

Short abstract
Recent studies have used cross-species comparisons of expression profiles to annotate gene functions, to draw evolutionary inferences concerning specific biological processes and to study the global properties of expression networks.
Abstract
The rapid accumulation of microarray data from multiple species provides unprecedented opportunities to study the evolution of biological systems. Recent studies have used cross-species comparisons of expression profiles to annotate gene functions, to draw evolutionary inferences concerning specific biological processes and to study the global properties of expression networks.
Combining sequence and expression information for functional annotation
The power of comparative genomic analysis relies on the assumption that important biological properties are often conserved across species. Cross-species sequence comparison has been widely used to infer gene function; but it is becoming apparent that sequence similarity is not always proportional to functional similarity [1,2]. In fact, Gene Ontology (GO) terms [3] distinguish between molecular and biological functions, and although the amino-acid sequence may imply that a gene possesses a particular molecular function, spatiotemporal expression data is required to infer biological function - which cellular or biological process the gene product participates in. To determine the function of a gene precisely, therefore, we need to investigate not only its sequence characteristics but also its expression characteristics. An increasing number of genetic studies indicate that the divergent functions of many duplicate genes are reflected in the divergence of expression patterns rather than in differences between their coding sequences [4,5]. On the other hand, changes in gene expression may often be associated with changes in function [6]. The expression pattern of a gene can thus serve as a sensitive indicator of its function. An early study in this regard was performed by Su et al. [7] who measured the correlation between the expression profiles of human and mouse ortholog pairs across 16 tissues (the dataset has now been extended to over 50 tissues and is available online as the Novartis Gene Expression Atlas [8]). This work identified several cases in which the ortholog pairs have dissimilar expression patterns, and the authors were able to infer, for example, that human and mouse collagen XV have different physiological functions.
Functional analysis of microarray data often begins with the determination of which genes are significantly co-expressed. Apparent co-expression of genes will often be 'real', but it can also occur by chance as a result of the noisiness of microarray data, the complexity of transcriptional programs or simply as a function of the enormous number of comparisons that are being made. Gene pairs exhibiting co-expression in multiple species and across a large number of arrays in each species are most likely to be functionally relevant. This is because co-regulation of a pair of genes over large evolutionary distances implies that divergence in their expression profiles is mechanistically and/or adaptively constrained, and because a high correlation of expression caused by chance or noisiness in the data in one species is unlikely to occur in another species. The evolutionary conservation of co-expression patterns thus provides functional information that is orthogonal and complementary to that provided by sequence data.
Two recent studies have integrated cross-species expression and sequence comparisons to infer gene functions [9,10]. In the first of these, Stuart et al. [9] compared the correlated patterns of gene expression in more than 3,182 DNA microarrays of tissues from humans, fruit flies, worms and yeast. As outlined in Figure 1a, they started by constructing lists of 'metagenes' on the basis of sequence information, where a metagene is defined as a set of genes from multiple organisms whose protein sequences are one another's best reciprocal BLAST hit; these are therefore strict clusters of orthologous genes. Pairs of metagenes were identified whose expression is significantly correlated in multiple organisms, suggesting that their co-expression has been conserved across evolution. Extending the concept, the authors then constructed gene co-expression networks in which vertices represent metagenes and edges represent interactions (significant co-expression) between two metagenes. They identified 12 regions within the network where components were highly inter-connected, and most of these components were enriched for metagenes involved in similar biological processes. This demonstrates an example of the 'guilt-by-association' principle placed in an evolutionary context [11]: if a gene is linked in the network to many genes that participate in the same biological process, it is reasonable to hypothesize that it also participates in that process. On the basis of this principle, Stuart et al. [9] hypothesized the involvement of five genes in cell proliferation, and validated these predictions by genetic manipulation and the use of additional microarray data. In addition, they found that the function of these five genes could be inferred much more easily from the multi-species co-expression network than from a network constructed with data from only a single organism.
Figure 1.
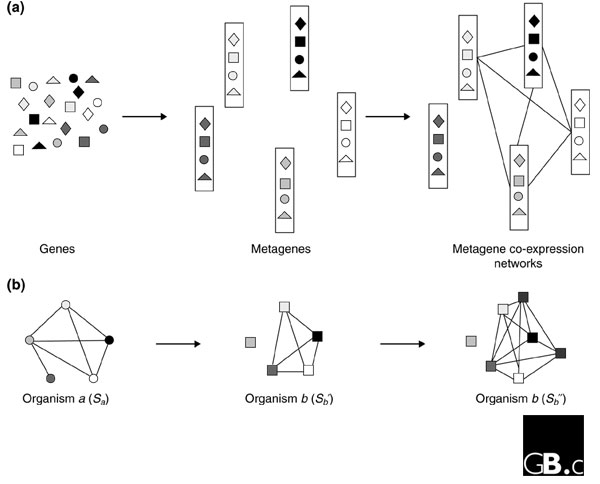
Combining sequence and expression data to make functional assignments. Each of five sets of orthologous genes is represented by a different shading, and each of four organisms is indicated by a different shape. Hence, genes with different shapes but the same shading are orthologs from different species. Edges (lines) in the networks represent co-expression between two genes. (a) The procedure used by Stuart et al. [9] to make functional annotations. Starting with genes from four organisms, they constructed 'metagenes', which are strict orthologous gene clusters. They then identified pairs of metagenes that are co-expressed in multiple organisms, leading to a metagene co-expression network. A set of metagenes that are densely connected to each other in the co-expression network are considered to share the same function. (b) The procedure used by Bergmann et al. [10] to identify functionally related genes across species. Starting from a set of co-expressed genes known to have the same function in organism a (Sa), the authors identify the set of sequence homologs in organism b (Sb') that are co-expressed. They then extend this co-expressed gene subset by including genes in organism b that show expression similarity but may not share sequence similarity (Sb'').
Bergmann et al. [10] used a slightly different procedure to combine sequence and expression analysis (Figure 1b), focusing on six species. They started with a set of co-expressed genes, Sa, known to be associated with a particular function in organism a, and identified the set of their sequence homologs, Sb, in organism b using BLAST. Only a subset Sb' of Sb was found to be co-expressed, and these genes were considered to be the functionally conserved homologs of Sa. Sb' was further expanded to Sb'' by including genes in organism b that are co-expressed with genes in Sb' but that do not share sequence similarity with genes in Sa. As an example, the authors started with a set of heat-shock genes in yeast, successfully identified a set of co-regulated heat-shock genes in Escherichia coli and Caenorhabditis elegans, and showed that half a dozen more co-regulated genes in these latter species also have functions in the heat-shock response even though their orthologs are not annotated in this way in yeast. The results demonstrate that the extent of co-regulation increases drastically from Sb to Sb' to Sb', leading to the conclusion that sequence-based functional annotation can be improved through the integration of expression data.
Cross-species comparison of global network properties
In addition to providing information about the function of individual genes, cross-species expression comparison can be used to analyze entire sets of genes to understand how system properties are conserved over evolution. Much has been written about the power-law connectivity of biological interaction networks: in protein interaction networks, rather than interactions occurring at random, it seems that certain key proteins have many more interactions with other proteins [12]. This pattern is thought to arise as a result of the way in which interaction networks grow by the addition of new elements to existing networks. It now appears that power-law connectivity is also observed in the correlations between pairs of genes, both in metagene co-expression networks [9] and in the expression networks of different organisms [10]. Certain features of gene-expression networks are likely to differ from those of protein networks, including the very high level of modularity seen in expression networks. There is also some suggestion that genes with high connectivity in a network are less dispensable to the organism and more likely to be evolutionarily conserved [10].
Many system properties have been shown to be different between species. Interestingly, Bergmann et al. [10] observed that most of the relations between functional modules differ between organisms. For example, heat-shock and protein-biosynthesis modules exhibit a strong negative correlation in the yeast and Drosophila expression data, but have a significant positive correlation in E. coli, C. elegans, Arabidopsis thaliana and human. In addition, genes involved in protein biosynthesis show tight co-regulation across a variety of conditions in yeast, but exhibit less significant co-expression in other organisms. This suggests that the transcriptional regulation of genes involved in protein biosynthesis plays a major role in the transcriptional program of unicellular organisms but a less dominant role in multicellular organisms. In some cases, the restriction of modules to one or two organisms reflects the modularity of tissue structure, for example in animal-specific signaling pathways and neuronal functions [9].
Cross-species comparison of specific biological processes
Besides the global expression modules, comparison of the expression patterns of genes involved in particular biological processes has the potential to provide more detailed and specific evolutionary information. This principle was first pursued by Alter et al. [13], who compared time points during the cell cycle between yeast and human using generalized singular value decomposition. This computational framework dissects expression patterns into those common to both species, as well as those that are exclusive to one dataset or the other. Another study by Rifkin et al. [14] investigated genome-wide expression variation between Drosophila simulans, Drosophila yakuba and four strains of Drosophila melanogaster during a major developmental transition - the start of metamorphosis. Extensive evolution of developmental gene expression was observed among these closely related species. Interestingly, both within the transcriptional network that controls metamorphosis and across the whole genome, the expression levels of transcription-factor genes appear to be more conserved than those of their downstream targets.
A more recent study compares genomic expression profiles during the aging process in D. melanogaster and C. elegans [15]. The comparison is based on shared patterns of regulation for orthologous genes. Specifically, McCarroll et al. [15] calculated the Pearson's correlation of the log-transformed relative expression change of orthologous genes between middle-aged adults and young adults in both species. Correlations ranging from 0.14 to 0.18 were shown to be statistically significant by permutation procedures despite the very different tissue structure and absolute ages of the two organisms. Furthermore, grouping of genes by GO categories led the authors to observe a conserved pattern of regulation that most notably includes several genes of oxidative metabolism. Nevertheless, most transcriptional changes were specific to worms or to flies; for example, the repression of genes encoding collagens and the induction of genes encoding histones, transposases and DNA and/or RNA helicases are specific to the aging process in worms, whereas the activation of expression of cytochrome P450s, glycosylases and peptidoglycan receptors are specific to aging in Drosophila. In an intriguing sideline, more detailed comparisons revealed that both the conserved global pattern of change in gene expression and the conserved repression of oxidative metabolism genes were abruptly implemented in early adulthood in both organisms. These results suggest that changes in gene expression observed in adults are not solely implemented in response to cumulative damage, as hypothesized in one common model of the aging process [16]. Instead, the timing of these conserved features of aging suggests that they are regulated by developmentally timed transcriptional regulation in young adults.
A critical assessment of the literature on the topic of cross-species comparisons of gene expression would probably start with the observation that some of the inferences are based on optimistic evaluation of very weak correlations. For example, where the correlations observed across species are reported [10,15], they are less than 0.2, so most of the variation is not explained by shared expression across species. It is not a question of whether the cup is half full or half empty: clearly there is just a small mouthful left to swallow, but the evolutionary elixir is an appealing one. Tasty enough, it seems, to justify the annotation of gene function and the inference of regulatory conservation. The statistical justification for this is that with thousands of data points, observation of even small correlations is highly unlikely, as indicated by Monte Carlo simulations resulting in p values less than 10-10 [14].
Strong inference from subtle evolutionary signals
A key potential difficulty in studies that use cross-species comparisons of gene expression is whether the available data for each organism provide a sufficient summary of the covariance structure of gene expression to facilitate reproducible comparison with other species. Current microarray data repositories are unbalanced in terms of the species represented: for example, data from human and yeast are much more abundant than those from fly and E. coli, and experimental conditions for each species also vary. To check the data sufficiency, Stuart et al. [9] randomly divided their compendium of datasets into two halves and evaluated the correspondence between expression networks. Just over 40% of the interactions they observed were significant in both halves, indicating that although these approaches are definitely sensitive to the number, and presumably nature, of conditions tested, there is still a strong enough signal to detect at least a portion of true interactions. McCarroll et al. [15] hint that there is likely to be considerable information to be found in detailed comparisons of specific biological processes, as they find some evidence for conservation of programs regulating larval and embryonic development in worm and fly, and of similar biological processes between more divergent organisms, such as sporulation in yeast and germline formation in C. elegans. Furthermore, there are certainly more sophisticated statistical approaches than simple evaluation of correlation between orthologous gene pairs that remain to be evaluated. These would include the incorporation of phylogenetic information and the use of Bayesian or mixture models to evaluate the significance of expression profiles in two or more species jointly.
Several studies using microarray expression data have suggested that there has been rapid divergence of expression between duplicated genes in human [17] and in yeast [6,18]. Although paralogs may diverge in expression more rapidly than do single-copy genes [19], data from studies comparing the expression levels of duplicated genes within a species [6,17,18] will provide some context for interpreting cross-species comparisons of gene-expression profiles [9,10]. More extensive datasets that evaluate gene expression in matched conditions (for example, similar genetic or environmental perturbations at the same developmental or life-history stages) are likely to improve the power of comparative studies. The signatures of conservation will probably remain subtle, but they will provide plenty of suggestions for hypothesis testing. A more detailed understanding of the conservation of regulatory systems will eventually also require careful attention to the mechanisms and patterns of transcriptional divergence, which after all lie at the heart of morphological, physiological and behavioral evolution.
References
- Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol. 2000;1:reviews0005.1–0005.10. doi: 10.1186/gb-2000-1-5-reviews0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol. 2000;297:233–249. doi: 10.1006/jmbi.2000.3550. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin XL, Lin YZ, Tang J. Relationships of human immunodeficiency virus protease with eukaryotic aspartic proteases. Methods Enzymol. 1994;241:195–224. doi: 10.1016/0076-6879(94)41066-6. [DOI] [PubMed] [Google Scholar]
- Hanks M, Wurst W, Anson-Cartwright L, Auerbach AB, Joyner AL. Rescue of the En-1 mutant phenotype by replacement of En-1 with En-2. Science. 1995;269:679–682. doi: 10.1126/science.7624797. [DOI] [PubMed] [Google Scholar]
- Wagner A. Decoupled evolution of coding region and mRNA expression patterns after gene duplication: implications for the neutralist-selectionist debate. Proc Natl Acad Sci USA. 2000;97:6579–6584. doi: 10.1073/pnas.110147097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GNF SymAtlas http://symatlas.gnf.org/SymAtlas/
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Bergmann S, Ihmels J, Barkai N. Similarities and differences in genome-wide expression data of six organisms. PLoS Biol. 2004;2:E9. doi: 10.1371/journal.pbio.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Microarrays - guilt by association. Science. 2003;302:240–241. doi: 10.1126/science.1090887. [DOI] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Alter O, Brown PO, Botstein D. Generalized singular value decomposition for comparative analysis of genome-scale expression data sets of two different organisms. Proc Natl Acad Sci USA. 2003;100:3351–3356. doi: 10.1073/pnas.0530258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin SA, Kim J, White KP. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 2003;33:138–144. doi: 10.1038/ng1086. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, Kenyon C, Bargmann CI, Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet. 2004;36:197–204. doi: 10.1038/ng1291. [DOI] [PubMed] [Google Scholar]
- Tower J. Aging mechanisms in fruit flies. Bioessays. 1996;18:799–807. doi: 10.1002/bies.950181006. [DOI] [PubMed] [Google Scholar]
- Makova KD, Li WH. Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res. 2003;13:1638–1645. doi: 10.1101/gr.1133803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Nicolae D, Lu HH, Li WH. Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet. 2002;18:609–613. doi: 10.1016/S0168-9525(02)02837-8. [DOI] [PubMed] [Google Scholar]
- Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154:459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]