

Abstract
Familial Mediterranean fever (FMF) is an autoinflammatory disease caused by MEditerranean FeVer gene (MEFV) mutations. In Japan, patients with FMF have been previously reported, including a mild or incomplete form. Several factors are presumed to contribute to the variable penetrance and to the phenotypic variability of FMF. We conducted the current study to investigate the correlation of variable clinical presentations and MEFV genotypic distributions in Japanese FMF patients.
We analyzed demographic, clinical, and genetic data for 311 FMF patients enrolled in the study. Clinically, we classified FMF into 2 phenotypes: 1) the “typical” form of FMF, and 2) the “atypical” form of FMF according to the Tel Hashomer criteria. Patients with the typical FMF phenotype had a higher frequency of febrile episodes, a shorter duration of febrile attacks, more frequent thoracic pain, abdominal pain, a family history of FMF, and MEFV exon 10 mutations. Conversely, patients with the atypical FMF phenotype had a lower frequency of fever episodes and more frequent arthritis in atypical distribution, myalgia, and MEFV exon 3 mutations. Multivariate analysis showed that the variable associated with typical FMF presentation was the presence of MEFV exon 10 mutations. Typical FMF phenotype frequencies were decreased in patients carrying 2 or a single low-penetrance mutations compared with those carrying 2 or a single high-penetrance mutations (M694I), with an opposite trend for the atypical FMF phenotype. In addition, patients having more than 2 MEFV mutations had a younger disease onset and a higher prevalence of thoracic pain than those carrying a single or no mutations. Thus, MEFV exon 10 mutations are associated with the more typical FMF phenotype. In contrast, more than half of the Japanese FMF patients without MEFV exon 10 mutations presented with an atypical FMF phenotype, indicating that Japanese FMF patients tend to be divided into 2 phenotypes by a variation of MEFV mutations.
INTRODUCTION
Familial Mediterranean fever (FMF) is an autosomal recessive autoinflammatory disease caused by MEditerranean FeVer gene (MEFV) mutations on chromosome 16,1,7 and characterized by periodic fever and serositis.24 The disease occurs most commonly in populations of eastern Mediterranean descent. However, patients of various ethnic origins are well documented, including several Japanese individuals.29,33 FMF can be classified as “typical” and “atypical” types based on clinical findings and genetic screening.13,20 As indicated in the Tel Hashomer criteria,13 a typical FMF attack is defined as episodes lasting 12 hours to 3 days with fever accompanied by peritonitis, pleuritis, or monoarthritis of hip, knee, or ankle. In contrast, an incomplete attack differs from the definition of a typical attack in the following features: temperature of less than 38°C; attack duration longer or shorter than specified periods (12 hours to 3 days), but not shorter than 6 hours or longer than a week; localized abdominal signs; or atypical distribution of arthritis.13 In addition, Ryan et al20 reported patients with symptoms of the syndrome of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis in atypical FMF. Although MEFV genotyping has enabled FMF to be confirmed in some cases, the diagnosis remains predominantly clinical since genotyping has shown that the disease is characterized by variable manifestations.19,28
To date, over 200 disease-associated mutations have been identified in MEFV, with the majority of mutations being missense changes and more than half clustering in exons 2 and 10.31 The presence of mutations on both MEFV alleles establishes a diagnosis of FMF, but in many cases only a single mutation or no mutation can be identified,12,14 resulting in a diagnostic dilemma. Additionally, some patients carrying MEFV mutations either as heterozygotes, compound heterozygotes, or complex alleles present with various atypical FMF clinical manifestations.17 In contrast to typical FMF cases in endemic areas, some FMF patients lack a classic clinical presentation, and a diagnosis is therefore difficult in populations where the disease is rare.3 Another problem is that the penetrance of a disease-causing mutation varies by MEFV mutations. Manifestation of FMF symptoms is mainly attributed to the high frequency of low-penetrance mutations (E148Q, V726A, etc) and the low frequency of high-penetrance mutations (M694V, M694I, etc).8 The low-penetrance pyrin mutation E148Q is found frequently in east Asia. The V726A mutation remains practically totally silent and nonpenetrant in Ashkenazi Jews.9
Although our knowledge of FMF is expanding rapidly,22 it remains very limited in Japan because of the rarity and phenotypic variability of Japanese FMF.16 To avoid the delayed disease diagnosis that might be associated with this, we conducted a nationwide survey and laboratory testing for the genetic diagnosis of FMF patients. We aimed to provide clear and comprehensive demographic data regarding Japanese patients with typical/incomplete FMF, and to analyze the impact of genetic factors on the disease phenotype in a large population of Japanese FMF patients.
METHODS
Patient Enrollment
A nationwide survey of FMF was conducted in cooperation with the Japan Research Committee on the Epidemiology of Intractable Diseases in 2009.16 Patients were diagnosed clinically according to the Tel Hashomer diagnostic criteria.13 Epidemiologic data (including sex, consanguinity of parents, familial history, and age of onset of inflammation signs) and main clinical data (including fever; thoracic, abdominal, articular, and cutaneous signs; duration and frequency of episodes; presence of amyloidosis; and response to colchicine) were recorded by the doctor in charge using a standard form. A diagnosis of FMF was made if the patient had 1 or more major criteria, or 2 or more minor criteria of the Tel Hashomer criteria.13 On the basis of the Tel Hashomer criteria, we divided the study subjects into 2 groups, typical FMF and atypical FMF. Typical FMF patients had the typical episode of peritonitis, pleuritis, monoarthritis, or fever alone as specified in the Tel Hashomer criteria. Atypical FMF patients had an “incomplete” attack. An attack was considered incomplete if it differed from the definition of a typical attack in only 1 or 2 of the following features: temperature less than 38°C; attack duration longer or shorter than specified periods (12 hours to 3 days), but not shorter than 6 hours or longer than a week; no signs of peritonitis during an abdominal attack, or signs were localized; atypical distribution of arthritis. The present study was approved by the ethical committees of Nagasaki Medical Center (No. 21015, 2009).
Mutational Analysis
Blood samples (2 mL) were collected from all subjects. Genomic DNA was extracted from whole blood by means of the Promega Wizard Genomic DNA Purification Kit (Promega, Madison, WI). Mutation analysis was performed by direct DNA sequencing. Polymerase chain reaction (PCR) was performed using the forward and reverse primers for each exon of the MEFV gene as described previously.27 PCR products were purified with the ExoSAP-IT (GE Healthcare Japan, Tokyo, Japan) and sequenced directly, using specific primers and BigDye Terminator v1.1 (Applied Biosystems, Tokyo, Japan). MEFV genetic analysis was approved by the Ethics Committee of Nagasaki Medical Center (No. 21003, 2009).
Statistical Analyses
We used SPSS software (SPSS Inc, Chicago, IL) to analyze the data. Results were expressed as the mean ± standard deviation for continuous variables. For quantitative data, analysis was performed using a Mann–Whitney U rank-sum test to compare 2 independent groups. Comparisons for categorical variables were evaluated using the chi-square test (or the Fisher exact test when appropriate).
We used logistic regression models to detect variables that affect disease phenotype. All possible variables were initially evaluated by univariate analysis, then suitable candidates for multivariate analysis were chosen that had a p value < 0.1 plus additional variables of known biological importance. The forward likelihood logistic regression approach was used to define variables that may affect the FMF phenotype. A p value < 0.05 was accepted as significant.
RESULTS
Demographic Features
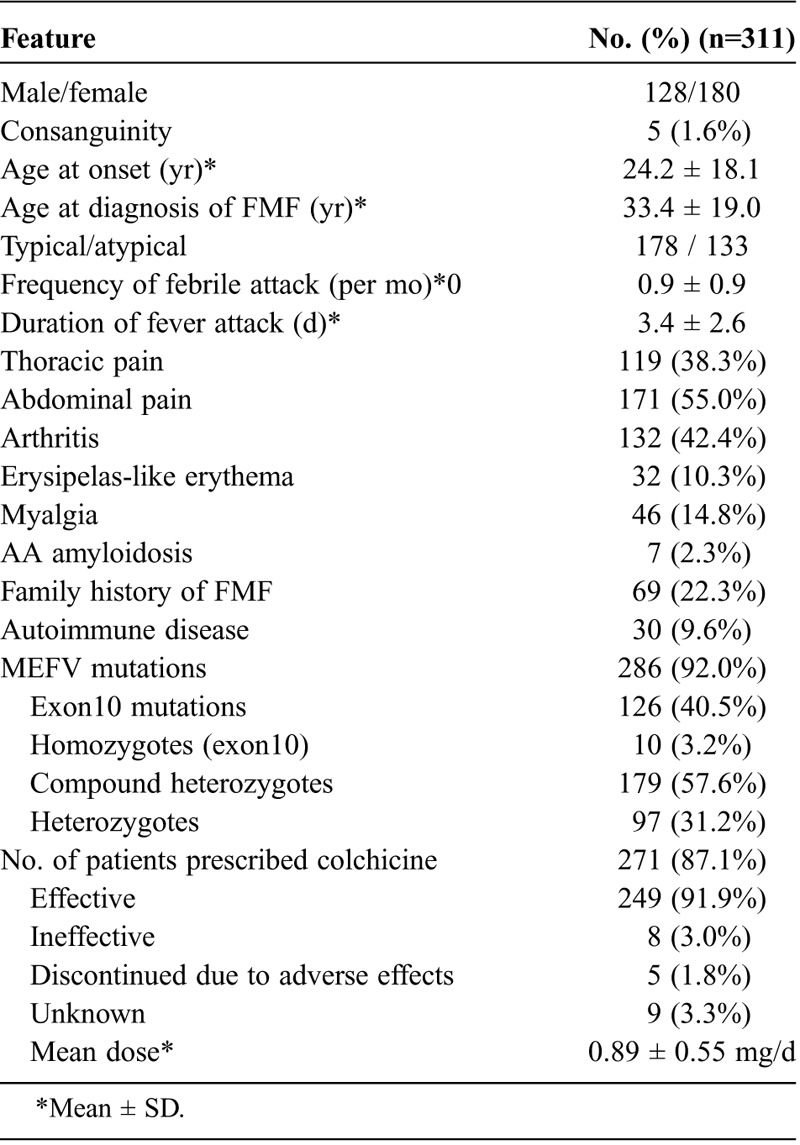
A total of 311 patients were enrolled in this study: 185 patients lived in East Japan and 126 patients lived in West Japan. Consanguinity was present in 1.6% of patients (Table 1). At the time of diagnosis, the mean age was 33.4 ± 19.0 years, and the mean time between disease onset and disease diagnosis was 9.0 ± 9.8 years. Demographic data showed that 242 patients (77.8%) had no family history suggestive of FMF. The main clinical findings were present at the following frequencies: abdominal pain (171, 55.0%), arthritis (132, 42.4%), thoracic pain (119, 38.3%), myalgia (46, 14.8%), erysipelas-like erythema (32, 10.3%), and AA amyloidosis (7, 2.3%). The association with autoimmune diseases was observed in 30 (9.6%) patients (rheumatoid arthritis 8; Adult-onset Still disease 4; Behçet disease 4; dermatomyositis 3; systemic lupus erythematosus 2; Basedow disease 1; idiopathic thrombocytopenic purpura 1; Kawasaki disease 1; Kikuchi disease 1; multiple sclerosis 1; polymyalgia rheumatica 1; Sjögren syndrome 2; ulcerative colitis 1). Colchicine was administered orally to 271 patients (87.1%), and a favorable therapeutic effect was seen in 249 patients (91.1%). Forty patients (12.9%) had not yet been treated with colchicine or information was not obtained in the survey. The mean dose of colchicine was 0.89 ± 0.55 mg/d. Our study population contained 59 pediatric patients (aged ≤16 yr) with FMF. The frequencies of the set of criteria for the diagnosis of FMF in childhood34 were as follows; fever (57/59, 96.6%), abdominal pain (38/59, 64.4%), chest pain (19/59, 32.2%), arthritis (19/59, 32.2%), and family history of FMF (13/59, 22.0%).
TABLE 1.
Demographic and Clinical Features of 311 Patients With FMF
Mutational Analysis
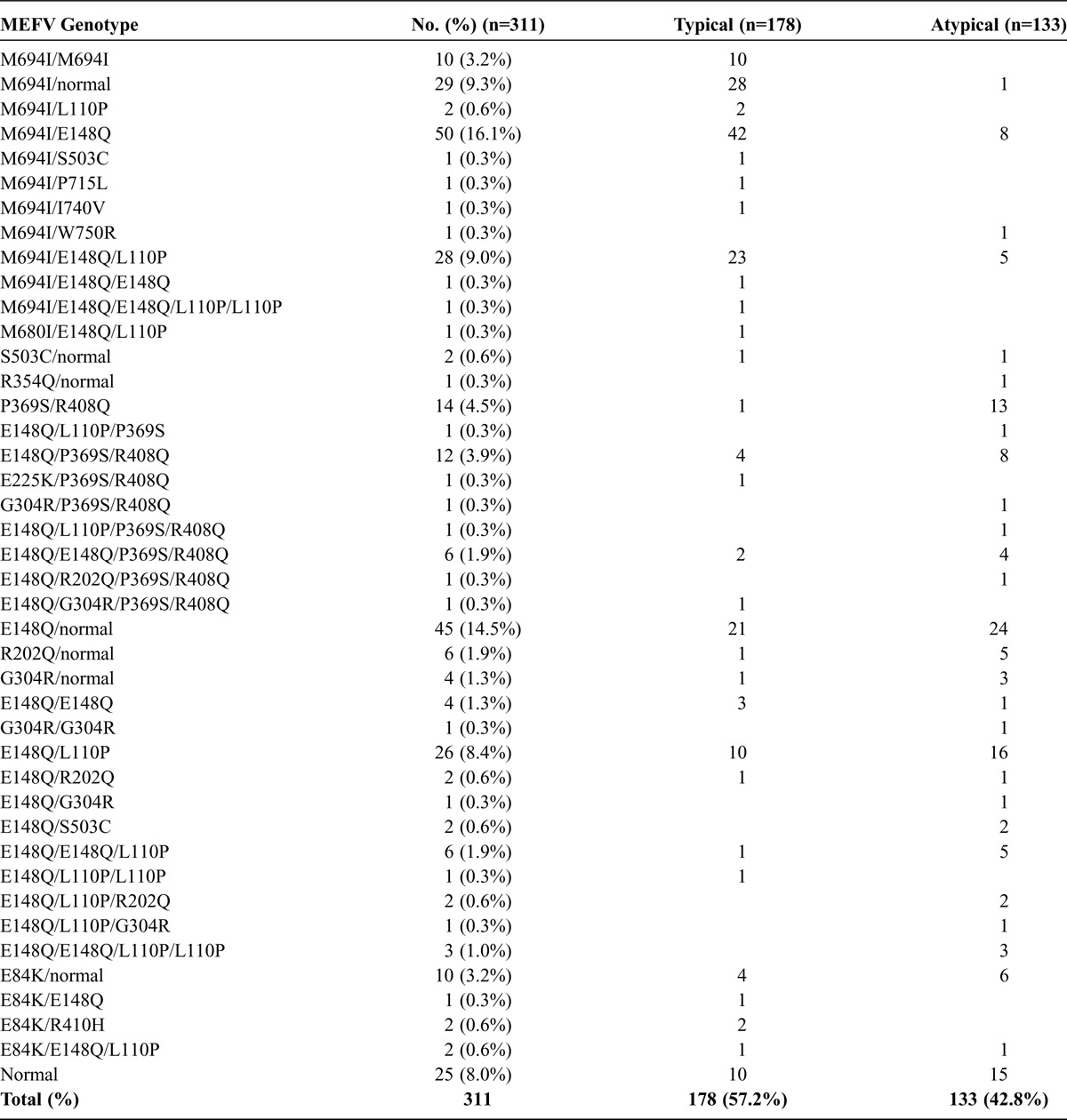
Of the total 311 FMF patients, 178 displayed a typical FMF phenotype, and 133 an atypical FMF phenotype according to the Tel Hashomer criteria.13 The distribution of the MEFV genotypes in patients with typical and atypical FMF is presented in Table 2. The detected mutations were heterogeneous and included M694I, E148Q, L110P, P369S, R408Q, and E84K. The allele frequencies of M6941, R408Q, P369S, G304R, R202Q, E148Q, and E84K were 21.7%, 5.9%, 6.1%. 1.6%, 1.8%. 35.4%, and 2.4%, respectively, in FMF patients, compared to those in healthy Japanese subjects of 0%, 3.3%, 4.0%. 2.7%, 3.3%. 23.3%, and 1.3%, respectively, as described previously.15 The allele frequencies were similar between the 2 groups except the M694I mutation.
TABLE 2.
MEFV Genotypes in FMF Patients With Typical or Atypical Phenotype
Clinical Features of Different FMF Phenotypes (Typical vs. Atypical)
The distribution of febrile attack durations is shown in Figure 1. Patients with atypical FMF had variable durations, compared to the limited durations of patients with typical FMF.
FIGURE 1.
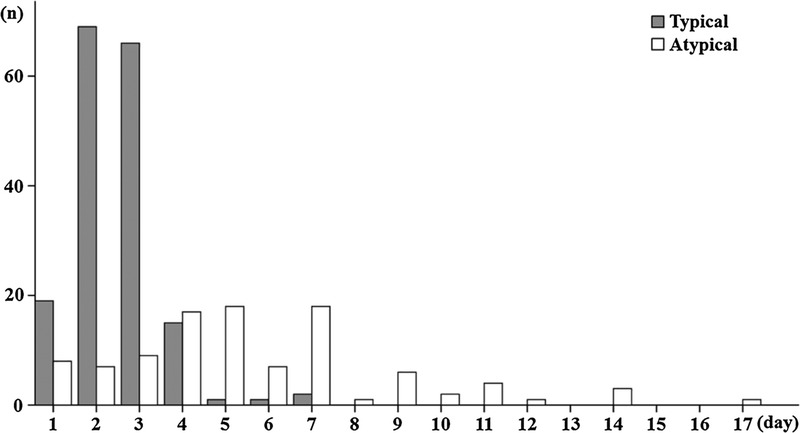
Distribution of febrile attack periods in patients with typical or atypical FMF phenotypes.
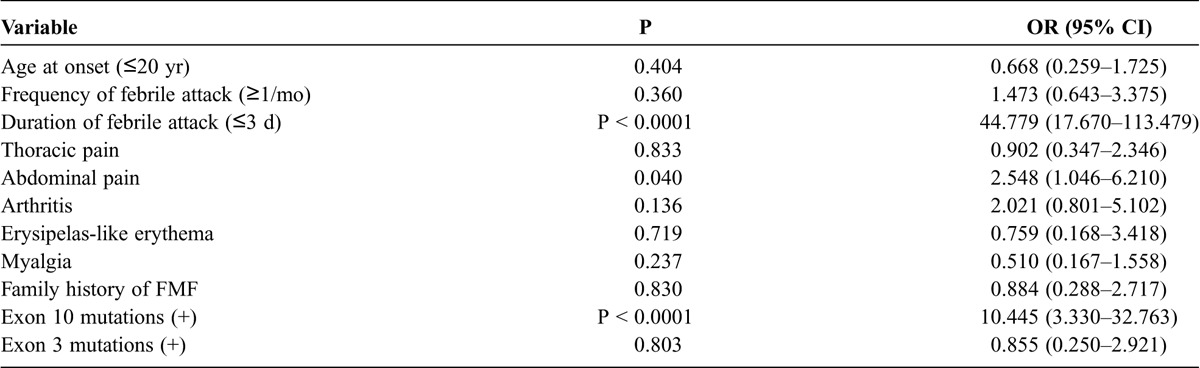
We compared the demographic and genetic data between patients with the different phenotypes (typical vs. atypical). Variables that were significantly different between the 2 groups in the univariate analysis are shown in Table 3. These factors were assessed by multivariate analysis to determine whether they independently affected the typical FMF phenotype. Our best fit model was obtained through logistic regression; we found that the presence of durations of febrile attacks ≤3 d (p < 0.0001; odds ratio [OR], 44.8; 95% confidence interval [CI], 17.7–113.5) and MEFV exon 10 mutations (p < 0.0001; OR, 10.4; 95% CI, 3.3–32.8) predicted the typical FMF phenotype (Table 4).
TABLE 3.
Comparison of Clinical Features in Patients With Typical or Atypical FMF Phenotype (Univariate Analysis)
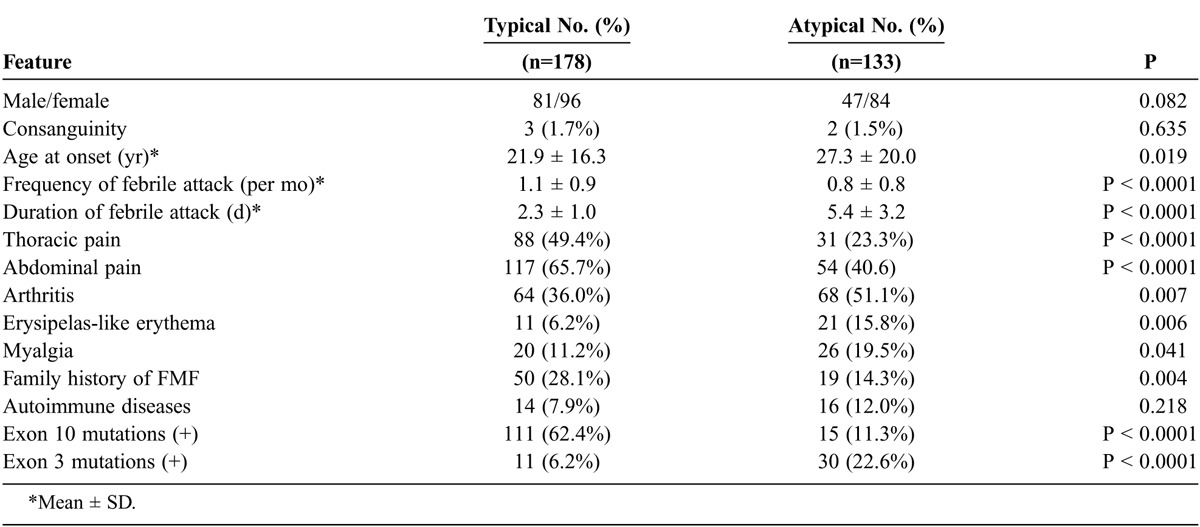
TABLE 4.
Comparison of Selected Variables for Typical FMF Phenotype (Multivariate Analysis)
Genotype/Phenotype Correlations
We analyzed the correlation between the phenotype (typical vs. atypical) and MEFV genotype using registered FMF patients. Japanese patients had a higher prevalence of the high-penetrance mutation M694I, and low-penetrance mutations in MEFV exons 1, 2, and 3 (E84K, L110P, E148Q, R202Q, G304R, P369S, R408Q). Based on these considerations, we further investigated the patient’s phenotype according to the presence of higher and/or lower penetrance of MEFV mutations. Most patients carrying 1 or 2 high-penetrance mutations had the typical FMF phenotype, while, by contrast, the typical FMF phenotype percentage was slightly decreased in patients carrying combined high- and low-penetrance mutations. In contrast to the patients carrying high-penetrance mutations, more than half of the patients carrying 1 or 2 low-penetrance mutations and no detectable mutation presented with the atypical FMF phenotype (Figure 2).
FIGURE 2.
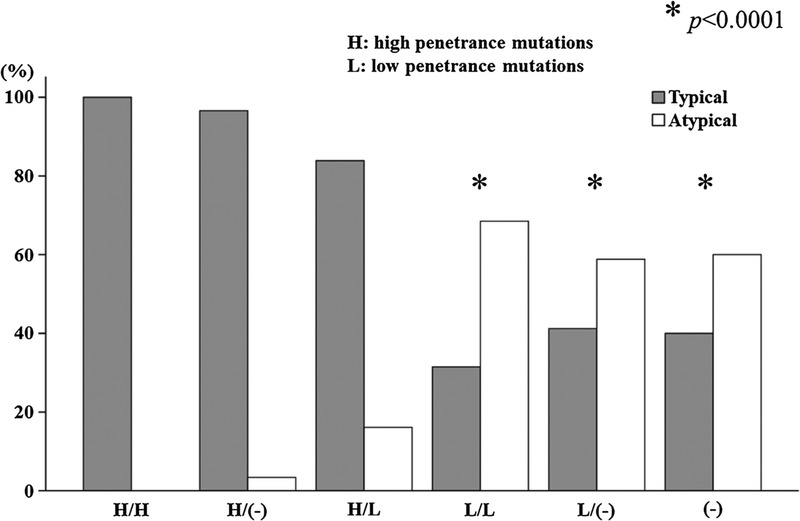
Prevalence of typical or atypical FMF phenotype in patients with high- (H) or low- (L) penetrance MEFV mutations. P values were assessed by Fisher exact test in comparison with patients having 2 high-penetrance (H/H) MEFV mutations.
Influence of the Number of MEFV Mutations on Clinical Phenotype
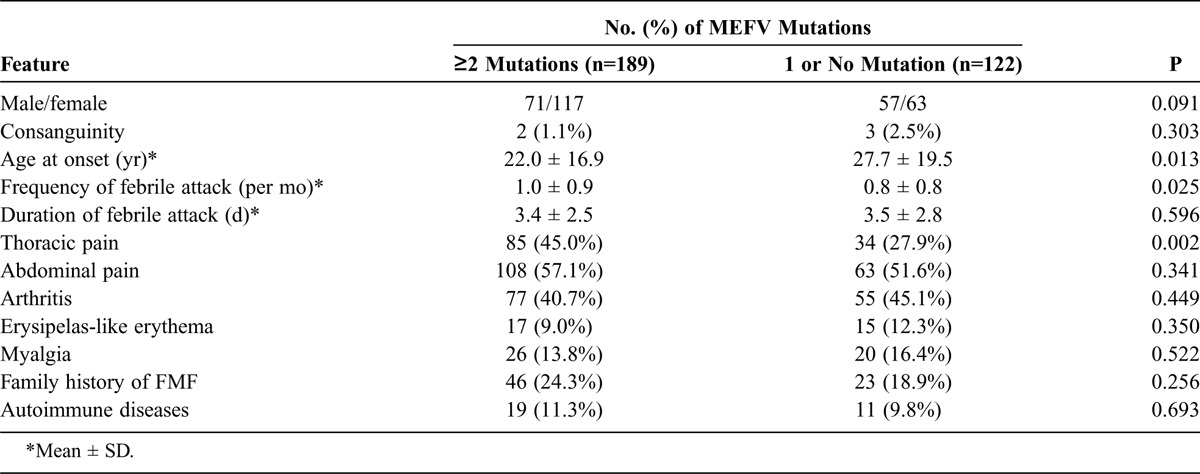
Although FMF is classically considered to be an autosomal recessive disease, the presence of a single mutation is often associated with a classic FMF phenotype, including the response to colchicine. We therefore analyzed the difference in clinical presentation between patients carrying 2 MEFV mutations and those with a single or no mutation. One hundred eighty-nine (60.8%) of 311 patients carried 2 or more than 2 MEFV mutations, 122 (39.2%) carried 1 or no detectable MEFV mutation. Most clinical manifestations did not differ between these 2 groups, except for the age of disease onset, the frequencies of febrile attack, and the presence of thoracic pain (Table 5).
TABLE 5.
Clinical Features of Patients With Different Numbers of MEFV Mutations
DISCUSSION
Since the identification of MEFV as the gene mutated in FMF,1,7 genetic analysis has become useful for confirming the diagnosis made traditionally by clinical findings.2,13 Analysis of FMF patients in various countries has revealed patients with homozygous and compound heterozygous mutations, others with single mutations, and some with none of the studied mutations.17,30 Indeed, we encountered Japanese FMF patients with only a single mutation or low-penetrance MEFV exon 2 or 3 mutations.16 This is especially common among populations with a high frequency of low-penetrance mutation carriers.30
In the current study, 43% of Japanese patients with FMF were classified as having atypical FMF according to the Tel Hashomer diagnostic criteria.13 These patients exhibited unique clinical manifestations, including some that are atypical FMF symptoms such as prolonged periods of febrile attacks and lower frequencies of serositis. Our data confirmed the correlation between the MEFV exon 10 mutations and the typical FMF phenotype, as well as that between MEFV exon 3 mutations and the atypical FMF phenotype in Japanese patients. Based on genetic analysis, the prevalence of the typical FMF phenotype was correlated with MEFV genotype, increasing from patients who carried no MEFV mutations or low-penetrance MEFV mutations to those who carried high-penetrance MEFV mutations. Multivariate analysis revealed that MEFV exon 10 mutations were significantly associated with the typical FMF phenotype in Japanese patients. This contrasts with a previous study in which only the M694V variant was associated with FMF disease severity.23
The main limitation of the current study is the lack of evaluation of disease severity; however, the MEFV genotype could be linked to the FMF clinical phenotype of Japanese patients as demonstrated in Middle Eastern countries.21 Although the dose effects of MEFV mutations to disease phenotype were minimal, the presence of a high-penetrance MEFV mutation (M694I) extensively affected the FMF phenotype. Our findings suggest that MEFV genotypes are useful to distinguish atypical from typical FMF patients. However, genetic testing has a limited diagnostic value, and the diagnosis of FMF remains clinical despite the understanding of the genotype-phenotype correlation. Nevertheless, our data shed light on the contribution of MEFV genotypes to the “FMF phenotype” in Japanese patients with FMF.
On the basis of the recessive mode of inheritance, FMF patients should inherit 2 mutations in the MEFV gene. However, several investigators have assumed that some heterozygous mutation carriers have clinically symptomatic FMF.4,5,18 Indeed, new studies have cast considerable doubt on whether FMF is a traditional autosomal recessive disease.26 Several possibilities including the presence of mutations in another autoinflammatory gene, epigenetic or post-translational modifications, and environmental factors have been suggested.5,17,32 Additionally, patients carrying MEFV mutations with low penetrance do not present with classical FMF and suffer from mild or atypical FMF characterized by episodic arthritis without fever, or febrile attacks without serositis.3,20,25 We suggest that appropriate molecular tests for MEFV could be useful for classifying the FMF phenotype in Japanese patients. Recent data from a pyrin knock-in mouse suggest that FMF is the result of gain-of-function mutations in pyrin that lead to interleukin-1β activation.11 This may account for FMF patients with only 1 identifiable MEFV mutation who present with FMF disease that is responsive to colchicine. This has been observed in patients with low-penetrance mutations in MEFV exons 1–3.
There has been some debate about the use of clinical versus genetic criteria, as the diagnosis of FMF cannot be made on the basis of genetic testing alone.6 Some patients with FMF diagnosed by clinical findings have only 1 demonstrable mutation, and a small number of patients have no identifiable mutations. In addition, the Tel Hashomer clinical criteria do not clearly distinguish typical from atypical FMF.10 Our results suggest that combined clinical investigations and molecular analysis are useful to discriminate the different phenotypes of FMF.
In summary, we confirmed that MEFV exon 10 mutations are associated with the more typical FMF phenotype. Conversely, more than half of the Japanese FMF patients without MEFV exon 10 mutations presented with an atypical FMF phenotype. Further studies involving a multicentric FMF registration are needed to establish a correlation between the MEFV genotype and various clinical phenotypes in different ethnic groups, which should be further explored in larger studies.
ACKNOWLEDGMENTS
The authors acknowledge the effective and dedicated participation of the following contributors: Munetoshi Nakashima, MD, and Akitomo Okada, MD (The Japanese Red Cross Nagasaki Hospital); Masataka Umeda, MD (Sasebo Chuo Hospital); Keita Fujikawa, MD (Isahaya Health Insurance General Hospital); Naoki Matsuoka, MD (Nagasaki Medical Hospital of Rheumatology).
Footnotes
Financial support and conflicts of interest: This work was supported by a Grant-in-Aid for Research on intractable diseases from Ministry of Health, Labour and Welfare of Japan. The authors have no other funding or conflicts of interest to disclose.
Abbreviations: CI = confidence interval, FMF = familial Mediterranean fever, IL-1β = interleukin 1 beta, OR = odds ratio.
REFERENCES
- 1.Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell. 1997; 90: 797– 807. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Chetrit E, Levy M. Familial Mediterranean fever. Lancet. 1998; 351: 659– 664. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Chetrit E, Peleg H, Aamar S, Heyman SN. The spectrum of MEFV clinical presentations—is it familial Mediterranean fever only? Rheumatology (Oxford). 2009; 48: 1455– 1459. [DOI] [PubMed] [Google Scholar]
- 4.Booth DR, Gillmore JD, Lachmann HJ, Booth SE, Bybee A, Soyturk M, Akar S, Pepys MB, Tunca M, Hawkins PN. The genetic basis of autosomal dominant familial Mediterranean fever. QJM. 2000; 93: 217– 221. [DOI] [PubMed] [Google Scholar]
- 5.Booty MG, Chae JJ, Masters SL, Remmers EF, Barham B, Le JM, Barron KS, Holland SM, Kastner DL, Aksentijevich I. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum. 2009; 60: 1851– 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Federici L, Rittore-Domingo C, Kone-Paut I, Jorgensen C, Rodiere M, Le Quellec A, Touitou I. A decision tree for genetic diagnosis of hereditary periodic fever in unselected patients. Ann Rheum Dis. 2006; 65: 1427– 1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997; 17: 25– 31. [DOI] [PubMed] [Google Scholar]
- 8.Gershoni-Baruch R, Brik R, Shinawi M, Livneh A. The differential contribution of MEFV mutant alleles to the clinical profile of familial Mediterranean fever. Eur J Hum Genet. 2002; 10: 145– 149. [DOI] [PubMed] [Google Scholar]
- 9.Gershoni-Baruch R, Shinawi M, Leah K, Badarnah K, Brik R. Familial Mediterranean fever: prevalence, penetrance and genetic drift. Eur J Hum Genet. 2001; 9: 634– 637. [DOI] [PubMed] [Google Scholar]
- 10.Grateau G, Pecheux C, Cazeneuve C, Cattan D, Dervichian M, Goossens M, Delpech M, Amselem S, Dode C. Clinical versus genetic diagnosis of familial Mediterranean fever. QJM. 2000; 93: 223– 229. [DOI] [PubMed] [Google Scholar]
- 11.Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, Katz SI, Kastner DL. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity. 2011; 34: 755– 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kone-Paut I, Hentgen V, Guillaume-Czitrom S, Compeyrot-Lacassagne S, Tran TA, Touitou I. The clinical spectrum of 94 patients carrying a single mutated MEFV allele. Rheumatology (Oxford). 2009; 48: 840– 842. [DOI] [PubMed] [Google Scholar]
- 13.Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, Migdal A, Padeh S, Pras M. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997; 40: 1879– 1885. [DOI] [PubMed] [Google Scholar]
- 14.Marek-Yagel D, Berkun Y, Padeh S, Abu A, Reznik-Wolf H, Livneh A, Pras M, Pras E. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum. 2009; 60: 1862– 1866. [DOI] [PubMed] [Google Scholar]
- 15.Migita K, Ida H, Moriuchi H, Agematsu K. Clinical relevance of MEFV gene mutations in Japanese patients with unexplained fever. J Rheumatol. 2012; 39: 875– 877. [DOI] [PubMed] [Google Scholar]
- 16.Migita K, Uehara R, Nakamura Y, Yasunami M, Tsuchiya-Suzuki A, Yazaki M, Nakamura A, Masumoto J, Yachie A, Furukawa H, Ishibashi H, Ida H, Yamazaki K, Kawakami A, Agematsu K. Familial Mediterranean fever in Japan. Medicine (Baltimore). 2012; 91: 337– 343. [DOI] [PubMed] [Google Scholar]
- 17.Moradian MM, Sarkisian T, Ajrapetyan H, Avanesian N. Genotype-phenotype studies in a large cohort of Armenian patients with familial Mediterranean fever suggest clinical disease with heterozygous MEFV mutations. J Hum Genet. 2010; 55: 389– 393. [DOI] [PubMed] [Google Scholar]
- 18.Ozen S. Changing concepts in familial Mediterranean fever: is it possible to have an autosomal-recessive disease with only one mutation? Arthritis Rheum. 2009; 60: 1575– 1577. [DOI] [PubMed] [Google Scholar]
- 19.Padeh S, Shinar Y, Pras E, Zemer D, Langevitz P, Pras M, Livneh A. Clinical and diagnostic value of genetic testing in 216 Israeli children with Familial Mediterranean fever. J Rheumatol. 2003; 30: 185– 190. [PubMed] [Google Scholar]
- 20.Ryan JG, Masters SL, Booty MG, Habal N, Alexander JD, Barham BK, Remmers EF, Barron KS, Kastner DL, Aksentijevich I. Clinical features and functional significance of the P369S/R408Q variant in pyrin, the familial Mediterranean fever protein. Ann Rheum Dis. 2010; 69: 1383– 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samuels J, Aksentijevich I, Torosyan Y, Centola M, Deng Z, Sood R, Kastner DL. Familial Mediterranean fever at the millennium. Clinical spectrum, ancient mutations, and a survey of 100 American referrals to the National Institutes of Health. Medicine (Baltimore). 1998; 77: 268– 297. [DOI] [PubMed] [Google Scholar]
- 22.Savic S, Dickie LJ, Battellino M, McDermott MF. Familial Mediterranean fever and related periodic fever syndromes/autoinflammatory diseases. Curr Opin Rheumatol. 2012; 24: 103– 112. [DOI] [PubMed] [Google Scholar]
- 23.Shinar Y, Livneh A, Langevitz P, Zaks N, Aksentijevich I, Koziol DE, Kastner DL, Pras M, Pras E. Genotype-phenotype assessment of common genotypes among patients with familial Mediterranean fever. J Rheumatol. 2000; 27: 1703– 1707. [PubMed] [Google Scholar]
- 24.Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. 1967; 43: 227– 253. [DOI] [PubMed] [Google Scholar]
- 25.Soriano A, Manna R. Familial Mediterranean fever: new phenotypes. Autoimmun Rev. 2012; 12: 31– 37. [DOI] [PubMed] [Google Scholar]
- 26.Stoffels M, Szperl A, Simon A, Netea MG, Plantinga TS, van Deuren M, Kamphuis S, Lachmann HJ, Cuppen E, Kloosterman WP, Frenkel J, van Diemen CC, Wijmenga C, van Gijn M, van der Meer JW. MEFV mutations affecting pyrin amino acid 577 cause autosomal dominant autoinflammatory disease. Ann Rheum Dis. 2014; 73: 455– 461. Epub 2013 Mar 16. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka M, Migita K, Miyashita T, Maeda Y, Nakamura M, Komori A, Ishibashi H, Eguchi K, Kikuchi M, Hirayama K, Yasunami M. Coexistence of familial Mediterranean fever and Sjogren’s syndrome in a Japanese patient. Clin Exp Rheumatol. 2007; 25: 792. [PubMed] [Google Scholar]
- 28.Tchernitchko D, Moutereau S, Legendre M, Delahaye A, Cazeneuve C, Lacombe C, Grateau G, Amselem S. MEFV analysis is of particularly weak diagnostic value for recurrent fevers in Western European Caucasian patients. Arthritis Rheum. 2005; 52: 3603– 3605. [DOI] [PubMed] [Google Scholar]
- 29.Tomiyama N, Higashiuesato Y, Oda T, Baba E, Harada M, Azuma M, Yamashita T, Uehara K, Miyazato A, Hatta K, Ohya Y, Iseki K, Jinno Y, Takishita S. MEFV mutation analysis of familial Mediterranean fever in Japan. Clin Exp Rheumatol. 2008; 26: 13– 17. [PubMed] [Google Scholar]
- 30.Touitou I. The spectrum of familial Mediterranean fever (FMF) mutations. Eur J Hum Genet. 2001; 9: 473– 483. [DOI] [PubMed] [Google Scholar]
- 31.Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, Shoham N, Aganna E, Hugot JP, Wise C, Waterham H, Pugnere D, Demaille J, Sarrauste de Menthiere C. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004; 24: 194– 198. [DOI] [PubMed] [Google Scholar]
- 32.Touitou I, Picot MC, Domingo C, Notarnicola C, Cattan D, Demaille J, Kone-Paut I. The MICA region determines the first modifier locus in familial Mediterranean fever. Arthritis Rheum. 2001; 44: 163– 169. [DOI] [PubMed] [Google Scholar]
- 33.Tsuchiya-Suzuki A, Yazaki M, Nakamura A, Yamazaki K, Agematsu K, Matsuda M, Ikeda S. Clinical and genetic features of familial Mediterranean fever in Japan. J Rheumatol. 2009; 36: 1671– 1676. [DOI] [PubMed] [Google Scholar]
- 34.Yalcinkaya F, Ozen S, Ozcakar ZB, Aktay N, Cakar N, Duzova A, Kasapcopur O, Elhan AH, Doganay B, Ekim M, Kara N, Uncu N, Bakkaloglu A. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford). 2009; 48: 395– 398. [DOI] [PubMed] [Google Scholar]