

Abstract
The diagnosis and treatment of patients with Sjögren syndrome (SS) with neuropathic pain pose several challenges. Patients with SS may experience unorthodox patterns of burning pain not conforming to a traditional “stocking-and-glove” distribution, which can affect the face, torso, and proximal extremities. This distribution of neuropathic pain may reflect mechanisms targeting the proximal-most element of the peripheral nervous system—the dorsal root ganglia (DRG). Skin biopsy can diagnose such a small-fiber neuropathy and is a surrogate marker of DRG neuronal cell loss. However, SS patients have been reported who have similar patterns of proximal neuropathic pain, despite having normal skin biopsy studies. In such cases, DRGs may be targeted by mechanisms not associated with neuronal cell loss. Therefore, alternative approaches are warranted to help characterize abnormal DRGs in SS patients with proximal neuropathic pain.
We performed a systematic review of the literature to define the frequency and spectrum of SS peripheral neuropathies, and to better understand the attribution of SS neuropathic pain to peripheral neuropathies. We found that the frequency of SS neuropathic pain exceeded the prevalence of peripheral neuropathies, and that painful peripheral neuropathies occurred less frequently than neuropathies not always associated with pain. We developed a novel magnetic resonance neurography (MRN) protocol to evaluate DRG abnormalities. Ten SS patients with proximal neuropathic pain were evaluated by this MRN protocol, as well as by punch skin biopsies evaluating for intraepidermal nerve fiber density (IENFD) of unmyelinated nerves. Five patients had radiographic evidence of DRG abnormalities. Patients with MRN DRG abnormalities had increased IENFD of unmyelinated nerves compared to patients without MRN DRG abnormalities (30.2 [interquartile range, 4.4] fibers/mm vs. 11.0 [4.1] fibers/mm, respectively; p = 0.03). Two of these 5 SS patients whose neuropathic pain resolved with intravenous immunoglobulin (IVIg) therapy had improvement of MRN DRG abnormalities.
We have developed a novel MRN protocol that can detect DRG abnormalities in SS patients with neuropathic pain who do not have markers of peripheral neuropathy. We found that SS patients with MRN DRG abnormalities had statistically significant, increased IENFD on skin biopsy studies, which may suggest a relationship between trophic mediators and neuropathic pain. Given that our literature review has demonstrated that many SS neuropathic pain patients do not have a neuropathy, our findings suggest an important niche for this MRN DRG technique in the evaluation of broader subsets of SS neuropathic pain patients who may not have underlying neuropathies. The improvement of MRN DRG abnormalities in patients with IVIg-induced remission of neuropathic pain suggests that our MRN protocol may be capturing reversible, immune-mediated mechanisms targeting the DRG.
INTRODUCTION
Sjögren syndrome (SS) affects up to 1%–2% of the adult population, causes symptoms of keratoconjunctivitis sicca (dry eyes) and xerostomia (dry mouth), and is a systemic autoimmune disease with different permutations of end-organ complications.21 Peripheral nervous system (PNS) manifestations of SS are among the most common extraglandular manifestations, and can be associated with severe neuropathic pain and psychosocial comorbidities.8 Such PNS complications are likely to be encountered by clinicians of different specialties, given that SS patients require ongoing care by primary care practitioners (for systemic, end-organ complications), ophthalmologists (for dry eyes), dentists and other oral health practitioners (for dry mouth), gynecologists (for vaginal dryness and dyspareunia), psychiatrists (for depression), otolaryngologists (for tracheal dryness and performance of lip biopsy), as well as neurologists and rheumatologists. Because of the requisite multifaceted and multidisciplinary care of SS patients, it is especially important for clinicians from all of these specialties to be familiar with the clinical spectrum, diagnostic approach, and etiopathogenic mechanisms of the PNS manifestations seen in SS patients.
The clinical manifestations of the neuropathies that occur in SS reflect disease-associated mechanisms that may target different anatomic structures. For example, SS is characterized by promiscuous B-cell dysregulation and the induction of both pathogenic and nonpathogenic antibodies.2,18,22,23,76 In this regard, one cause of mononeuritis multiplex may be a vasculitic neuropathy associated with cryoglobulinemia and subsequent immune-complex deposition in the vasa nervorum.58 In addition, SS is characterized by the homing and infiltration of lymphocytes to different end organs.22,37,69,70 There is accordingly a spectrum of neuropathies characterized by lymphocytic infiltration of the dorsal root ganglia (DRG) and other ganglionic elements, including the sensory neuronopathies (characterized by proprioceptive loss that can affect entire limbs), painful small-fiber neuropathies (see below), facial pain disorders (such as trigeminal neuropathies), and subtypes of autonomic neuropathies.8,13,28,51,58,64 The evaluation for peripheral neuropathies may require an exhaustive assessment including elicitation of symptoms, neurologic examination, and appropriate ancillary studies.
However, we and others have reported on a diagnostic dilemma frequently encountered by all medical specialists involved in the care of SS patients. Specifically, up to 40% of SS patients may at some time report neuropathic pain,34,65 even in the absence of objective markers of an axonal neuropathy,7,17,65,67 and even after alternative causes of neuropathic pain have been excluded (such as central nervous system disorders, radiculopathies).17,67 There are several potential explanations for why SS patients may report vivid descriptors of neuropathic pain (for example, “burning,” “tearing,” “raking”), without seeming to have supportive features of a peripheral neuropathy.
First, the limitations of electrodiagnostic studies may not be appreciated. Small-fiber neuropathies cause severe neuropathic pain, which may occur in 5%–10% of SS patients,58 but target unmyelinated nerves that cannot be assessed by electrodiagnostic studies.41 In such cases, additional studies such as skin biopsy are warranted to diagnose a neuropathy.47
Second, diagnosing small-fiber neuropathies in SS patients poses unique challenges. Neuropathic pain may be readily ascribed to a PNS etiology when presenting in a familiar “stocking-and-glove” pattern. Such a pattern reflects mechanisms that symmetrically target the distal-most small-fiber nerves—and is therefore referred to as a length-dependent, small-fiber neuropathy. In contrast, SS patients may have an entirely different pattern of neuropathic pain, occurring in a distribution that can affect the face, torso, and proximal-most extremities12—and is therefore referred to as a “non-length-dependent,” small-fiber neuropathy. Yet because of this unorthodox distribution of neuropathic pain, SS patients with such proximal neuropathic pain may be misdiagnosed as having fibromyalgia or even psychiatric disease. However, skin biopsy studies have suggested that such patterns of proximal neuropathic pain reflect neuronal cell loss affecting the proximal-most element of the PNS—the DRG.10,12,26,30,38,43,55 Biopsies have revealed extensive lymphocytic infiltration of the DRG in case series and small numbers of patients with SS neuropathy.33,38,55 Therefore, SS patients who complain of such diffuse neuropathic pain, and who may be misdiagnosed as having fibromyalgia or suspected as having a nonorganic pain syndrome, can experience a “real” small-fiber neuropathy that is peripheral, immune-mediated, and directed against the DRG.
Finally, a third reason for unexplained neuropathic pain in SS patients is that such pain may reflect mechanisms that are not associated with degeneration of neurons. For example, peripheral neuropathy implies a structural lesion that either causes transection of axons, or leads to neuronal cell death of the DRG. However, neuropathic pain can also result in the absence of structural insults to the peripheral nerve. Instead, functional as opposed to neurotoxic mechanisms can be directed against completely viable DRGs. Examples of how neuropathic pain syndromes may occur without an underlying peripheral neuropathy include mechanisms of DRG neuronal hyperexcitability, and the enhanced chemosensitivity of DRGs to pronocioceptive cytokines.4,60,63,79 This emerging recognition that neuropathic pain may actually require the sustained viability as opposed to degeneration of DRGs has been identified in various neuropathic pain disorders,5,14,40 but has not been extensively considered in SS.
SS patients may also experience neuropathic pain due to similar mechanisms that target viable DRGs, and therefore lack traditional biomarkers of a peripheral neuropathy. Alternative approaches are warranted to characterize abnormal DRGs in SS patients with neuropathic pain. Neuroimaging of the DRGs represents a potentially valuable approach to characterize DRG abnormalities. In other PNS disorders, high-resolution magnetic resonance neurography (MRN) has emerged as a noninvasive modality complementing electrodiagnostic and other ancillary studies.71 Similarly, dedicated MRN studies of the DRGs would represent a valuable opportunity to characterize abnormal DRGs in SS patients with neuropathic pain. However, current neuroimaging approaches cannot assess such DRG abnormalities. Given that the DRGs normally demonstrate enhancement due to the lack of a surrounding blood-nerve barrier, such enhancement cannot be distinguished from contiguous anatomic elements.
To circumvent these challenges, we here report on a novel, high-resolution MRN technique (MRN DRG protocol), which evaluates radiographic parameters of DRG abnormalities. In the current study we applied this MRN DRG protocol to a case series of 10 SS patients with a proximal distribution of neuropathic pain, and additionally defined the association of abnormal MRN findings with skin biopsy studies.
We conducted a systematic literature review and found that SS patients frequently experience neuropathic pain in the absence of a painful neuropathy; this reinforces how our findings may be applicable to larger subsets of SS patients who experience neuropathic pain but lack such neurotoxic indicators of a neuropathy. Finally, we consider the implications of our findings in the context of the emerging neuropathic pain literature, wherein other authors similarly have identified how functional as opposed to DRG neurotoxic mechanisms are associated with a wide range of genetic, infectious, and immune-mediated neuropathic pain disorders.
PATIENTS AND METHODS
Patients
This was an observational cohort study, characterizing neuropathic pain in a case series of 10 SS patients. Patients were enrolled from the Johns Hopkins Neuro-Rheumatology Clinic. This Neuro-Rheumatology Clinic evaluates patients suspected of having neurologic complications of systemic rheumatic diseases. Patients were recruited between July 2010 and March 2012. This study was approved by the Johns Hopkins University School of Medicine Institutional Review Board. All patients provided informed consent to participate in the study.
All patients underwent a uniform neurologic assessment, including a standardized neurologic examination performed by 1 of the authors (JB), who is both a board-certified neurologist and rheumatologist, along with nerve-conduction studies and punch skin biopsies.
Inclusion Criteria
Inclusion criteria were SS and neuropathic pain. SS syndrome: patients needed to satisfy the 2002 revised American European classification criteria of primary SS.74 Neuropathic pain: we defined neuropathic pain using a standardized approach based on neuropathic pain questionnaires, neurologic examinations, electrophysiologic studies, and punch skin biopsies. Specifically, patients with neuropathic pain were required to have a score of ≥12 on the s-LANSS neuropathy pain questionnaire, which is a validated questionnaire including descriptors of burning as well as other “positive” descriptors.6 In addition, patients were required to have abnormalities to superficial pain modalities (including pinprick and/or temperature) in symptomatic regions on neurologic examination, and to have electrophysiologic studies revealing no evidence of axonal neuropathies or demyelinating neuropathies. Similar to previous studies, this permitted us to evaluate a clinically homogenous cohort including patients with predominantly burning as opposed to nonpainful dysesthesias, and with examination deficits to small-fiber modalities indicative of unmyelinated C-fiber dysfunction affecting small-fiber nerves.10,12,26,30,38,43,55 Finally, we studied SS patients who experienced neuropathic pain in a proximal distribution as opposed to a distal, stocking-and-glove pattern, because the proximal distribution has been associated with DRG abnormalities.12 A motivating feature of the current study was to determine whether the integrated approach of applying our MRN DRG protocol with skin biopsy enables us to radiographically characterize abnormal DRGs even in neuropathic pain patients without traditional markers of an underlying neuropathy. Therefore, we recruited patients with the above-described clinical features supportive of small-fiber DRG dysfunction, irrespective of whether skin biopsy was indicative of an underlying non-length-dependent neuropathy, and interpreting radiologists were blinded to the results of skin biopsy studies. We designated such patients as having a “proximal” distribution of neuropathic pain, as opposed to a “non-length-dependent” pain distribution, which implies the presence of an underlying small-fiber neuropathy.
Exclusion Criteria and Associated Tests
We used the following exclusion criteria: 1) alternative causes of proximal neuropathic pain were excluded by 2-hour glucose tolerance test (assessing for glucose intolerance and diabetes), vitamin B12, screening for infections (including hepatitis B, hepatitis C, human immunodeficiency virus [HIV]), screening for paraneoplastic antibodies by the Mayo Clinic Paraneoplastic Panel, antigliadin/antiendomysial IgA antibodies for celiac sprue, radiation, or other toxic exposures, and/or medications associated with neuropathic pain. 2) Other causes of DRG enlargement: patients were required to lack clinical, electrodiagnostic, and/or neuroimaging evidence of other disorders that can be associated with DRG enlargement, including demyelinating neuropathies (particularly chronic inflammatory demyelinating neuropathy), genetic disorders such as Charcot-Marie-Tooth disease, and diabetes (also excluded because of exclusion criterion #1, above). In addition, patients were required to lack electrodiagnostic evidence of a radiculopathy as a cause of asymmetric neuropathic pain associated with DRG enlargement, and were required to lack magnetic resonance imaging (MRI) evidence of foraminal stenosis in symptomatically affected regions.
Skin Biopsies for Evaluation of Small-Fiber Neuropathy
We performed skin biopsies as previously described.49 Immunostaining against the panaxonal protein (PGP) 9.5 and quantifying the intraepidermal nerve fiber density (IENFD) of unmyelinated, small-fiber nerves were done by standardized techniques, as previously described.49 The IENFD was evaluated according to previously validated counting rules, performed by a trained technician, who was blinded to the clinical assessment and evaluation.
MR Neurography With DRG Protocol
All patients underwent optimized MRN with and without intravenous contrast (gadolinium DTPA) administration, with high-resolution (in-plane resolution—sub 1 mm) sequences to characterize the DRGs. We used a combination of 2D and 3D imaging sequences, encompassing axial T1W, axial T2 SPAIR (Spectral Adiabatic Inversion Recovery, Siemens, Erlangen, Germany) TSE (turbo spin echo), Sagittal STIR (short Tau inversion recovery), Coronal 3D isotropic STIR SPACE (Sampling Perfection with Application optimized Contrasts using varying flip angle evolutions), Coronal 3D DW PSIF (diffusion weighted reversed fast imaging in steady state free precession), and postcontrast Sagittal 3D fat suppressed T1 VIBE (Volume Interpolated Breath-hold Examination) for detection and characterization of normal and abnormal DRGs. All images were scored by 2 radiologists (AC, JC), each with 15 years of radiology experience. The radiologists were informed only that these were SS patients with neuropathic pain. They were otherwise blinded with regard to intensity of pain, distribution of symptoms, electrodiagnostic studies, skin biopsy results, modality of treatment, and response to treatment. At each vertebral level, the presence of any DRG abnormality was categorically defined by 1 or more of the following: 1) increase in the size of DRGs; 2) increase in the T2 signal of DRGs; 3) increase in the enhancement characteristics of DRGs—relative to the contralateral level and/or levels above and below imaging abnormalities.
Statistical Analysis
The data were analyzed and summarized using appropriate descriptive statistics for continuous and categorical data. The association of DRG abnormalities with demographic features, clinical attributes of neuropathic pain, autoantibodies, and skin biopsies was evaluated by the Fisher exact test or chi-square analysis for categorical variables, and by Wilcoxon rank sum test or the Student t-test for continuous variables. For all analyses, p values <0.05 (2-tailed) were considered statistically significant. We used the STATA 11.0 statistical program (College Station, TX) to analyze the data.
Literature Review
Purpose of Literature Review and Inclusion Criteria
To characterize the clinical characteristics, diagnostic evaluation, and potential pathogenic mechanisms in SS patients with neuropathic pain, we conducted a systematic review of the literature. We searched the PubMed database (National Library of Medicine, Bethesda, MD) using the following keywords: “Sjögren’s syndrome,” “neuropathy,” and “neuropathic pain.” Concerning PNS manifestations: we found only 1 study that characterized the frequency of neuropathic pain in SS patients irrespective of whether there was an underlying peripheral neuropathy.65 We included this study because the frequency of neuropathic pain was assessed as part of a rigorous protocol that included the administration of well-characterized pain questionnaires in all consecutively evaluated patients. With the exception of this study, we considered that there was a systemic perspective in the literature to only characterize neuropathic pain in SS patients for whom neuropathic pain was necessarily attributed to peripheral neuropathies. Given that only distinct subtypes of neuropathies are invariably associated with neuropathic pain, we required that identification of peripheral neuropathies be objectively defined. We therefore included only studies that used clearly articulated study protocols and sought to rigorously characterize peripheral neuropathies using objective ancillary studies. This included abnormal electrodiagnostic studies, and/or appropriate ancillary studies in cohorts with small-fiber neuropathies (including skin biopsy and quantitative sensory testing).
Concerning the diagnosis of SS: several iterations of SS diagnostic criteria have been refined over the past decades.74 We required certain core features that have been preserved in the different criteria, including the following: 1) functional studies corroborating symptoms of keratoconjunctivitis sicca and/or xerostomia; 2) evidence of autoimmunity based on the presence of characteristic antibody specificities, such as anti-Ro/SS-A and/or anti-La/SS-B antibody specificities, and/or focal lymphocytic sialadenitis on lip biopsy.24,74,75
Exclusion Criteria
Secondary SS: we excluded cohorts in which patients were identified as having an alternative primary inflammatory disease (such as, systemic lupus erythematosus, rheumatoid arthritis), and patients with SS considered a subordinated and “secondary” disease process, because such cohorts may include a heterogeneous variety of PNS disorders not unique to SS patients.
RESULTS
Case Vignette: A Patient With MRN Radiographic Improvement of DRG Abnormalities Associated With Clinical Response to Intravenous Immunoglobulin (IVIg) Therapy
A 42-year-old, right-handed man with SS was referred for multifocal, proximal neuropathic pain. Eighteen months prior to evaluation, he developed burning pain compared to “being stung repeatedly by a swarm of bees,” in both legs but also in the face. The severity of such pain was described as 8/10 on a visual analog scale. The patient had deficits to superficial pain modalities of pinprick and temperature in symptomatic regions, and electrodiagnostic studies did not reveal evidence of an axonal or demyelinating neuropathy.
The patient underwent MRN studies with DRG protocol (Figure 1a). As indicated, there was enlargement of the DRGs at the symptomatic L5 and S1 vertebral levels, compared to the DRGs at the L4 and more rostral vertebral levels in the lumbar spine.
FIGURE 1.
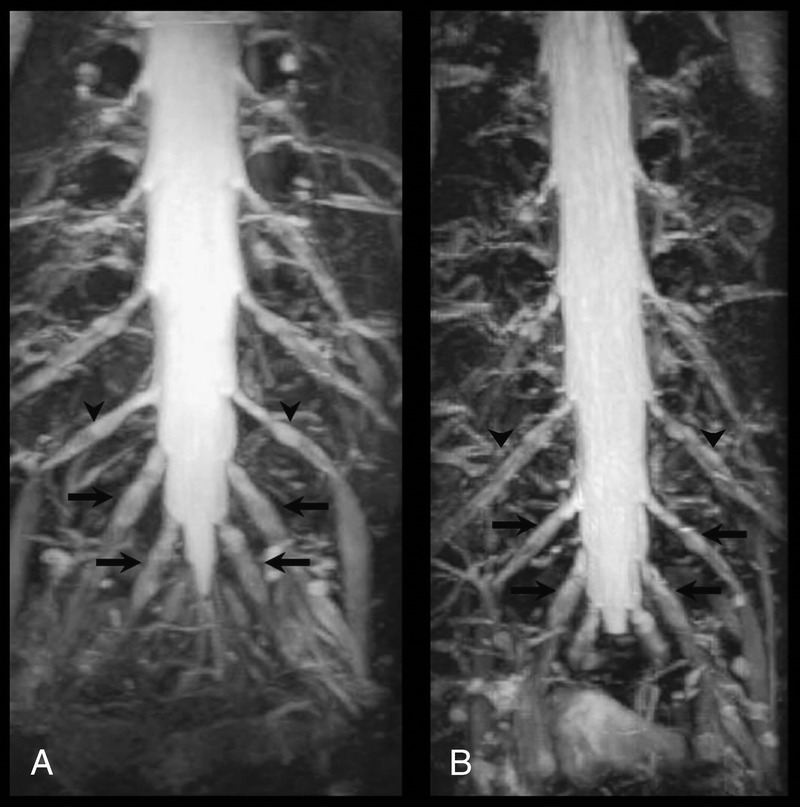
MRN DRG protocol in a SS patient with neuropathic pain before and after IVIg therapy. On this Coronal 3D isotropic STIR SPACE sequence, disproportionately enlarged DRGs are indicated by arrows, and normal sized DRGs are indicated by arrowheads. A) Before IVIg therapy, L5 and S1 DRGs (arrows) are disproportionately enlarged compared to L4 (arrowheads) and more rostral DRGs. B) After IVIg therapy, L5 and S1 DRGs (arrows) are no longer disproportionately enlarged compared to L4 and more rostral DRGs (arrowheads).
Prior to his evaluation at our institution, the patient had no improvement in the severity of neuropathic pain after being treated with several months of prednisone at 60 mg/d and oral cyclophosphamide at 2 mg/kg, given over 5 consecutive days. We initiated treatment with IVIg administered at a cumulative monthly dose of 2 g/kg, given over 5 consecutive days. During this period of IVIg therapy, there was no concomitant use of other immunomodulatory agents. The patient reported decreased severity of neuropathic pain after 2 months, and when evaluated 6 months after continuing IVIg treatment, the pain severity had decreased to a 1/10 on a visual analog scale. He underwent repeat MRN studies with DRG protocol after this 6-month period of IVIg-induced clinical improvement (Figure 1b).
Prior to IVIg therapy, the L5 and S1 DRGs were disproportionately enlarged compared to L4 and more rostral DRGs. After IVIg therapy, there was a disproportionate decrease in the size of the previously enlarged L5 and S1 DRG—such that the L5 and S1 DRGs no longer appear enlarged compared to the L4 DRG and to the DRGs at more rostral levels. Such changes notably paralleled the clinical improvement associated with IVIg-induced reduction in neuropathic pain severity. The patient was maintained on IVIg therapy for 18 months, with no further recurrence of neuropathic pain.
Demographic, Clinical, and Immunologic Characteristics of SS Patients
During the study period, this MRN DRG protocol was performed in 10 SS patients who met the inclusion criteria. The demographic, clinical, and immunologic characteristics of the SS patients are summarized in Table 1. Six patients reported neuropathic pain that antedated the emergence of sicca symptoms. The median age of onset of neuropathic pain was 43 years (range, 25–60 yr), and of sicca symptoms was 48 years (range, 18–63 yr). Eight patients were white and 2 were African-American. The prevalence of male sex, 5 of our 10 patients, is higher than the prevalence reported in the general SS population (where 90% of SS patients are females).22 Similar to prior studies on SS patients with PNS manifestations,8,67 we noted that our SS patients had a lower frequency of autoantibodies and other surrogate markers of B-cell activation compared to patients in other SS cohort studies not enriched with PNS disease.22 Only 3 patients had anti-Ro52 antibodies, 6 patients had anti-Ro60 antibodies, 2 patients had anti-La/SS-B antibodies, and 2 patients had anti-rheumatoid factor antibodies. Only 1 patient had a polyclonal gammopathy, no patient had cryoglobulins, and no patient had decreased levels of complements. Table 1 also describes the clinical characteristics of pain, with 9 of 10 patients describing burning pain, and with patients experiencing moderate pain severity, reported as a median of 5.5 on a visual analog scale.
TABLE 1.
Demographic, Clinical, and Immunologic Characteristics of SS Patients With Proximal Neuropathic Pain

Figure 2 illustrates the distribution of pain, clinical and electrodiagnostic findings, skin biopsy studies, results of MRN DRG protocol, severity of pain, and therapeutic response to symptomatic and/or immunomodulatory therapy. As stipulated by our exclusion criteria, none of our patients had clinical or electrodiagnostic evidence of either an axonal or demyelinating neuropathy. Instead, 2 patients (Patient 7 and Patient 9) developed a neuropathy termed a “neuronopathy,” which developed within 6 months after onset of neuropathic pain. Such a neuronopathy is known to be associated with DRG neuronal cell loss, and is not associated with axonal or demyelinating nerve injury.8,55 Patient 9 was the only SS patient to have a small-fiber neuropathy and skin biopsy indicators of smaller-sized DRG neuronal cell loss. (The association of abnormal MRN studies with skin biopsies is discussed below).
FIGURE 2.
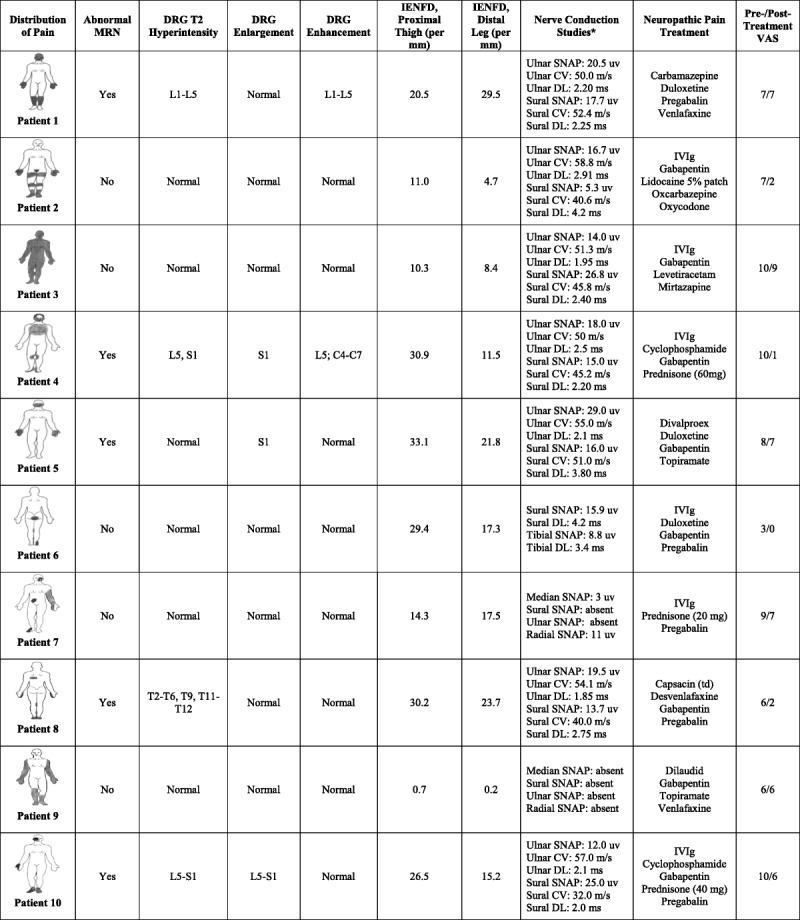
Distribution of pain, results of MRN DRG protocol, skin biopsy studies, electrodiagnostic findings, and therapeutic responses to symptomatic and/or immunomodulatory therapy.
Distribution of Neuropathic Pain and Associated MRN Findings
We performed the MRN DRG protocol on all SS patients with neuropathic pain. Figure 2 illustrates the distribution of pain and associated MRN findings. As indicated, all patients had a distribution of neuropathic pain dissimilar from a traditional stocking-and-glove pattern. Affected regions included the proximal extremities, face, and/or torso. The patient described in the above vignette corresponds to Patient 10 in Figure 2.
Nine patients underwent MRN at the lumbo-sacral spine; a single patient concomitantly underwent MRN of the cervical spine; and guided by severe, neuropathic pain primarily restricted to the torso, a single patient underwent MRN protocol at the thoracic spine. Altogether, 5 patients had MRN indicators of DRG abnormalities. Four patients had hyperintensity of the DRGs on T2 signal; 3 patients had enlargement of the DRGs; and 2 patients had enhancement of the DRGs. There were no statistically significant differences in the demographic, clinical, and autoantibody characteristics between SS patients with and without MRN DRG abnormalities.
Skin Biopsy Findings in Patients With Abnormal DRGs on MRN DRG Studies Compared to Patients With Normal DRGs
Skin biopsy is a biomarker of a small-fiber neuropathy, with findings of decreased IENFD of unmyelinated nerves diagnostic of a small-fiber neuropathy.48,49 In addition, in patients with proximal neuropathic pain, decreased IENFD at the proximal thigh is a marker of proximal-most DRG neuronal degeneration. Such patients are referred to as having a non-length-dependent, small-fiber neuropathy. We sought to define whether patients with MRN DRG abnormalities had discriminating skin biopsy findings, compared to patients without MRN DRG abnormalities.
We noted that our SS patients with MRN DRG abnormalities had statistically significant increased IENFD of unmyelinated nerves at the proximal thigh. Specifically, patients with MRN DRG abnormalities had increased IENFD of unmyelinated nerves at the proximal thigh (median [interquartile range]) compared to patients without DRG abnormalities (30.2 [4.4] fibers/mm vs. 11.0 [4.1] fibers/mm; p = 0.03). This finding of increased IENFDs in patients with abnormal MRN DRG studies may reflect the influence of trophic mediators, and is further discussed below.50,66
Symptomatic Response to IVIg With Follow-Up MRN Studies Showing Interval Improvement in DRG Abnormalities
Prior studies have reported that SS neuropathic pain may be uniquely responsive to IVIg, although it is highly intractable to other potent immunosuppressive therapy (such as cyclophosphamide).39,52,56,77 Therefore, we compared the clinical response in 6 patients who received IVIg with that in 4 patients who received only symptomatic therapy (due to refusal of insurance companies to reimburse for IVIg treatment).
Altogether, patients who received IVIg had decreased severity of pain compared with patients with symptomatic therapy. Specifically, patients who received IVIg had a mean visual analog scale score that decreased from 8.2 to 4.0 (p = 0.02), compared with patients treated with only symptomatic therapies, whose mean score decreased from 6.8 to 5.5 (p = 0.28). One patient treated with IVIg developed intolerable headaches requiring the discontinuation of IVIg, with pain severity relapsing back to pre-IVIg levels. In the 5 patients who did not have complications stemming from IVIg, monthly treatment at doses of 2 g/kg was associated with no complications and with sustained improvement in pain severity. The 4 patients treated with symptomatic therapy had pain severity that was mainly unresponsive to polysymptomatic approaches, including 3 patients who had been treated with 5 or more symptomatic agents.
Two patients who presented with IVIg-induced clinical improvement in neuropathic pain had baseline abnormal MRN DRG studies and had follow-up MRN studies. Both of these patients had follow-up MRN DRG studies that demonstrated interval improvement in MRN DRG abnormalities. These follow-up studies were interpreted by radiologists blinded to IVIg-induced clinical improvement.
LITERATURE REVIEW
Identification of Studies
From our search of the PubMed database we identified 31 potentially eligible studies. There was only a single study reporting on the frequency of neuropathic pain in SS patients, which was assessed as part of a rigorous protocol that included the administration of well-validated neuropathic pain questionnaires in all consecutively studied patients.65 In addition to this study, we identified 30 cohort studies reporting on peripheral neuropathies in SS, of which 18 cohort studies satisfied our inclusion criteria for defining SS and objectively characterizing peripheral neuropathies. We excluded 12 studies, due to inclusion of patients with other primary rheumatic diseases7,11; ascertainment of SS using criteria that did not uniformly require supportive features of autoimmunity27,32,44,46,53; ascertainment of PNS manifestations without detailed description of objective ancillary studies45,54,72; and systematic screening of SS patients who lacked any neurologic symptoms and findings.16,29
The 18 studies that satisfied our inclusion criteria are listed in Table 2. We distinguished between the 10 studies reporting on unselected cohorts of SS patients1,11,25,31,34,35,58,61,67,68 and the 8 studies reporting on selected cohorts of SS patients.12,15,19,28,42,55,62,73 In general, unselected cohort studies permit evaluation of the comparative frequency and spectrum of PNS manifestations, whereas selected cohort studies provide more detailed characterization of a limited subset of PNS manifestations.
TABLE 2.
Spectrum and Frequency of Peripheral Neuropathies in Patients With SS, From the Literature Review
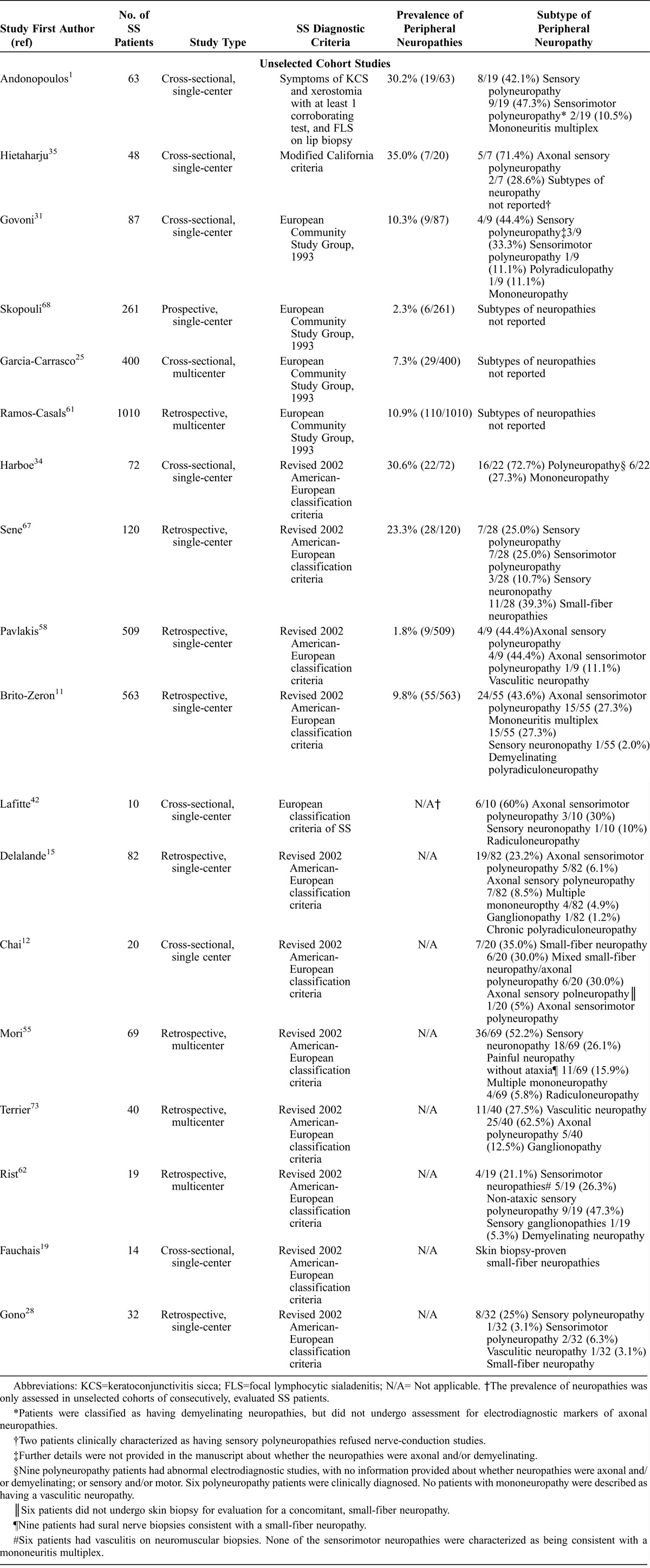
As indicated above, we wanted to identify a framework whereby our MRN DRG protocol may be applicable to SS patients with neuropathic pain. Table 2 details the spectrum of the PNS manifestations of SS, the relative frequency of distinct neuropathies in both unselected and selected cohort studies, as well as ancillary studies used in the evaluation of distinct PNS manifestations. The overall goal of this literature review was to consider the potential relationship of neuropathic pain to peripheral neuropathies, and then to juxtapose clinical features of our cohort with cohorts described in the literature.
Frequency of Neuropathic Pain
The initial step was to consider the extent to which SS patients have neuropathic pain that can be attributed to peripheral neuropathies. The frequency of neuropathic pain was recently reported to be 40% in SS patients, in whom the presence of neuropathic pain, fatigue, and other psychosocial morbidities was rigorously assessed by well-validated pain and disability questionnaires.65 This study also extensively administered questionnaires for chronic disease and other pain syndromes (such as fibromyalgia), which helped to ensure that the validity of neuropathic pain questionnaires was not confounded by the presence of other undetected pain disorders. However, this study did not seek to characterize the presence of peripheral neuropathies using electrodiagnostic and skin biopsy studies. Therefore, to understand whether neuropathic pain was frequently or uncommonly attributable to peripheral neuropathies, we characterized the frequency of peripheral neuropathies in unselected cohort studies.
Prevalence of Peripheral Neuropathies in SS
We found strikingly disparate frequencies of SS peripheral neuropathies in different cohort studies, which varied between 2% and 35% of patients.35,59 However, Table 2 provides additional insight into why the frequency of peripheral neuropathies varied substantially among different cohorts. In particular, such variability stems from how SS was defined (that is, with 5 different diagnostic criteria of SS being utilized), how neuropathies were ascertained, and differences related to spectrum and ascertainment bias in smaller cohorts. The cohort studies reporting a comparatively higher frequency of peripheral neuropathies, occurring in more than 30% of patients, were described in cohorts that defined SS using older diagnostic criteria that have since been revised35 and did not necessarily require all patients to have markers of autoimmunity,1 and therefore potentially included patients with other autoimmune diseases or noninflammatory causes of sicca symptoms. Furthermore, the smallest studies which reported the highest frequency of neuropathies (approximately 30%) were most susceptible to ascertainment bias, and may have overestimated the prevalence of neuropathies.1,35
In contrast, of the 6 largest studies including between 120 and 1010 SS patients,11,25,58,61,67,68 5 studies reported that the prevalence of neuropathies was only 2%–11%.11,25,58,61,68 The remaining study by Sene et al,67 which described peripheral neuropathies in 23% of patients (28 of 120 patients, with 2 patients with isolated trigeminal neuropathies not categorized in our analysis as having peripheral neuropathies), may have comprised patients with more severe disease as the cohort was recruited shortly after hospitalization.
Those studies that used updated SS diagnostic criteria, required all SS patients to have supportive features of autoimmunity, and characterized peripheral neuropathies in larger numbers of patients reported neuropathies in 2%–10% of patients. An important implication of this finding is that such a low frequency of peripheral neuropathies (≤10%) cannot completely account for the substantially higher frequency of neuropathic pain described in up to 40% of SS patients.19,65
Decreased Frequency of Painful Neuropathies, Compared to Neuropathies Not Necessarily Associated With Neuropathic Pain
Although the above analysis indicates that peripheral neuropathies are described in 2%–10% of SS patients, it is important to consider that peripheral neuropathies are not necessarily painful. As a subsequent step to define the potential attribution of SS neuropathic pain to peripheral neuropathies, we reviewed the frequency of painful compared with nonpainful neuropathies. In doing so, we considered that the only peripheral neuropathies invariably associated with neuropathic pain are mononeuritis multiplex and small-fiber neuropathies. In contrast, the sensory manifestations of polyneuropathies may be conveyed using “negative” sensory descriptors (for example, “numbness”), as opposed to “positive” descriptors of neuropathic pain (such as “burning” or “tearing” pain). Therefore, we compared the frequency of the painful neuropathies—mononeuritis multiplex and small-fiber neuropathies—with polyneuropathies, which are not invariably associated with neuropathic pain.
Such painful neuropathies are reported at substantially lower frequencies than polyneuropathies (Table 2). Reports of unselected cohort studies described sensory or sensorimotor polyneuropathies as constituting between 44% and 71% of SS-associated peripheral neuropathies.1,11,31,35,58,67 In contrast, mononeuritis multiplex was the least commonly reported neuropathy in 6 of 7 unselected cohort studies reporting on subtypes of neuropathies (≤11%), and was not even identified in 4 of these 7 studies.31,34,35,67 Altogether, unselected cohort studies reported that the overall prevalence of vasculitic neuropathies in SS patients is only between 0 and 3%.1,11,25,31,34,35,58,61,67,68
As noted above, small-fiber neuropathy is the only peripheral neuropathy other than vasculitic neuropathies that uniformly presents with neuropathic pain. Small-fiber neuropathies are invariably painful; they target the thinly myelinated A-delta fibers and unmyelinated C-fiber studies, but cannot be detected by routine electrodiagnostic studies.41,47 Therefore, the diagnosis of small-fiber neuropathies requires alternative diagnostic techniques such as skin biopsy.49 However, of the 10 unselected cohort studies, a striking finding is that only a single study reported on SS patients with small-fiber neuropathies.67 Reasons for such an omission of small-fiber neuropathies in unselected cohort studies may include lack of awareness of small-fiber neuropathy as a distinct PNS entity in SS, and inaccessibility to diagnostic techniques required in the detection of small-fiber neuropathies.
In contrast, the identification of SS small-fiber neuropathies was noted in 4 of the 8 studies reporting on selected SS patients,12,19,28,55 which likely reflects greater awareness of small-fiber neuropathy as a distinct PNS entity of SS, as well as available expertise to perform supportive, ancillary studies.
Distinguishing Features of the Current Case Series of SS Patients Compared to the Spectrum of SS PNS Disease Reported in the Literature
Of all the cohort studies characterizing SS small-fiber neuropathies,12,19,28,55,67 only the study of Chai et al12 reported a clinical spectrum of neuropathic pain that resembled that of the patients in our study. In particular, the study of Chai et al and our study similarly characterized neuropathic pain occurring in a proximal distribution, not conforming to a distal, stocking-and-glove pattern. However, whereas Chai et al12 reported on SS neuropathic pain associated with skin biopsy markers of DRG cell loss and an underlying neuropathy, we report on radiographic features of DRG abnormalities in SS patients without underlying markers of a neuropathy.
Summary of Literature Review
Our literature review indicates that the frequency of neuropathic pain in SS patients exceeds the prevalence of peripheral neuropathies, and suggests that SS patients with neuropathic pain have alternative mechanisms not associated with peripheral neuropathies.
DISCUSSION
We describe here the application of a novel MRN DRG protocol to a well-characterized cohort of 10 SS patients with neuropathic pain, all of whom underwent a standardized and rigorous evaluation including the administration of neuropathic pain questionnaires, clinical examination, electrodiagnostic studies, and punch skin biopsies. There were several significant findings. First, we characterized MRN DRG abnormalities in 5 SS patients with neuropathic pain, all of whom had normal skin biopsy studies and otherwise would not have been considered as having objective evidence of PNS disease. Second, we defined that patients with abnormal MRN DRG studies had statistically significant increased IENFD on skin biopsy compared to patients with normal MRN DRG studies. Third, these findings are of special interest given that our literature review demonstrated that most SS patients with neuropathic pain do not have neurotoxic markers of a peripheral neuropathy. Given that we detected that MRN DRG changes were not associated with an underlying peripheral neuropathy, our findings suggest a unique and important niche for this MRN DRG technique in the evaluation of this broader subset of SS patients with neuropathic pain but without underlying neuropathies. Finally, we consider below the implications of our findings in the context of an emerging neuropathic pain literature, where others have similarly identified how functional as opposed to DRG neurotoxic mechanisms are associated with a wide range of genetic, infectious, and immune-mediated neuropathic pain disorders.
An important, multidisciplinary implication of the current study is how our MRN DRG protocol can characterize DRG abnormalities in SS patients with a proximal distribution of pain, who may otherwise be considered to have a nonorganic pain disorder. Although neuropathic pain is well recognized when occurring in a familiar stocking-and-glove distribution, SS patients with unorthodox distributions of pain are frequently thought not to have an objective, anatomical basis for the pain (Figure 2). Skin biopsy can provide objective evidence that 1 such cause of neuropathic pain is a small-fiber neuropathy associated with DRG neuronal cell death.10,12,26,30,38,43,55 Our MRN DRG protocol can serve as an additional biomarker to substantiate that SS patients with multifocal neuropathic pain have a “real” neuropathic pain disorder, associated with neuroimaging findings of DRG abnormalities.
We took the novel approach of integrating our MRN DRG protocol with skin biopsy studies. We determined that patients with abnormal MRN DRG abnormalities had statistically significant increased IENFD, compared to patients with normal MRN DRG studies. These findings suggest that abnormal MRN DRG studies may reflect mechanisms associated with trophic as opposed to neurotoxic mediators.
In particular, trophic mediators such as nerve-growth factor can exert pronociceptive influences at the level of the DRG, by increasing DRG neuronal hyperexcitability, chemosensitivity, and mechanosensitivity.60,63,79 Such neurotrophins may also have immunomodulatory actions, and have been reported to be upregulated in inflammatory disorders such as SS.66 Given the increasingly large therapeutic armamentarium to target such trophic influences,50 this association between abnormal MRN DRG studies and increased nerve-fiber density may have therapeutic as well as mechanistic implications; further studies using this paired neuroimaging-pathologic approach are justified.
We emphasize the context in which we are interpreting these skin biopsy findings. In clinical practice, skin biopsy is a diagnostic biomarker of a small-fiber neuropathy and is only defined as being “abnormal” when the IENFD is below the fifth percentile compared to normative controls. Prior studies have not considered whether there is a comparable threshold for defining skin biopsy as being abnormal, based on “supra-normal” values of IENFD. Therefore, in the current study we are careful to characterize and distinguish patients with MRN DRG abnormalities as having increased IENFD only relative to patients without MRN DRG abnormalities; and we have purposefully not designated such patients as having abnormal skin biopsies. If we had just defined all patients with values of IENFDs above the fifth percentile of normative controls as having normal studies, such a characterization would have masked important differences between the patients with MRN DRG abnormalities and those without. By quantifying and comparing IENFD in patients with and without MRN DRG abnormalities, we have demonstrated distinct and statistically significant differences that likely have mechanistic implications.
Our findings that SS patients with abnormal MRN DRG studies may lack neurotoxic markers of a neuropathy are consistent with recent reports of different genetic,14,36 immune-mediated,40 infectious,57,78 and iatrogenic causes of neuropathic pain.20
For instance, gain-of-function, genetic mutations affecting the DRG-associated, Nav1.7 nociceptive channel have been identified in neuropathic pain disorders without markers of a neuropathy.36 Recently, a patient with widespread neuropathic pain was described as having a novel mutation affecting the DRG-associated Nav1.7 DRG channel, but lacking markers of a neuropathy, and with in-vitro studies demonstrating mechanisms of neuropathic pain driven by DRG neuronal hyperexcitability.14 Similarly, a recent study described anti-neuronal antibodies targeting the voltage-gated potassium channel antibody in patients with chronic pain.40 That cohort included neuropathic pain patients with attributes similar to the SS patients in the current study, including widespread and burning neuropathic pain not associated with skin biopsy or other markers of a neuropathy. While such anti-neuronal antibodies are known to target axons, it was hypothesized that such antibodies may also be directed against DRGs.5 Reactivation of the varicella zoster virus in the DRG may cause postherpetic neuralgia, which can cause burning pain and tactile allodynia similar to that experienced by SS patients in the current study. Whereas the most common pathologic finding is DRG hemorrhagic necrosis, other patients without skin biopsy findings of a neuropathy may have DRG inflammatory changes without necrosis.57,78
Patients with other inflammatory disorders have been described as having a burden of neuropathic pain not explained by an underlying neuropathy. In a cohort of neuro-sarcoidosis patients, two-thirds of patients with small-fiber symptoms and clinically suspected as having a small-fiber neuropathy had normal skin biopsy studies.3 Finally, whereas chemotherapy-associated neuropathic pain may be neurotoxic to DRGs at high doses, lower doses may not be neurotoxic to DRGs and are therefore not associated with markers of a neuropathy.20
Information from these reports illustrates how neuropathic pain may occur in the absence of a neuropathy, and therefore especially suggests why our MRN DRG studies represent a potentially valuable diagnostic niche in the evaluation of SS neuropathic pain. Our collective findings suggest a diagnostic approach to SS neuropathic pain patients, and is described as follows.
Diagnostic Algorithm With MRN DRG Neuroimaging for Evaluating Neuropathic Pain in SS Patients
The initial evaluation of potential neuropathic pain hinges on the symmetry and distribution of symptoms. As discussed above, the likelihood that neuropathic symptoms stem from an underlying neuropathy is enhanced when patients describe symptoms occurring in a well-recognized, stocking-and-glove distribution. Clinical and electrodiagnostic assessment can evaluate for larger-fiber demyelinating or axonal neuropathies. In contrast to patients with small-fiber dysfunction, patients with larger-fiber neuropathies may describe numbness as well as neuropathic pain, and have electrodiagnostic studies indicative of an axonal and/or a demyelinating polyneuropathy. In such cases, no further assessment is necessary. Skin biopsy studies can be used in the evaluation of patients with potential small-fiber dysfunction,49 who usually complain of burning pain and may have neurologic examinations showing selective deficits to small-fiber modalities (such as pin-prick and temperature).41 However, as demonstrated in the current study, patients with suspected small-fiber dysfunction can have normal skin biopsy studies but abnormal MRN DRG studies. Our findings support that the MRN DRG protocol may provide evidence of DRG abnormalities in such SS neuropathic pain patients without skin biopsy markers of a peripheral neuropathy. This suggested algorithm can now be evaluated in future studies.
We also add to the literature about the efficacy of IVIg in SS patients experiencing neuropathic pain without electrophysiologic features of axonal neuropathies.39,52,56,77 We report that 2 SS neuropathic pain patients with MRN DRG abnormalities who clinically responded to IVIg had repeat MRN studies noting interval improvement in DRG abnormalities. This reduction of neuropathic pain associated with improvement of MRN DRG abnormalities, described in the patient vignette, provides indirect but tantalizing evidence that MRN may be capturing immune-mediated mechanisms that may be targeted by IVIg immunomodulatory therapy.
We emphasize that our findings should be interpreted with caution, given the small number of patients treated with IVIg. As far as we know, our findings also represent the first example whereby IVIg-associated improvement in neuropathic pain has been longitudinally associated with interval improvement of any ancillary study. Yet we acknowledge that neuroimaging markers of PNS disease may fluctuate over time, independent of clinical response and treatment intervention. These preliminary findings in our case series suggest that larger studies evaluating the efficacy of IVIg are now warranted, and that the natural history of MRN DRG abnormalities needs to be longitudinally assessed in IVIg-naive patients.
An important point is whether the multiple versus individual indicators of DRG abnormalities characterized in the current study are associated with a more severe clinical profile, associated with distinct electrodiagnostic or skin biopsy findings, or augur responsiveness or intractability to treatment. Within the context of this study, we could not detect any such associations. The lack of any associations may reflect the small number of patients evaluated in this case series, and would be an important and intriguing aspect to explore in future studies. Whereas we predominantly characterized MRN DRG abnormalities in patients with neuropathic pain having normal skin biopsy studies, prior studies have primarily reported that SS patients with such proximal neuropathic pain have abnormal skin biopsy studies, which can be associated with abnormal electrodiagnostic studies showing a sensory neuronopathy (and associated with larger-sized DRG neuronal cell death). Only 1 of the current patients had small-fiber neuropathy, and only 2 patients had sensory neuronopathies. This may reflect referral bias, given that patients were primarily referred to the Neuro-Rheumatology Clinic for evaluation of a systemic rheumatic disease, whereas referral to tertiary-care neuromuscular cohorts may be instead driven by severity of neurologic symptoms (that is, increased severity of vasculitic neuropathies in selected compared with unselected cohort studies; see Table 2). Although we found that patients with lower IENFD on skin biopsy lacked MRN DRG abnormalities, further application of our MRN DRG protocol to SS patients with small-fiber neuropathy would be useful.
Several limitations of the current study need to be considered. First, we acknowledge that our findings were characterized in a small number of patients, and we have therefore been careful to emphasize our findings as occurring in the context of a case series. Given the nature of this case series, we have presented our findings in the context of a comprehensive review of the SS PNS literature, which suggests how our patients may represent an overlooked spectrum of SS PNS disease.
We acknowledge that the indicators we used to characterize the DRGs, based on hyperintensity, enlargement, and/or enhancement, were interpreted by radiologists with extensive expertise (15 years of experience each), and may initially be difficult to interpret for less-experienced radiologists. However, such challenges are not unlike limitations currently faced by the application of various imaging modalities in rheumatic diseases, in which the qualitative distinction between abnormal and normal radiographic findings depends on the experience of interpreting radiologists.9 In such examples, radiographic approaches that can assess quantitative changes may permit more valid radiographic interpretation and distinction between abnormal and normal tissue. Our MRN study is ideal for such quantitative approaches. Therefore, we are now planning diffusion tensor imaging (DTI) studies to characterize the size of DRGs quantitatively. Assessment for disproportionate enhancement can be subtle, given that the DRGs normally enhance. The use of radiographically unaffected nerves for comparison also needs to be substantiated in this disease that has diffuse systemic effects. However, we note that SS patients may have neuropathies exquisitely localized to a single DRG or sensory ganglia (such as trigeminal neuropathy), even in the context of highly active systemic disease that jeopardizes other end-organs. Finally, the more diffuse and multifocal pain in the torso and upper extremities could not be fully evaluated in this initial study, which primarily evaluated DRGs in the lumbar spine.
In conclusion, we have developed a novel MRN approach that circumvents traditional barriers of imaging abnormal DRGs, and have demonstrated abnormal MRN DRG studies in SS patients with neuropathic pain. These findings in our case series provide exciting directions for future studies with larger numbers of patients.
Footnotes
Abbreviations: CNS = central nervous system, DRG = dorsal root ganglia, IENFD = intraepidermal nerve fiber density, IVIg = intravenous immunoglobulin, MRI = magnetic resonance imaging, MRN = magnetic resonance neurography, PNS = peripheral nervous system, SS = Sjögren syndrome, VAS = visual analog scale.
Financial support and conflicts of interest: funding for this research was provided in part by the Jerome L. Greene Foundation; a grant from the Radiological Society of North America (KCW); and National Institutes of Health grant P30 AR053503 from the National Institute of Arthritis And Musculoskeletal And Skin Diseases. The following authors reported relationships (personal or institutional) with the listed entities, unrelated to the present work: KCW: Society for Imaging Informatics in Medicine; MML Investors Services, DexNote LLC; JC: Carestream, General Electric, Siemens, Best Doctors, BioClinica, Medtronic, Pfizer, Toshiba; AC: Siemens, Integra, GEAUR. The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Andonopoulos AP, Lagos G, Drosos AA, Moutsopoulos HM. The spectrum of neurological involvement in Sjogren’s syndrome. Br J Rheumatol. 1990; 29: 21– 23. [DOI] [PubMed] [Google Scholar]
- 2.Andonopoulos AP, Lagos G, Drosos AA, Moutsopoulos HM. Neurologic involvement in primary Sjogren’s syndrome: a preliminary report. J Autoimmun. 1989; 2: 485– 488. [DOI] [PubMed] [Google Scholar]
- 3.Bakkers M, Merkies IS, Lauria G, Devigili G, Penza P, Lombardi R, Hermans MC, van Nes SI, De Baets M, Faber CG. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology. 2009; 73: 1142– 1148. [DOI] [PubMed] [Google Scholar]
- 4.Baron R. Mechanisms of disease: neuropathic pain—a clinical perspective. Nat Clin Pract Neurol. 2006; 2: 95– 106. [DOI] [PubMed] [Google Scholar]
- 5.Bennett DL, Vincent A. Autoimmune pain: an emerging concept. Neurology. 2012; 79: 1080– 1081. [DOI] [PubMed] [Google Scholar]
- 6.Bennett MI, Smith BH, Torrance N, Potter J. The S-LANSS score for identifying pain of predominantly neuropathic origin: validation for use in clinical and postal research. J Pain. 2005; 6: 149– 158. [DOI] [PubMed] [Google Scholar]
- 7.Birnbaum J. Peripheral neuropathy is an uncommon cause of neuropathic symptoms in Sjogrens patients [abstract]. Arthritis Rheum. 2009; 60( Suppl 10): 506. [Google Scholar]
- 8.Birnbaum J. Peripheral nervous system manifestations of Sjogren syndrome: clinical patterns, diagnostic paradigms, etiopathogenesis, and therapeutic strategies. Neurologist. 2010; 16: 287– 297. [DOI] [PubMed] [Google Scholar]
- 9.Bliddal H, Boesen M, Christensen R, Kubassova O, Torp-Pedersen S. Imaging as a follow-up tool in clinical trials and clinical practice. Best Pract Res Clin Rheumatol. 2008; 22: 1109– 1126. [DOI] [PubMed] [Google Scholar]
- 10.Brannagan TH, III, Hays AP, Chin SS, Sander HW, Chin RL, Magda P, Green PH, Latov N. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol. 2005; 62: 1574– 1578. [DOI] [PubMed] [Google Scholar]
- 11.Brito-Zeron P, Akasbi M, Bosch X, Bove A, Perez-De-Lis M, Diaz-Lagares C, Retamozo S, Gandia M, Perez-Alvarez R, Soto-Cardenas MJ, Siso A, Valls-Sole J, Graus F, Ramos-Casals M. Classification and characterisation of peripheral neuropathies in 102 patients with primary Sjogren’s syndrome. Clin Exp Rheumatol. 2013; 31: 103– 110. [PubMed] [Google Scholar]
- 12.Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjogren syndrome. Neurology. 2005; 65: 925– 927. [DOI] [PubMed] [Google Scholar]
- 13.Chai J, Logigian EL. Neurological manifestations of primary Sjogren’s syndrome. Curr Opin Neurol. 2010; 13: 509– 513. [DOI] [PubMed] [Google Scholar]
- 14.Dabby R, Sadeh M, Gilad R, Lampl Y, Cohen S, Inbar S, Leshinsky-Silver E. Chronic non-paroxysmal neuropathic pain—Novel phenotype of mutation in the sodium channel SCN9A gene. J Neurol Sci. 2011; 301: 90– 92. [DOI] [PubMed] [Google Scholar]
- 15.Delalande S, de Seze J, Fauchais AL, Hachulla E, Stojkovic T, Ferriby D, Dubucquoi S, Pruvo JP, Vermersch P, Hatron PY. Neurologic manifestations in primary Sjogren syndrome: a study of 82 patients. Medicine (Baltimore). 2004; 83: 280– 291. [DOI] [PubMed] [Google Scholar]
- 16.Drosos AA, Andonopoulos AP, Lagos G, Angelopoulos NV, Moutsopoulos HM. Neuropsychiatric abnormalities in primary Sjogren’s syndrome. Clin Exp Rheumatol. 1989; 7: 207– 209. [PubMed] [Google Scholar]
- 17.Fauchais AL, Magy L, Vidal E. Central and peripheral neurological complications of primary Sjogren’s syndrome. Presse Med. 2012; 41: e485– e493. [DOI] [PubMed] [Google Scholar]
- 18.Fauchais AL, Martel C, Gondran G, Lambert M, Launay D, Jauberteau MO, Hachulla E, Vidal E, Hatron PY. Immunological profile in primary Sjogren syndrome: clinical significance, prognosis and long-term evolution to other auto-immune disease. Autoimmun Rev. 2010; 9: 595– 599. [DOI] [PubMed] [Google Scholar]
- 19.Fauchais AL, Richard L, Gondran G, Ghorab K, Palat S, Bezanahary H, Loustaud-Ratti V, Ly K, Jauberteau MO, Vallat JM, Vidal E, Magy L. Small fibre neuropathy in primary Sjogren syndrome. Rev Med Interne. 2011; 32: 142– 148. [DOI] [PubMed] [Google Scholar]
- 20.Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006; 122: 245– 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox PC, Bowman SJ, Segal B, Vivino FB, Murukutla N, Choueiri K, Ogale S, McLean L. Oral involvement in primary Sjogren syndrome. J Am Dent Assoc. 2008; 139: 1592– 1601. [DOI] [PubMed] [Google Scholar]
- 22.Fox RI. Sjogren’s syndrome. Lancet. 2005; 366: 321– 331. [DOI] [PubMed] [Google Scholar]
- 23.Fox RI, Michelson P. Approaches to the treatment of Sjogren’s syndrome. J Rheumatol Suppl. 2000; 61: 15– 21. [PubMed] [Google Scholar]
- 24.Fox RI, Robinson CA, Curd JG, Kozin F, Howell FV. Sjogren’s syndrome. Proposed criteria for classification. Arthritis Rheum. 1986; 29: 577– 585. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Carrasco M, Ramos-Casals M, Rosas J, Pallares L, Calvo-Alen J, Cervera R, Font J, Ingelmo M. Primary Sjogren syndrome: clinical and immunologic disease patterns in a cohort of 400 patients. Medicine (Baltimore). 2002; 81: 270– 280. [DOI] [PubMed] [Google Scholar]
- 26.Gemignani F, Ferrari G, Vitetta F, Giovanelli M, Macaluso C, Marbini A. Non-length-dependent small fibre neuropathy. Confocal microscopy study of the corneal innervation. J Neurol Neurosurg Psychiatry. 2010; 81: 731– 733. [DOI] [PubMed] [Google Scholar]
- 27.Gemignani F, Marbini A, Pavesi G, Di Vittorio S, Manganelli P, Cenacchi G, Mancia D. Peripheral neuropathy associated with primary Sjogren’s syndrome. J Neurol Neurosurg Psychiatry. 1994; 57: 983– 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gono T, Kawaguchi Y, Katsumata Y, Takagi K, Tochimoto A, Baba S, Okamoto Y, Ota Y, Yamanaka H. Clinical manifestations of neurological involvement in primary Sjogren’s syndrome. Clin Rheumatol. 2011; 30: 485– 490. [DOI] [PubMed] [Google Scholar]
- 29.Goransson LG, Herigstad A, Tjensvoll AB, Harboe E, Mellgren SI, Omdal R. Peripheral neuropathy in primary sjogren syndrome: a population-based study. Arch Neurol. 2006; 63: 1612– 1615. [DOI] [PubMed] [Google Scholar]
- 30.Gorson KC, Herrmann DN, Thiagarajan R, Brannagan TH, Chin RL, Kinsella LJ, Ropper AH. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry. 2008; 79: 163– 169. [DOI] [PubMed] [Google Scholar]
- 31.Govoni M, Bajocchi G, Rizzo N, Tola MR, Caniatti L, Tugnoli V, Colamussi P, Trotta F. Neurological involvement in primary Sjogren’s syndrome: clinical and instrumental evaluation in a cohort of Italian patients. Clin Rheumatol. 1999; 18: 299– 303. [DOI] [PubMed] [Google Scholar]
- 32.Grant IA, Hunder GG, Homburger HA, Dyck PJ. Peripheral neuropathy associated with sicca complex. Neurology. 1997; 48: 855– 862. [DOI] [PubMed] [Google Scholar]
- 33.Griffin JW, Cornblath DR, Alexander E, Campbell J, Low PA, Bird S, Feldman EL. Ataxic sensory neuropathy and dorsal root ganglionitis associated with Sjogren’s syndrome. Ann Neurol. 1990; 27: 304– 315. [DOI] [PubMed] [Google Scholar]
- 34.Harboe E, Tjensvoll AB, Maroni S, Goransson LG, Greve OJ, Beyer MK, Herigstad A, Kvaloy JT, Omdal R. Neuropsychiatric syndromes in patients with systemic lupus erythematosus and primary Sjogren syndrome: a comparative population-based study. Ann Rheum Dis. 2009; 68: 1541– 1546. [DOI] [PubMed] [Google Scholar]
- 35.Hietaharju A, Yli-Kerttula U, Hakkinen V, Frey H. Nervous system manifestations in Sjogren’s syndrome. Acta Neurol Scand. 1990; 81: 144– 152. [DOI] [PubMed] [Google Scholar]
- 36.Hoeijmakers JG, Merkies IS, Gerrits MM, Waxman SG, Faber CG. Genetic aspects of sodium channelopathy in small fiber neuropathy. Clin Genet. 2012; 82: 351– 358. [DOI] [PubMed] [Google Scholar]
- 37.Karabiyik A, Peck AB, Nguyen CQ. The important role of T cells and receptor expression in Sjogren’s syndrome. Scand J Immunol. 2013; 78: 157– 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawagashira Y, Koike H, Fujioka Y, Hashimoto R, Tomita M, Morozumi S, Iijima M, Katsuno M, Tanaka F, Sobue G. Differential, size-dependent sensory neuron involvement in the painful and ataxic forms of primary Sjogren’s syndrome-associated neuropathy. J Neurol Sci. 2012; 15: 139– 146. [DOI] [PubMed] [Google Scholar]
- 39.Kizawa M, Mori K, Iijima M, Koike H, Hattori N, Sobue G. Intravenous immunoglobulin treatment in painful sensory neuropathy without sensory ataxia associated with Sjogren’s syndrome. J Neurol Neurosurg Psychiatry. 2006; 77: 967– 969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klein CJ, Lennon VA, Aston PA, McKeon A, Pittock SJ. Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology. 2012; 79: 1136– 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lacomis D. Small-fiber neuropathy. Muscle Nerve. 2002; 26: 173– 188. [DOI] [PubMed] [Google Scholar]
- 42.Lafitte C, Amoura Z, Cacoub P, Pradat-Diehl P, Picq C, Salachas F, Leger JM, Piette JC, Delattre JY. Neurological complications of primary Sjogren’s syndrome. J Neurol. 2001; 248: 577– 584. [DOI] [PubMed] [Google Scholar]
- 43.Lauria G, Devigili G. Skin biopsy as a diagnostic tool in peripheral neuropathy. Nat Clin Pract Neurol. 2007; 3: 546– 557. [DOI] [PubMed] [Google Scholar]
- 44.Lopate G, Pestronk A, Al-Lozi M, Lynch T, Florence J, Miller T, Levine T, Rampy T, Beson B, Ramneantu I. Peripheral neuropathy in an outpatient cohort of patients with Sjogren’s syndrome. Muscle Nerve. 2006; 33: 672– 676. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Lavin M, Vaughan JH, Tan EM. Autoantibodies and the spectrum of Sjogren’s syndrome. Ann Intern Med. 1979; 91: 185– 190. [DOI] [PubMed] [Google Scholar]
- 46.Mauch E, Volk C, Kratzsch G, Krapf H, Kornhuber HH, Laufen H, Hummel KJ. Neurological and neuropsychiatric dysfunction in primary Sjogren’s syndrome. Acta Neurol Scand. 1994; 89: 31– 35. [PubMed] [Google Scholar]
- 47.McArthur JC. Painful small fiber neuropathies. Continuum (Minneap Minn). 2012; 18: 106– 125. [DOI] [PubMed] [Google Scholar]
- 48.McArthur JC, Griffin JW. Another tool for the neurologist’s toolbox. Ann Neurol. 2005; 57: 163– 167. [DOI] [PubMed] [Google Scholar]
- 49.McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol. 1998; 55: 1513– 1520. [DOI] [PubMed] [Google Scholar]
- 50.McKelvey L, Shorten GD, O’Keeffe GW. Nerve growth factor-mediated regulation of pain signalling and proposed new intervention strategies in clinical pain management. J Neurochem. 2013; 124: 276– 289. [DOI] [PubMed] [Google Scholar]
- 51.Mellgren SI, Goransson LG, Omdal R. Primary Sjogren’s syndrome associated neuropathy. Can J Neurol Sci. 2007; 34: 280– 287. [DOI] [PubMed] [Google Scholar]
- 52.Molina JA, Benito-Leon J, Bermejo F, Jimenez-Jimenez FJ, Olivan J. Intravenous immunoglobulin therapy in sensory neuropathy associated with Sjogren’s syndrome. J Neurol Neurosurg Psychiatry. 1996; 60: 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Molina R, Provost TT, Arnett FC, Bias WB, Hochberg MC, Wilson RW, Alexander EL. Primary Sjogren’s syndrome in men. Clinical, serologic, and immunogenetic features. Am J Med. 1986; 80: 23– 31. [DOI] [PubMed] [Google Scholar]
- 54.Moll JW, Markusse HM, Pijnenburg JJ, Vecht CJ, Henzen-Logmans SC. Antineuronal antibodies in patients with neurologic complications of primary Sjogren’s syndrome. Neurology. 1993; 43: 2574– 2581. [DOI] [PubMed] [Google Scholar]
- 55.Mori K, Iijima M, Koike H, Hattori N, Tanaka F, Watanabe H, Katsuno M, Fujita A, Aiba I, Ogata A, Saito T, Asakura K, Yoshida M, Hirayama M, Sobue G. The wide spectrum of clinical manifestations in Sjogren’s syndrome-associated neuropathy. Brain. 2005; 128( 11): 2518– 2534. [DOI] [PubMed] [Google Scholar]
- 56.Morozumi S, Kawagashira Y, Iijima M, Koike H, Hattori N, Katsuno M, Tanaka F, Sobue G. Intravenous immunoglobulin treatment for painful sensory neuropathy associated with Sjogren’s syndrome. J Neurol Sci. 2009; 279: 57– 61. [DOI] [PubMed] [Google Scholar]
- 57.Oaklander AL. The density of remaining nerve endings in human skin with and without postherpetic neuralgia after shingles. Pain. 2001; 92: 139– 145. [DOI] [PubMed] [Google Scholar]
- 58.Pavlakis PP, Alexopoulos H, Kosmidis ML, Mamali I, Moutsopoulos HM, Tzioufas AG, Dalakas MC. Peripheral neuropathies in Sjogren’s syndrome: a critical update on clinical features and pathogenetic mechanisms. J Autoimmun. 2012; 39: 27– 33. [DOI] [PubMed] [Google Scholar]
- 59.Pavlakis PP, Alexopoulos H, Kosmidis ML, Stamboulis E, Routsias JG, Tzartos SJ, Tzioufas AG, Moutsopoulos HM, Dalakas MC. Peripheral neuropathies in Sjogren syndrome: a new reappraisal. J Neurol Neurosurg Psychiatry. 2011; 82: 798– 802. [DOI] [PubMed] [Google Scholar]
- 60.Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006; 29: 507– 538. [DOI] [PubMed] [Google Scholar]
- 61.Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, Del Pino-Montes J, Calvo-Alen J, Jimenez-Alonso J, Mico ML, Beltran J, Belenguer R, Pallares L, GEMESS Study Group. Primary Sjogren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore). 2008; 87: 210– 219. [DOI] [PubMed] [Google Scholar]
- 62.Rist S, Sellam J, Hachulla E, Sordet C, Puechal X, Hatron PY, Benhamou CL, Sibilia J, Mariette X, Club Rhumatismes et Inflammation. Experience of intravenous immunoglobulin therapy in neuropathy associated with primary Sjogren’s syndrome: a national multicentric retrospective study. Arthritis Care Res (Hoboken). 2011; 63: 1339– 1344. [DOI] [PubMed] [Google Scholar]
- 63.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007; 10: 1361– 1368. [DOI] [PubMed] [Google Scholar]
- 64.Segal B, Carpenter A, Walk D. Involvement of nervous system pathways in primary Sjogren’s syndrome. Rheum Dis Clin North Am. 2008; 34: 885– 906, viii. [DOI] [PubMed] [Google Scholar]
- 65.Segal BM, Pogatchnik B, Henn L, Rudser K, Sivils KM. Pain severity and neuropathic pain symptoms in primary Sjogren’s syndrome: a comparison study of seropositive and seronegative Sjogren’s syndrome. Arthritis Care Res (Hoboken). 2013; 65: 1291– 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seidel MF, Herguijuela M, Forkert R, Otten U. Nerve growth factor in rheumatic diseases. Semin Arthritis Rheum. 2010; 4: 109– 126. [DOI] [PubMed] [Google Scholar]
- 67.Sene D, Jallouli M, Lefaucheur JP, Saadoun D, Costedoat-Chalumeau N, Maisonobe T, Diemert MC, Musset L, Haroche J, Piette JC, Amoura Z, Cacoub P. Peripheral neuropathies associated with primary Sjogren syndrome: immunologic profiles of nonataxic sensory neuropathy and sensorimotor neuropathy. Medicine (Baltimore). 2011; 90: 133– 138. [DOI] [PubMed] [Google Scholar]
- 68.Skopouli FN, Dafni U, Ioannidis JP, Moutsopoulos HM. Clinical evolution, and morbidity and mortality of primary Sjogren’s syndrome. Semin Arthritis Rheum. 2000; 29: 296– 304. [DOI] [PubMed] [Google Scholar]
- 69.Skopouli FN, Fox PC, Galanopoulou V, Atkinson JC, Jaffe ES, Moutsopoulos HM. T cell subpopulations in the labial minor salivary gland histopathologic lesion of Sjogren’s syndrome. J Rheumatol. 1991; 18: 210– 214. [PubMed] [Google Scholar]
- 70.Skopouli FN, Moutsopoulos HM. Autoimmune epitheliitis: Sjogren’s syndrome. Clin Exp Rheumatol. 1994; 12: S9– S11. [PubMed] [Google Scholar]
- 71.Soldatos T, Andreisek G, Thawait GK, Guggenberger R, Williams EH, Carrino JA, Chhabra A. High-resolution 3-T MR neurography of the lumbosacral plexus. Radiographics. 2013; 33: 967– 987. [DOI] [PubMed] [Google Scholar]
- 72.Sutcliffe N, Inanc M, Speight P, Isenberg D. Predictors of lymphoma development in primary Sjogren’s syndrome. Semin Arthritis Rheum. 1998; 28: 80– 87. [DOI] [PubMed] [Google Scholar]
- 73.Terrier B, Lacroix C, Guillevin L, Hatron PY, Dhote R, Maillot F, Diot E, Sarrot-Reynauld F, Sordet C, Dubourg O, Meyer L, Mariette X, Gottenberg JE. Club Rhumatismes et Inflammation. Diagnostic and prognostic relevance of neuromuscular biopsy in primary Sjogren’s syndrome-related neuropathy. Arthritis Rheum. 2007; 57: 1520– 1529. [DOI] [PubMed] [Google Scholar]
- 74.Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS, Pillemer SR, Talal N, Weisman MH, European Study Group on Classification Criteria for Sjogren’s Syndrome. Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002; 61: 554– 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vitali C, Bombardieri S, Moutsopoulos HM, Balestrieri G, Bencivelli W, Bernstein RM, Bjerrum KB, Braga S, Coll J, de Vita S. Preliminary criteria for the classification of Sjogren’s syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum. 1993; 36: 340– 347. [DOI] [PubMed] [Google Scholar]
- 76.Voulgarelis M, Tzioufas AG. Pathogenetic mechanisms in the initiation and perpetuation of Sjogren’s syndrome. Nat Rev Rheumatol. 2010; 6: 529– 537. [DOI] [PubMed] [Google Scholar]
- 77.Wakasugi D, Kato T, Gono T, Ito E, Nodera H, Kawaguchi Y, Yamanaka H, Hara M. Extreme efficacy of intravenous immunoglobulin therapy for severe burning pain in a patient with small fiber neuropathy associated with primary Sjogren’s syndrome. Mod Rheumatol. 2009; 19: 437– 440. [DOI] [PubMed] [Google Scholar]
- 78.Watson CP, Deck JH, Morshead C, Van der Kooy D, Evans RJ. Post-herpetic neuralgia: further post-mortem studies of cases with and without pain. Pain. 1991; 44: 105– 117. [DOI] [PubMed] [Google Scholar]
- 79.Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001; 429: 23– 37. [DOI] [PubMed] [Google Scholar]