phenotype [46]; and the systematic deletion approach has been claimed to be five-fold more sensitive then a microarray gene-expression analysis for finding genes involved in pathways of interest [11].
Table 1.
Screens performed so far on the yeast deletion strain collection and transposon-insertion collection
| Condition, treatment or phenotype | Number of genes responding to the treatments* | References |
| Phenotype screens of the deletion collection | ||
| Fitness after growth on minimal medium, high salt, low salt, galactose, sorbitol, pH 8, heat shock, cold shock, or hydrogen peroxide | ? | [10] |
| Growth on non-fermentable carbon substrates | 466 | [11] |
| Response to DNA-damaging agents, ionizing radiation, UV radiation, cisplatin, or hydrogen peroxide | >170 | [13-16] |
| Chemical DNA damage (methane sulfonate sensitivity) | 103 | [17] |
| Enhanced sporulation proficiency | 102 | [18,19] |
| Sporulation defects | 261 | [18,19] |
| Post-germination defects | 158 | [18,19] |
| Genes essential for sporulation | 334 | [18,19] |
| Saline response | About 500 | [12] |
| Suppression of the accumulation of mutations | 33 | [20] |
| Non-homologous DNA end-joining | About 30 | [21] |
| Ty1 retrotransposition cycle | 101 | [22] |
| Response to the introduction of human Huntingtin or α-synuclein fragments (unfolded protein response) | 52/86 | [23] |
| Karmellae assembly | 120 | [24] |
| Bipolar bud-site selection | 127 | [25] |
| Cell-size distribution | 500 | [26,27] |
| Altered cell morphology | ? | [28] |
| Meiotic chromosomal segregation | ? | [29] |
| Vacuolar protein sorting | 146 | [30] |
| Host genes affecting replication of a positive-strand RNA virus | 100 | [31] |
| Glycogen storage (low, high) | 324, 242 | [32] |
| Sensitivity of the deletion collection to drugs or chemicals | ||
| Rapamycin | 106 | [43] |
| PS-341 | ? | [38] |
| Bleomycin (hypersensitivity, resistance) | 231, 5 | [37,40] |
| Amoxillin, penicillin, rifampin, vancomycin, (oxy)tetracycline, or gentamicin | 23 | [42] |
| Anticancer, antifungal agents, statins, alverine citrate, dyclonine, or nystatin | About 10 | [10,41] |
| K1 killer toxin, hygromycin B, calcofluor or sodium dodecyl sulfate | 268 | [44] |
| 6-Azauracil | 42 | [45] |
| Wortmannin (resistance, sensitivity) | 591, 476 | [39] |
| Phenotypes of transposon-insertion libraries | ||
| Phenotypes under 20 conditions, expression levels and subcellular localization (TRIPLES database) | ? | [55] |
| High salt | 31 | [58] |
| Survival of hypo-osmotic shock and growth at 15°C | 10 | [58] |
| Ethanol tolerance and cell-wall modifications | 5 | [59] |
| Nitrogen starvation, invasive or filamentous growth and sporulation | About 105 | [60] |
*The number of open reading frames responding to the relevant screen is reported whenever this information could be deduced from the relevant article; ? indicates that the information could not be deduced.
Some new phenotypes have emerged from these analyses, although many known phenotypes have not yet been explored [47]. As an example of recent novel information provided by the systematic-deletion approach, over 100 new genes resulting in a defective bipolar budding pattern [25] have been identified. Other examples are the identification of a new putative bleomycin transporter [37]; the NEJ1 gene involved in non-homologous DNA end-joining and claimed to be one of the guardians of the cancer cells [21]; and 52 genes, the deletion of which causes lethality in yeast transfected with a fragment of heterologous human Huntingtin [23]. The latter set, which includes genes involved in lipid metabolism, vesicle transport and of unknown function, may help to define the still poorly defined pathway termed the unfolded protein response [23].
Uncertainties in the results obtained from systematic deletion mutants
The yeast deletion collection allows powerful novel screens and has revealed many novel components of complex metabolic and developmental pathways. The data produced should be examined with great care, however, as there are several problems with the use of the collection (for further details, see two reviews by Elisabeth Winzeler [46,48]). A first issue is that the collection is incomplete, as a few hundred genes were missed in the first annotation of the genome that was used as the basis of the deletions (see updates of yeast genome data in Saccharomyces genome database (SGD) [49]). Moreover, relative amounts of some deleted strains may have been distributed unequally during the initial creation of the pools and further amplified [4]. Secondly, there can be problems with mutations or variations elsewhere in the genome that affect the phenotype. For instance, because the transformation procedure that was used to create the deletion library is a mutagenic event, mutations in sites some distance away from the intended deletion might be present in some strains [50]. Also, up to 8% of the deleted strains are known to retain a wild-type copy of the targeted gene, presumably because of aneuploidy or some duplication event [50]. Even in diploid strains, mutations that apparently have no phenotype may in fact modify the phenotype of other mutations in subtle ways (a phenomenon termed haploinsufficiency) [51].
When deletions are moved into a different genetic background, their phenotypes may change; for instance, up to 18% of synthetic-lethal interactions were not confirmed in different backgrounds [33,46]. Another source of discrepancy might be genetic variation among the individual isolates of the strains that are amplified under given selective conditions. For instance, the phenotype for a given mutant strain can be masked by extracellular complementation by a protein product of another strain in the pool.
Other factors are related to the use of the barcodes. Their presence may modify the phenotype or, conversely, the stress created by the knockout procedure may mutagenize the barcode. About 17% of the 4,600 homozygous strains in the collection have a hybridization signal similar to that of the background. Moreover, trivial technical factors may affect the hybridization data, for example when the wrong piece of DNA is deposited on the microarray or when the tag modifies the secondary structures and thus causes poor hybridization. Also, some published datasets have too few repetitions of experiments to be statistically valid.
These and other problems concerning the specificity and sensitivity of the screen used must be taken into account when interpreting data obtained using the yeast deletion collection. As a gross estimate, over one third of the data may consist of false positives or false negatives. As a golden rule, therefore, the data from a primary screen of the collection should always be confirmed by independent approaches.
Collections of strains mutated using transposons
Transposons are mobile genetic elements that can be used to disrupt genes in a non-targeted fashion. Insertional mutagenesis using transposons can disrupt non-coding as well as coding regions and can lead to partial loss or gain of function; it is thus complementary to deletion of genes [52]. The 'mini-Mu' transposon modified for insertional mutagenesis in yeast was pioneered by Daignan-Fornier and Bolotin-Fukuhara [53]. Ross-Macdonald et al. [52] used a version of the same transposon including a reporter gene (lacZ), an epitope tag (encoding hemagglutinin) and selectable markers for yeast and bacteria to perform random transposon mutagenesis. The approach uses a minitransposon that was introduced into the yeast genome by a two step method: a yeast genomic DNA library was mutagenized in Escherichia coli using the minitransposon; the library plasmids were then cut to excise the yeast genomic DNA with the transposon insertion, which was transformed into diploid yeast, replacing one chromosomal copy by homologous recombination [52]. This transposon-mutagenesis system can be used for the analysis of gene expression, detection of subcellular localization and identification of phenotypes caused by the insertion, and the transposon can be deleted using the Cre-LoxP recombinase system, removing the selectable marker. The insertion point in the genome can be determined for each mutant by sequencing outwards from the transposon sequence into the surrounding yeast sequences. The article describing the first such screen [52] has been cited 209 times in the five years since its publication [6]. The collection of transposon-inserted strains created in this work [52] is available free of charge from Michael Snyder's laboratory [54].
A database called 'transposon-insertion phenotypes, localization and expression in Saccharomyces cerevisiae' (TRIPLES) [55,56] contains the results obtained from exploiting the mutants obtained by the transposon-insertion approach. A total of 240,768 yeast insertional mutants were screened for transposon-encoded β-galactosidase activity, and a collection of 28,428 yeast strains was generated that produce β-galactosidase under conditions of vegetative growth and/or sporulation. The insertion point of 23,191 of these strains has been sequenced, identifying 3,750 different yeast genes, and the subcellular localization of 2,744 different yeast proteins has been documented (A. Kumar, personal communication).
The transposon-insertion collection [52,54] has been used to identify genes involved in sporulation and vegetative growth. Analysis of the collection has also identified 137 new genes that were not originally annotated by the yeast genome sequencing consortium [57]. Ferreira et al. [58] have used the collection - which was produced in the S288C strain used to sequence the yeast genome [52] - to mutagenize the W303-1A strain (which is known to be more responsive to gene inactivation than the S288C). Ferreira et al. [58] identified genes required for growth in high-salt medium (61 insertions in 31 genes) and survival of hypo-osmotic shock and growth at 15°C (31 insertion in 10 genes). A similar approach was also used by Takahashi et al. [59] to screen in other strains for ethanol tolerance and modification of the cell wall, and by Suzuki et al. [60] to screen for the effects of insertions on the response to nitrogen starvation, on invasive or filamentous growth and on sporulation.
Other approaches for insertional mutagenesis have also been applied to yeast. Firstly, instead of bacterial mini-Mu transposons, T-DNA from Agrobacterium tumefaciens has been used to mutagenize yeast on a genome-wide scale [61]. A second approach is the use of non-homologous insertion of DNA cassettes not derived from mobile elements. This process, which exploits the cellular machinery that normally repairs DNA double-strand breaks by non-homologous end joining, is increased by gamma irradiation [62]. Thirdly, Blanc and Adams [63] used mutations resulting from insertion of the Ty1 transposon to identify yeast mutations that generate evolutionarily significant phenotypes by causing small but positive increments of fitness. Finally, an application of transposon mutagenesis called direct allele replacement technology (DART) allows rapid transfer of any insertion allele into any strain [64]. A transposon library consisting of a collection of plasmids containing yeast genomic DNA with transposon insertions is sequenced to identify the exact insertion point in the yeast genomic DNA [64]. After excision from the plasmid, the yeast genomic DNA containing the transposon is used to transform a yeast strain of choice by homologous recombination. The procedure was validated by identification of 29 insertions into 17 genes involved in apical growth [64].
Insertional mutagenesis using transposons has several potential advantages over targeted deletion: for instance, insertion occurs in the non-coding as well as in the coding segments, so regulatory regions and other non-genie regions can be disrupted. Moreover, depending on the site of insertion, transposon mutagenesis may lead to partial loss of function or gain of function and hence to the identification of novel functions that would not be found from studies of complete knockouts of genes [52]. Conditional alleles may be generated, as well as mutants in promoter or terminal regions. Also, apart from phenotype analysis of the mutation, the level of expression of the targeted gene can be measured in vivo and the subcellular localization of its product can be determined. A disadvantage, however, is that transposon insertions are not random, and this method may therefore never cover all the genes in the genome. Also, several of the problems that were mentioned above for the analysis of the yeast deletion collection apply equally to the transposon mutant collections, including the observation that phenotypes are often very strongly background-dependent.
Other genome-wide mutant collections and their uses
Several databases have been developed to catalog the subcellular localizations of yeast proteins as identified by fluorescence microscopy. The yeast protein localization database [65,66] describes the results obtained using a library of yeast genes fused to a green fluorescent protein (GFP) reporter. The TRIPLES database [55,56] includes the use of the transposon-insertion libraries to determine protein localization for 5,504 insertions. The yeast GFP fusion localization database [67,68] presents the localization of 4,156 proteins into 22 distinct subcellular locations, as determined using a library of GFP-tagged proteins compared with reference strains expressing proteins of known localization tagged with red fluorescent protein (the strains used are available from Invitrogen [9]).
In a recent study [69], an exhaustive global analysis of protein expression in yeast was reported. Each open reading frame was marked by an insertion cassette consisting of a modified version of the tandem affinity purification (TAP) tag, a yeast selectable marker to drive homologous recombination, and regions homologous to yeast genes. The level of expression of each protein was determined by very sensitive western-blotting analysis that could detect less than 50 protein molecules per cell. The level of expression of 4,251 gene products was identified in exponential growth conditions [69].
Like the sequencing of the yeast genome, the construction of the single and double deletion libraries and that of the major transposon insertion library has required a considerable investment by the international yeast research community. The three original papers [4,33,52] presenting the libraries have a total of 83 authors, funded by public agencies from the USA, from the European Union and from Canada. Was this effort worthwhile?
Over the last five years, the three seminal papers have been cited more than 950 times [6]. We have found over 50 experimental papers reporting on different phenotypic screens carried out with these genome-wide libraries. More than 100 experimental conditions have been tested, representing near a million individual mutant screens. We estimate that more than 5,000 novel phenotypic traits have been assigned to yeast genes of known or unknown molecular functions. This undoubtedly represents a considerable amount of progress towards the ultimate goal of a full description of the functions and interactions of all the molecular components of a basic eukaryotic cell. Several recent improvements of the use of the original genome-wide mutant libraries have been reported, such as the DART [64] and SLAM [35] approaches described above. New genome-wide libraries using more sensitive protein tagging are being developed [69] for global measurements of protein levels or subcellular expression. In all this work, yeast continues to be a very convenient test-bed for the development of novel high-throughput tools that rely on the availability of the complete genome sequence.
But a word of caution is appropriate. As was the case for other high-throughput tools that were also pioneered using yeast, such as two-hybrid protein-interaction screening, DNA-hybridization microarrays and proteomic analyses, the information provided by large screens of genome-wide libraries contains an appreciable number of false-positive and false-negative data points. It is therefore essential to confirm each result by several independent approaches. Ultimately, we will have to return to 'reductionist' biochemical approaches to demonstrate fully the molecular function suggested by a large primary screen.
Finally, there is an urgent need to build databases to collect and organize all the data obtained from all the large screening approaches. The SGD [49] and the Munich information center for protein sequences (MIPS) database [70] are making good progress in this respect. The authors of these databases should, however, be encouraged to further develop procedures that take into account the difficult assessment of the uncertainties associated with much of the data. Here again, yeast is expected to become a pioneer.
References
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. Life with 6000 genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Aert R, Agostini-Carbone ML, Ahmed A, Aigle M, Alberghina L, Albermann K, Albers M, Aldea M, Alexandraki D, et al. The yeast genome directory. Nature. 1997;387(Suppl):1–105. [Google Scholar]
- Goffeau A. Four years of post-genomic life with 6,000 yeast genes. FEBS Lett. 2000;480:37–41. doi: 10.1016/S0014-5793(00)01775-0. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Saccharomyces Genome Deletion Project http://sequence-www.stanford.edu/group/yeast_deletion_project/deletions3.html
- Web of Science http://www.isinet.com/products/citation/wos/
- European Saccharomyces cerevisiae Archive for Functional Analysis (Euroscarf) http://www.uni-frankfurt.de/fb15/mikro/euroscarf/index.html
- About the ATCC-LGC Promochem Partnership http://www.lgcpromochem.com/atcc/
- Invitrogen - Clones http://clones.invitrogen.com/
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Steinmetz LM, Scharfe C, Deutschbauer AM, Mokranjac D, Herman ZS, Jones T, Chu AM, Giaever G, Prokisch H, Oefner PJ, Davis RW. Systematic screen for human disease genes in yeast. Nat Genet. 2002;31:400–404. doi: 10.1038/ng929. [DOI] [PubMed] [Google Scholar]
- Warringer J, Ericson E, Fernandez L, Nerman O, Blomberg A. High-resolution yeast phenomics resolves different physiological features in the saline response. Proc Natl Acad Sci USA. 2003;100:15724–15729. doi: 10.1073/pnas.2435976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, Snipe JR, Resnick MA. Genes required for ionizing radiation resistance in yeast. Nat Genet. 2001;29:426–434. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- Birrell GW, Giaever G, Chu AM, Davis RW, Brown JM. A genomewide screen in Saccharomyces cerevisiae for genes affecting UV radiation sensitivity. Proc Natl Acad Sci USA. 2001;98:12608–12613. doi: 10.1073/pnas.231366398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Game JC, Birrell GW, Brown JA, Shibata T, Baccari C, Chu AM, Williamson MS, Brown JM. Use of a genome-wide approach to identify new genes that control resistance of Saccharomyces cerevisiae to ionizing radiation. Radiat Res. 2003;160:14–24. doi: 10.1667/rr3019. [DOI] [PubMed] [Google Scholar]
- Birrell GW, Brown JA, Wu HI, Giaever G, Chu AM, Davis RW, Brown JM. Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. Proc Natl Acad Sci USA. 2002;99:8778–8783. doi: 10.1073/pnas.132275199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA. 2002;99:16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99:15530–15535. doi: 10.1073/pnas.202604399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyenihi AH, Saunders WS. Large-scale functional genomic analysis of sporulation and meiosis in Saccharomyces cerevisiae. Genetics. 2003;163:47–54. doi: 10.1093/genetics/163.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ME, Rio AG, Nicolas A, Kolodner RD. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci USA. 2003;100:11529–11534. doi: 10.1073/pnas.2035018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SL, Shoemaker DD, Boeke JD. A DNA microarray-based genetic screen for nonhomologous end-joining mutants in Saccharomyces cerevisiae. Science. 2001;294:2552–2556. doi: 10.1126/science.1065672. [DOI] [PubMed] [Google Scholar]
- Griffith JL, Coleman LE, Raymond AS, Goodson SG, Pittard WS, Tsui C, Devine SE. Functional genomics reveals relationships between the retrovirus-like Ty1 element and its host Saccharomyces cerevisiae. Genetics. 2003;164:867–879. doi: 10.1093/genetics/164.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- Wright R, Parrish ML, Cadera E, Larson L, Matson CK, Garrett-Engele P, Armour C, Lum PY, Shoemaker DD. Parallel analysis of tagged deletion mutants efficiently identifies genes involved in endoplasmic reticulum biogenesis. Yeast. 2003;20:881–892. doi: 10.1002/yea.994. [DOI] [PubMed] [Google Scholar]
- Ni L, Snyder M. A genomic study of the bipolar bud site selection pattern in Saccharomyces cerevisiae. Mol Biol Cell. 2001;12:2147–2170. doi: 10.1091/mbc.12.7.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M. Systematic identification of pathways that couple cell growth and division in yeast. Science. 2002;297:395–400. doi: 10.1126/science.1070850. [DOI] [PubMed] [Google Scholar]
- Zhang J, Schneider C, Ottmers L, Rodriguez R, Day A, Markwardt J, Schneider BL. Genomic scale mutant hunt identifies cell size homeostasis genes in S. cerevisiae. Curr Biol. 2002;12:1992–2001. doi: 10.1016/S0960-9822(02)01305-2. [DOI] [PubMed] [Google Scholar]
- Saito TL, Ohtani M, Sawai H, Sano F, Saka A, Watanabe D, Yukawa M, Ohya Y, Morishita S. SCMD: Saccharomyces cerevisiae morphological database. Nucleic Acids Res. 2004;32 Database issue:D319–D322. doi: 10.1093/nar/gkh113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston AL, Tham WH, Shah H, Amon A. A genome-wide screen identifies genes required for centromeric cohesion. Science. 2004;303:1367–1370. doi: 10.1126/science.1094220. [DOI] [PubMed] [Google Scholar]
- Bonangelino CJ, Chavez EM, Bonifacino JS. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:2486–2501. doi: 10.1091/mbc.02-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner DB, Lindenbach BD, Grdzelishvili VZ, Noueiry AO, Paul SM, Ahlquist P. Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus. Proc Natl Acad Sci USA. 2003;100:15764–15769. doi: 10.1073/pnas.2536857100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WA, Wang Z, Roach PJ. Systematic identification of the genes affecting glycogen storage in the yeast Saccharomyces cerevisiae: implication of the vacuole as a determinant of glycogen level. Mol Cell Proteomics. 2002;1:232–242. doi: 10.1074/mcp.M100024-MCP200. [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Ooi SL, Shoemaker DD, Boeke JD. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat Genet. 2003;35:277–286. doi: 10.1038/ng1258. [DOI] [PubMed] [Google Scholar]
- Jorgensen P, Nelson B, Robinson MD, Chen Y, Andrews B, Tyers M, Boone C. High-resolution genetic mapping with ordered arrays of Saccharomyces cerevisiae deletion mutants. Genetics. 2002;162:1091–1099. doi: 10.1093/genetics/162.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouida M, Page N, Leduc A, Peter M, Ramotar D. A genome-wide screen in Saccharomyces cerevisiae reveals altered transport as a mechanism of resistance to the anticancer drug bleomycin. Cancer Res. 2004;64:1102–1109. doi: 10.1158/0008-5472.can-03-2729. [DOI] [PubMed] [Google Scholar]
- Fleming JA, Lightcap ES, Sadis S, Thoroddsen V, Bulawa CE, Blackman RK. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc Natl Acad Sci USA. 2002;99:1461–1466. doi: 10.1073/pnas.032516399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zewail A, Xie MW, Xing Y, Lin L, Zhang PF, Zou W, Saxe JP, Huang J. Novel functions of the phosphatidylinositol metabolic pathway discovered by a chemical genomics screen with wortmannin. Proc Natl Acad Sci USA. 2003;100:3345–3350. doi: 10.1073/pnas.0530118100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouida M, Tounekti O, Leduc A, Belhadj O, Mir L, Ramotar D. Isolation and characterization of Saccharomyces cerevisiae mutants with enhanced resistance to the anticancer drug bleomycin. Curr Genet. 2004;45:265–272. doi: 10.1007/s00294-004-0492-x. [DOI] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci USA. 2004;101:793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn AS, Avery SV. Genome-wide screening of Saccharomyces cerevisiae to identify genes required for antibiotic insusceptibility of eukaryotes. Antimicrob Agents Chemother. 2003;47:676–681. doi: 10.1128/AAC.47.2.676-681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TF, Carvalho J, Riles L, Zheng XF. A chemical genomics approach toward understanding the global functions of the target of rapamycin protein (TOR). Proc Natl Acad Sci USA. 2000;97:13227–13232. doi: 10.1073/pnas.240444197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page N, Gerard-Vincent M, Menard P, Beaulieu M, Azuma M, Dijkgraaf GJ, Li H, Marcoux J, Nguyen T, Dowse T, et al. A Saccharomyces cerevisiae genome-wide mutant screen for altered sensitivity to K1 killer toxin. Genetics. 2003;163:875–894. doi: 10.1093/genetics/163.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riles L, Shaw RJ, Johnston M, Reines D. Large-scale screening of yeast mutants for sensitivity to the IMP dehydrogenase inhibitor 6-azauracil. Yeast. 2004;21:241–248. doi: 10.1002/yea.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunenfelder B, Winzeler EA. Treasures and traps in genomewide data sets: case examples from yeast. Nat Rev Genet. 2002;3:653–661. doi: 10.1038/nrg886. [DOI] [PubMed] [Google Scholar]
- Hampsey M. A review of phenotypes in Saccharomyces cerevisiae. Yeast. 1997;13:1099–1133. doi: 10.1002/(SICI)1097-0061(19970930)13:12<1099::AID-YEA177>3.3.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Que QQ, Winzeler EA. Large-scale mutagenesis and functional genomics in yeast. Funct Integr Genomics. 2002;2:193–198. doi: 10.1007/s10142-002-0057-3. [DOI] [PubMed] [Google Scholar]
- Saccharomyces Genome Database http://www.yeastgenome.org/
- Hughes TR, Roberts CJ, Dai H, Jones AR, Meyer MR, Slade D, Burchard J, Dow S, Ward TR, Kidd MJ, et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet. 2000;25:333–337. doi: 10.1038/77116. [DOI] [PubMed] [Google Scholar]
- Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21:278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, Jansen R, Cheung KH, Sheehan A, Symoniatis D, Umansky L, et al. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;402:413–418. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- Daignan-Fornier B, Bolotin-Fukuhara M. In vivo functional characterization of a yeast nucleotide sequence: construction of a mini-Mu derivative adapted to yeast. Gene. 1988;62:45–54. doi: 10.1016/0378-1119(88)90578-1. [DOI] [PubMed] [Google Scholar]
- Yale Genome Analysis Center http://ygac.med.yale.edu
- Kumar A, Cheung KH, Tosches N, Masiar P, Liu Y, Miller P, Snyder M. The TRIPLES database: a community resource for yeast molecular biology. Nucleic Acids Res. 2002;30:73–75. doi: 10.1093/nar/30.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRIPLES - a database of Transposon-Insertion Phenotypes, Localization, and Expression in Saccharomyces http://ygac.med.yale.edu/triples/triples.htm
- Kumar A, Harrison PM, Cheung KH, Lan N, Echols N, Bertone P, Miller P, Gerstein MB, Snyder M. An integrated approach for finding overlooked genes in yeast. Nat Biotechnol. 2002;20:58–63. doi: 10.1038/nbt0102-58. [DOI] [PubMed] [Google Scholar]
- de Jesus Ferreira MC, Bao X, Laize V, Hohmann S. Transposon mutagenesis reveals novel loci affecting tolerance to salt stress and growth at low temperature. Curr Genet. 2001;40:27–39. doi: 10.1007/s002940100237. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Shimoi H, Ito K. Identification of genes required for growth under ethanol stress using transposon mutagenesis in Saccharomyces cerevisiae. Mol Genet Genomics. 2001;265:1112–1119. doi: 10.1007/s004380100510. [DOI] [PubMed] [Google Scholar]
- Suzuki C, Hori Y, Kashiwagi Y. Screening and characterization of transposon-insertion mutants in a pseudohyphal strain of Saccharomyces cerevisiae. Yeast. 2003;20:407–415. doi: 10.1002/yea.970. [DOI] [PubMed] [Google Scholar]
- Bundock P, van Attikum H, den Dulk-Ras A, Hooykaas PJ. Insertional mutagenesis in yeasts using T-DNA from Agrobacterium tumefaciens. Yeast. 2002;19:529–536. doi: 10.1002/yea.858. [DOI] [PubMed] [Google Scholar]
- Kiechle M, Manivasakam P, Eckardt-Schupp F, Schiestl RH, Friedl AA. Promoter-trapping in Saccharomyces cerevisiae by radiation-assisted fragment insertion. Nucleic Acids Res. 2002;30:e136. doi: 10.1093/nar/gnf136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc VM, Adams J. Evolution in Saccharomyces cerevisiae: identification of mutations increasing fitness in laboratory populations. Genetics. 2003;165:975–983. doi: 10.1093/genetics/165.3.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidlingmaier S, Snyder M. Large-scale identification of genes important for apical growth in Saccharomyces cerevisiae by directed allele replacement technology (DART) screening. Funct Integr Genomics. 2002;1:345–356. doi: 10.1007/s10142-001-0043-1. [DOI] [PubMed] [Google Scholar]
- Habeler G, Natter K, Thallinger GG, Crawford ME, Kohlwein SD, Trajanoski Z. YPL.db: the yeast protein localization database. Nucleic Acids Res. 2002;30:80–83. doi: 10.1093/nar/30.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeast Protein Localization Database http://genome.tugraz.at/YPL/YPL.html
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Yeast GFP Fusion Localization Database http://yeastgfp.ucsf.edu
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- MIPS http://mips.gsf.de/
- Kumar A, Snyder M. Emerging technologies in yeast genomics. Nat Rev Genet. 2001;2:302–312. doi: 10.1038/35066084. [DOI] [PubMed] [Google Scholar]