

Abstract
Background
The efficacy of lapatinib is limited by the development of acquired resistance. The aim of this study was to investigate the role of estrogen receptor (ER) signaling compensatory activation in acquired resistance to lapatinib in breast cancer cells BT474 and the related mechanism.
Methods
Acquired resistant cell model resistant (r)BT474 was generated with an increasing concentration of lapatinib. Real-time polymerase chain reaction and Western blotting were used to determine the changes of human epidermal growth factor receptor (HER)2 and ER pathways in breast cancer cell BT474 after treatment with lapatinib and the distinction between BT474 and rBT474. Methyl thiazolyl tetrazolium and colony formation assays were employed to detect the proliferation of rBT474 and BT474 cells treated with lapatinib and/or an ER inhibitor, fulvestrant, respectively.
Results
Lapatinib could inhibit phosphorylation of HER2 and induce expression of forkhead-box protein O3a and progesterone receptor. Acquired resistant cell model rBT474 could grow in the presence of 5 μM lapatinib, with an apoptosis rate of only 5%. Significant inhibition of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (AKT) pathway and the activation of the mitogen-activated protein kinases (MAPK) and ER pathways were detected in rBT474, compared with BT474. Furthermore, the expressions of Src phosphorylation and caveolin-1 were also upregulated. The viability of rBT474 was markedly suppressed by the lapatinib/fulvestrant combination in vitro, confirmed by the BT474 xenograft model.
Conclusion
ER signaling compensatory activation may partly contribute to lapatinib acquired resistance in HER2-overexpressing/ERα-positive breast cancer cells, which might be related to PI3K/AKT inhibition and MAPK pathway activation.
Keywords: Acquired resistance, breast cancer, ER, HER2, Lapatinib
Introduction
Lapatinib (Tykerb, GlaxoSmithKline, Middlesex, UK) is a molecular targeting drug for treating human epidermal growth factor receptor (HER)2-positive breast cancer, and is the second HER2 inhibitor to be approved by the United States Food and Drug Administration after trastuzumab (Herceptin, Roche, San Francisco, CA, USA). Lapatinib is also the first dual epidermal growth factor receptor (EGFR)/HER2 inhibitor capable of inhibiting the tyrosine kinase activity of EGFR.1 Lapatinib can cross the blood-brain barrier because of its small molecular weight and lipotropy, and can be satisfactorily absorbed by oral administration. Clinical research has reported that lapatinib can reduce the risk of brain metastases in breast cancer patients with trastuzumab resistance.2 Moreover, lapatinib has weak cardiotoxicity. However, like other molecular targeting drugs, primary and acquired resistance dramatically limit the efficiency of lapatinib. A few studies have demonstrated an effective response of only 24% when lapatinib was used alone, and a majority of treated patients developed acquired resistance within six months.3 To improve the curative efficiency of lapatinib, clarifying the mechanism of acquired resistance of lapatinib is crucial. However, only a few studies have confirmed that the survival of cells with acquired resistance not completely dependent on the HER2 signal pathway, but activation of some other compensatory pathways. Most studies report that the HER2 pathway interacts with the ER pathway; nevertheless, the significance and specific mechanism of compensatory activation of the ER pathway in lapatinib drug resistance need to be further clarified. To explore the influence of lapatinib on the ER pathway, and to distinguish the differences of the HER2/EGFR and ER signaling pathways between sensitive and resistant cell lines, we established an acquired resistance model with a clinical concentration of lapatinib in vitro. ER pathway activation was identified when there was acquired drug resistance to lapatinib, and the molecular mechanism is discussed in vitro.
Materials and methods
Cell line and reagents
Breast cancer cell line BT474 was purchased from Type Culture Collection (Chinese Academy of Sciences, Shanghai, China). Lapatinib and fulvestrant (Faslodex, Astra Zeneca, Wilmington DE, USA) were purchased from Selleck Chemicals (Houston, TX, USA). RPMI-1640 culture medium and fetal bovine serum (FBS) were purchased from Life Technologies (Grand Island, NY, USA). Anti-protein kinase B (AKT), anti-phospho-AKT (Ser-437), anti-extracellular-signal-related-kinase (ERK), anti-phospho-ERK, and goat anti-rabbit immunoglobin (Ig)G were purchased from Proteintech (Wuhan, China). Trizol was purchased from Invitrogen (Carlsbad, CA, USA). Radioimmunoprecipitation assay lysis buffer was purchased from Beyotime (Jiangsu, China); Moloney Murine Leuemia Virus Reverse Transcriptase cDNA Synthesis and SYBR Premix Ex Taq Kits were purchased from TaKaRa (Dalian, China); Methyl thiazolyl tetrazolium (MTT) was purchased from Sigma (St. Louis, MO, USA); Giemsa stain was purchased from Bolight Biological Co., Ltd (Shanghai, China); and polymerase chain reaction (PCR) primer was synthesized by Jierui Bioengineering Co., Ltd. (Shanghai, China)
Establishment of lapatinib acquired drug resistance model
BT474 cells were cultured in RPMI 1640 medium with 10% FBS. Cells were cultured in 5% CO2 at 37°C. Cells were treated with lapatinib, with an increased concentration from 0.25 to 2 μM for 12 weeks. Resistant (r)BT474 was then obtained by treatment with complete medium containing 5 μM lapatinib. rBT474 cells were cultured with RPMI 1640 medium with 10% FBS and 5 μM lapatinib in 5% CO2 at 37°C in an incubator.
Ribonucleic acid extraction and real time-polymerase chain reaction
According to Trizol instructions, total ribonucleic acid (RNA) was extracted from BT474 and rBT474 cells respectively, using oligo dT as primer and M-MLV as reverse transcriptase; cDNA was then obtained through reverse transcription. The relative expression quantity of messenger (m)RNA of each gene was detected by ABI Real-time PCR analyzer 7500 (Foster City, CA, USA) according to SYBR Premix Ex Taq Kit (Takara, Shiga, Japan) instructions. The primer sequences of HER2 were as follows: forward primer, 5′-GGA TGT GCG GCT CGT ACA C-3,′ reverse primer, 5′-TAA TTT TGA CAT GGT TGG GAC TCT T-3.′ The primer sequences of ERα were as follows: forward primer, 5′-CTG CCC TGG AGA CCA CAA AT-3,′ reverse primer, 5′-GGA CTT TAC TGC TGA TGT TGT TCT TT-3.′ The primer sequences of progesterone receptor (PR) were as follows: forward primer, 5′-TTC ACC AGG TCA AGA CAT ACA GTT G-3,′ reverse primer, 5′-TTG TCA TGT CCT GCA TAG ATC ACA-3.′ The primer sequences of forkhead-box protein (FOX)O3a were as follows: forward primer, 5′-TCT ACG AGT GGA TGG TGC GTT-3,′ reverse primer, 5′-CGA CTA TGC AGT GAC AGG TTG TG-3.′ The primer sequences of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as internal controls: forward primer 5′-GGT CGT ATT GGG CGC CTG GTC ACC-3,′ reverse primer,5′-CAC ACC CAT GAC GAA CAT GGG GGC-3.′
Western blotting assay
The cells were collected and washed by cold phosphate buffered saline (PBS) twice and RIPA lysis buffer was added to lyse the cells and collect proteins. Protein quantification was analyzed by BCA Assay (Beyotime, Jiangsu, China). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was used to separate protein samples. The samples were then transferred from gel to a nitrocellulose membrane, which was blocked in 5% fat-free milk for two hours at room temperature. The membrane was incubated with primary antibody at 4°C overnight, washed with tris-buffered saline plus tween 20 (TBST) three times, and then incubated with a related second antibody for two hours. Finally, an enhanced chemiluminescence kit (Millipore, Temecula, CA, USA) was applied to detect the immunocomplexes.
Apoptosis detection
BT474 and rBT474 cells were disposed with 5 μM lapatinib for six hours. Cells were then collected and washed twice with PBS, and 500 μl Binding Buffer was added to suspend the cells. The cells were then mixed with 5 μl Annexin V-fluorescein isothiocyanate and 5 μl propium iodide in the dark for 10 minutes at room temperature. A BD FACSCalibur Flow Cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) was used for detection. The result was analysed by Flowjo software ver. 10.0.0 (Ashland, OR, USA).
Cell proliferation examination
The logarithmic phases of BT474 and rBT474 cells were plated at a density of 5 × 103 cell/well into the 96-well plate. The cells were divided into four groups: rBT474, rBT474 + fulvestrant (10 nM), BT474, and BT474 + fulvestrant (10 nM). Each group had five duplicated wells. Both cells were treated with different concentrations of lapatinib for 48 hours. Then, 20 μl MTT (5 mg/ml) solution was added to each well and cells were incubated in the dark at 37°C for four hours. Media was discarded and 150 μl dimethyl sulfoxide (DMSO) was added to each well and then shaken for 10 minutes. After the solution completely dissolved, the absorbance at 570 nm (A570) was measured using a microplate reader. The inhibition ratio of each group was calculated using the formula: cell viability (%) = experimental group A570/control group A570 × 100%. The absorbance of the rBT474 group without the drug at 570 nm was used as the control. All results were repeated three times.
Colony formation assay
BT474 cells were divided into four groups and were plated into a six-well plate at a density of 300 cells/well. Each well was cultivated with RPMI 1640 medium containing 0.1% DMSO, 1 μM lapatinib, 10 nM fulvestrant, and 1 μM lapatinib/10 nM fulvestrant, respectively, for 21 days. The cells were then washed twice by PBS, fixed by methyl alcohol for 15 minutes, and giemsa staining was used to count the living cell clones. Cloning efficiency was determined with the formula: (%) = (copy numbers/cell population in 1% DMSO group) × 100%. All results were repeated for three times.
Studies with xenografts
The Tongji University Institutional Animal Care and Use Committee approved our animal studies. In brief, BT474 cells were injected into female athymic nude mice, which were given slow-release estrogen pellets (Innovative Research of America, Sarasota, FL, USA) subcutaneously, as previous described.4 After tumors reached 200 mm3, treatment commenced with lapatinib (100 mg/kg) by daily oral gavage and/or fulvestrant (5 mg/week) by subcutaneous injection. The short (a) and long (b) diameters of tumors were measured by digital vernier caliper every three days, tumor volume was calculated by a2b/2, and growth curve was depicted. At the end of the trial after the nude mice were sacrificed, tumors were stripped and tumor weight was recorded and analyzed.
Statistical analysis
SPSS 14.0 software (SSPS Inc., Chicago, IL, USA) was used for statistic analysis and the data were shown as mean ± standard deviation using analysis of variance. The significant difference between each mean was determined by Student’s t-test, and P < 0.05 was judged as the standard of statistical significance.
Results
The cell model of lapatinib acquired resistance
According to some clinical pharmacokinetic studies regarding lapatinib, the average maximum blood concentration of the patients (Cmax) was 2.43 μg/ml (1.57–3.77 μg/ml or 2.70–6.49 μM).5 In this study, mediums containing different concentrations of lapatinib were applied to the incubation of BT474 for 21 days (concentration of lapatinib gradually increased from 0.25 μM to 5 μM). The lapatinib acquired resistance cell, rBT474, was obtained from the survival cells in the medium with 5 μM lapatinib. The obvious reduction of BT474 was observed under microscope after 24 hours of 5 μM lapatinib treatment. However, the number of rBT474 cells had changed slightly (Fig. 1a). Flow cytometry also showed that the apoptosis rate of rBT474 treated with 5 μM lapatinib for 12 hours (11.42 ± 3.67%) was remarkably lower than BT474 (65.64 ± 2.14%) (Fig. 1b), P < 0.01, implying the success of the establishment of acquired resistance cells rBT47 (Fig. 1c).
Figure 1.

Identification of the lapatinib acquired drug resistance model rBT474. (a) Comparison of the cell number of BT474 and rBT474 under microscope after treatment with 5 μM lapatinib for 24 hours. (b) The result of flow cytometry detection of BT474 and rBT474. (c) The difference in apoptosis rate between BT474 and rBT474 (n = 3). Lap, lapatinib.  , Lap(−);
, Lap(−);  , Lap(+).
, Lap(+).
Estrogen receptor (ER)α and human epidermal growth factor (HER)2 pathways were influenced by lapatinib in BT474 cells
The ERα pathway interacted with HER2. To observe the impact of lapatinib on the ERα pathway in breast cancer cells, HER2 overexpression and ERα(+) breast cancer cell line BT474 were chosen for this study. Real-time PCR and Western blot were used to detect changes of the ERα and HER2 pathways in the sensitive strain cell BT474 after 24 hours of treatment with 1 μM lapatinib. The results implied a reduction of phosphorylation of HER2, AKT, and ERK with lapatinib. The HER2 pathway was notably inhibited by lapatinib. However, the ER pathway was activated, and the expression of mRNA and the protein level of ERα, PR, and FOXO3a increased (Fig. 2a,b). A higher concentration of lapatinib activated PR and FOXO3, increasing their expression level (P < 0.05).
Figure 2.
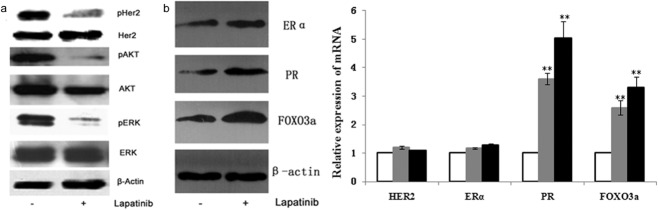
Impact of lapatinib on the estrogen receptor (ER)α and human epidermal growth factor receptor (HER)2 pathways in the sensitive strain cell BT474. (a) & (b) The of phosphorylation of HER2, protein kinase B (AKT), and extracellular-signal-related-kinase (ERK) of BT474 were reduced with 1 μM lapatinib, but the ER pathway was activated, which was manifested by the increased expression of ERα, PR, and forkhead-box (FOX)O3a at messenger ribonucleic acid (mRNA) and protein level (P < 0.05). Lap, lapatinib.  , dimethyl sulfoxide (DMSO);
, dimethyl sulfoxide (DMSO);  , Lap (0.5 μM);
, Lap (0.5 μM);  , Lap (1 μM).
, Lap (1 μM).
Difference in activation of ERα and HER2 pathways between acquired resistance cells rBT474 and BT474
Western blot was used to compare any change in the activity of the ERα and HER2 pathways between sensitive strain BT474 and acquired resistance cell rBT474. The results showed that compared to the untreated BT474, in rBT474 treated with the 5 μM lapatinib, the activation of the ER pathway was manifested by increased expression levels of ERα and PR (Fig. 3a). The inhibition of HER2 and downstream phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3k)/AKT pathways and the activation of the mitogen-activated protein kinase (MAPK)-ERK pathway lead to a decreased expression of phosphorylated HER2 and phosphorylated AKT, but an increased expression of phosphorylated ERK. (Fig. 3b) The results show that the activation of FOXO3a caused by the inhibition of AKT may promote the expression level of ERα. Moreover, some studies have determined that the inhibition of HER2 may induce vicarious activation of Src, which can affect the MAPK pathway, and the caveolin-1 controlled by MAPK can activate ERα.6–8 The detection of Src in the MAPK-ERK signal pathway upstream and caveolin-1 downstream showed that the phosphorylation of Src and caveolin-1 were increased, and as a result, the phosphorylation of Src activated the MAPK-ERK pathway, and then activated caveolin-1, which has the capacity to enhance the activation of transcription (Fig. 3c).
Figure 3.
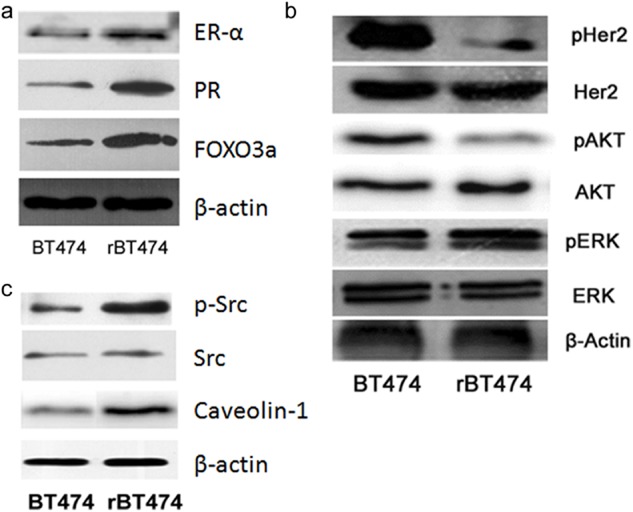
The difference in activation of estrogen receptor (ER)α and human epidermal growth factor receptor (HER)2 pathways between acquired-resistance cell rBT474 and BT474. (a) A comparison of rBT474 treated with 5 μM lapatinib and untreated BT474 cells; the expression of ERα, PR, and forkhead-box (FOX)O3a obviously increased. (b) The expression of phosphorylation of HER2 and protein kinase B (AKT) decreased in rBT474, but there was an increase in the expression of phosphorylation of extracellular-signal-related-kinase (ERK). (c) Increasing the expression of phosphorylation Src and caveolin-1 in rBT474 implied that the phosphorylation of Src activated the mitogen-activated protein kinase (MAPK)-ERK pathway, and then activated caveolin-1, which has the capacity to enhance the activation of transcription.
Inhibition of ERα pathway can reverse acquired resistance to lapatinib
Our results implied that the vicarious activation of the ERα pathway is involved in the molecular mechanism of lapatinib drug resistance. Thus, we inferred that using the ERα pathway inhibitor may recover sensitivity to lapatinib in rBT474. Fulvestrant can degrade ERα effectively with no agonist effects, which works by downregulating the estrogen receptor.9 Fulvestrant was chosen to inhibit the ERα pathway and MTT was used to detect whether rBT474 recovered sensitivity to lapatinib. Both BT474 and rBT474 were divided into two groups, one treated with 10 nM fulvestrant and the other as the control, followed by treatment with 0 μM, 0.5 μM, 1 μM, 5 μM lapatinib and detection after 48 hours. As a result, the cell viability of rBT474 treated only with lapatinib was decreased at the higher concentration (1 μM) and the cell survival rate was 80.40 ± 9.44% (Fig. 4). However, the cell survival rate of rBT474 remarkably decreased with the higher concentration of lapatinib after treatment with 10 nM fulvestrant (43.96 ± 3.79%), notably lower than the group without fulvestrant (77.84 ± 6.37%), P < 0.01. The cell viability of BT474, after treatment with only lapatinib or adding fulvestrant both saw a decrease in concentration in a dose-dependent manner. Moreover, with 5 μM lapatinib, the cell viability of BT474 treated with a combination of lapatinib and fulvestrant decreased more than the group with lapatinib alone, (14.58 ± 2.3% vs. 31.41 ± 1.7%), P < 0.05 Treatment with both lapatinib and fulvestrant had a remarkably synergistic inhibition on breast cancer cells BT474 and rBT474, and enhanced the sensitivity to lapatinib in rBT474.
Figure 4.
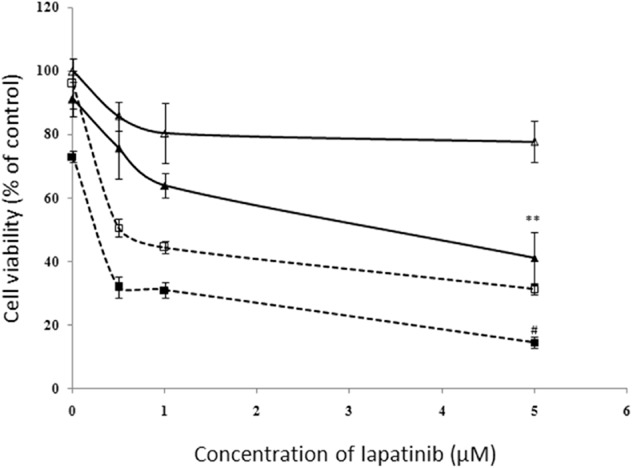
Effects of lapatinib and fulvestrant on the growth of BT474 and rBT474. The cell viability of both of BT474 and rBT474 were decreased markedly when treatment of lapatinib and fulvestrant were combined; BT474 was more sensitive to both of the drugs than rBT474. Ful, fulvestrant.  , rBT474;
, rBT474;  , rBT474+Ful;
, rBT474+Ful;  , BT474;
, BT474;  , BT474+Ful.
, BT474+Ful.
Double target treatment can prevent acquired resistance to lapatinib in BT474
The rate of cell colony formation, as well as the survival rate of cell inoculation, indicated the cloning number of survival cells after inoculation and adherence. Not all cells after adherence can proliferate and clone, while cloning cells can adhere and proliferate. The rate of cell colony formation reflected the group dependence and proliferation of cells. BT474 was divided into four groups and incubated with RPMI 1640 containing 0.1% DMSO, 1 μM lapatinib, 10 nM fulvestrant, 1 μM lapatinib/10 nM fulvestrant, respectively, for 21 days. The results revealed the cloning number of the group treated with 1 μM lapatinib and 10 nM fulvestrant was evidently lower than the other groups treated with DMSO, lapatinib, and fulvestrant, and the rates of cell cloning were 10.67 ± 3.06%, 95.67 ± 4.04%, 60.67 ± 4.73%, and 70.33 ± 5.51%, respectively. The rates of cell cloning demonstrate that treatment with both lapatinib and fulvestrant can notably inhibit cell cloning of BT474 and might prevent acquired resistance to lapatinib (Fig 5).
Figure 5.
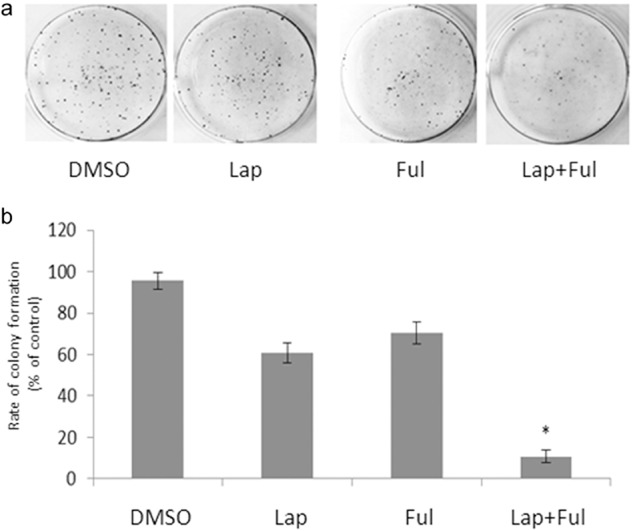
The combination of lapatinib and fulvestrant influenced the colony formation of BT474. In a comparison of the effect of 0.1% dimethyl sulfoxide (DMSO), 1 μM lapatinib, 10 nM fulvestrant and 1 μM lapatinib+ 10 nM fulvestrant on BT474 colony formation, the cloning number of 1 μM lapatinib, 10 nM fulvestrant was evidently lower than the combination of DMSO, lapatinib, and fulvestrant; rates of cell cloning were 10.67 ± 3.06%, 95.67 ± 4.04%, 60.67 ± 4.73%, and 70.33 ± 5.51%, respectively.
Lapatinib and ER inhibitor fulvestrant synergize against HER2-overexpressing xenografts
We found that the upregulation of ER activity was acquired as the cells developed resistance to lapatinib. Thus, we hypothesized that the addition of an ER inhibitor to lapatinib would prevent or delay the development of drug resistance and might further suppress tumor growth compared to lapatinib alone. To test this, mice bearing BT474 xenografts were randomized to therapy with vehicle controls (DMSO), lapatinib, fulvestrant or a combination of both drugs for 30 days. According to the growth curve of the established BT474 xenografts (Fig. 6a), lapatinib had a significant inhibition effect on tumor growth (P < 0.05) in the labatinib only group, while fulvestrant alone had little activity compared with the control mice (P > 0.05). Tumors treated with the combination method exhibited a statistical reduction in tumor volume compared with other groups starting at one week of therapy (Student’s t-test, P < 0.05, Fig. 6a). At the end of therapy, the tumor volume in the combined group was 421 ± 45 mm3, while in the vehicle group the volume was 1366 ± 115 mm3 (P < 0.05). When tumors were stripped from the nude mice, tumor weight was calculated in company with the results from the xenograft growth curve. The tumor weight in the fulvestrant group (0.22 ± 0.04 g) was a little lighter than the vehicle (0.29 ± 0.07 g), but with no significant difference (P > 0.05), while in the lapatinib group, tumor weight (0.16 ± 0.13 g) was more significantly reduced than the vehicle group, and the inhibition rate was 45.8%. Further, in the combination group, tumor weight was down to 0.10 ± 0.04 g and the inhibition rate was up to 66.4%, (Fig. 6b). This data indicated that the combination method had a superior effect on tumor growth compared with single drug treatment.
Figure 6.
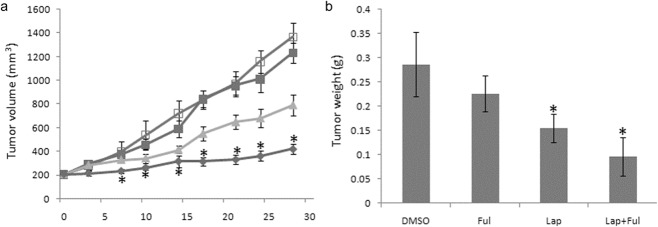
Lapatinib and the estrogen receptor (ER) inhibitor fulvestran synergize against human epidermal growth factor receptor (HER)2-overexpressing xenografts. BT474 xenografts were established in female athymic nude mice. When tumors reached a volume ≥200 mm3, mice were randomly allocated to four treatment groups: vehicle (dimethyl sulfoxide [DMSO]) alone, lapatinib (100 mg/kg daily by orogastric gavage), fulvestran (5 mg/mice week by subcutaneous injection), or a combination. (a) Tumors were measured every three days. Mean tumor volume (n = 10) in mm3 ± standard error of the mean (SEM) for each treatment group is displayed. Mice tumors were stripped at the end of the treatment and weighed. The average weight ± SEM of mice in each treatment group is displayed. (bars: mean ± S.E.M.; asterisks: *, P < 0.05, compared with DMSO group; Student’s t-test). Ful, fulvestrant; Lap, lapatinib.  , DMSO;
, DMSO;  , Ful;
, Ful;  , Lap;
, Lap;  , Lap+Ful.
, Lap+Ful.
Discussion
Human epidermal growth factor receptor 2 is highly expressed in 25% of breast cancer cases, and HER2(+) breast cancer is highly malignant, invasive, and has a poor prognosis.10,11 In current studies, one focus is upon developing a molecular targeting drug to alter the HER2 gene. Fortunately, trastuzumab and lapatinib have been approved for clinical therapy. Lapatinib, as a result of its excellent performance, has been given special attention since the beginning. Lapatinib was demonstrated to be able to interact with both HER1 and HER2, indicating that it is efficient for trastuzumab-resistant breast cancer cells. The molecular weight of lapatinib is small; therefore it can pass the blood-brain barrier, reducing the risk of brain metastases. Furthermore, it has low cardiotoxicity. However, in clinical studies, its efficiency was found to be only 24%, as primary and acquired resistance seriously limited its performance.12 Some early-stage clinical trials have shown that patients appeared drug resistant in half a year of treatment, because of the acquired resistance. 13,14 Therefore, it is important to clarify the mechanism of acquired resistance of lapatinib; however, few studies have focused on this area. Some evidence has confirmed that the mutation of the PIKC3A gene caused PI3K signal pathway over-activation, which could lead to the primary drug resistance of lapatinib.15 The receptors of TK kinase, Brk, which is expressed in company with HER2, can enhance cell proliferation signaling through both Ras/MAPK and E/cdk2 pathways. This process partly explains the primary drug resistance of lapatinib.16 Currently, the general view of the mechanism of acquired drug resistance supports that lapatinib is the most successful inhibitor of the HER2 pathway, thus causing the primary interaction among the pathways to be changed, which leads to the activation of other compensatory channels. As a result the cancer cells no longer depend only upon the HER2 pathway; therefore, other pathways, such as ER, AXL, and fibroblast growth factor receptor 2, can open.3,17–20 A few clinical trials have applied the drug combination to inhibit the HER2 and ER pathways in order to prolong disease-free survival of HER2+/ER+ patients.21 However, there has been discrepancy as to the condition of stimulation of the HER2 and ERα pathways in the mechanism of lapatinib acquired drug resistance; for example, a study found that the PIK3K/AKT pathway was activated as compensation in drug-resistant strains.
In this study, we built HER2(+) and ER(+) breast cancer cells, BT474, as models to detect the influence of lapatinib on HER2 and ER pathways. We found that lapatinb was able to inhibit the activation of the HER2 pathway, leading to reduced phosphorylation levels of HER2, AKT, and ERK; our results correspond with earlier studies.22 In addition, lapatinib can induce the ERα pathway to be activated to cause the expression of PR (the target gene of ERα), to observably increase. We obtained rBt474 cell lines from BT474 and tested the difference of the activated condition of HER2 and ER pathways between sensitive BT474 and rBT474. Results showed that activation of the ER pathway existed in rBT474 cells, the PI3K/AKT pathway was obviously inhibited, the phosphorylation of AKT was reduced, and the expression of downstream protein, FOXO3a, was increased, which subsequently upregulated the expression of ER.23 Meanwhile the MAPK pathway was activated and the phosphorylation levels of ERK increased. Furthermore, the protein expression of Scr and caveolin-1 were also analyzed, which were representative molecules and belonged to the upstream and downstream ER-MAPK pathways.24 Results showed that the expressions of Src and caveolin-1 were increased, and HER-2 was inhibited by lapatinib long term to cause to Scr compensatory activation. This leads the ERK-MAPK pathway to be elicited to increase the expression of caveolin-1, and because of this process, the independent ERα ligand transfers into the nucleus and is activated, and PR and FOXO3a are stimulated to express. Because of this whole process, cells are able to regain the ability to proliferate. In this study, we have shown that this acquired drug resistance pathway is one possibility, although there are other signal pathways that exist in different kinds of persister cells.
Soeneshein et al. have proven that FOXO3a is an activating transcription factor of ER, the expression of ER has a positive correlation with the level of activated FOXO3a in the breast cancer cell line, and heterologous FOXO3a elicits the level of ER protein and the activation of promoter; whereas RNA interference was used to downregulate the expression of FOXO3a to decrease ER levels.23 However, an activated PI3K/AKT pathway leads to the inactivation of FOXO3a via phosphorylation, and, as a result, the expression of ER is reduced.6 In rBT474 cells, the PI3K/AKT pathway is inhibited, which causes a reverse of the FOXO3a inhibited situation, and the ER pathway can, therefore, be activated. In addition, research has increasingly demonstrated that the MAPK pathway plays a key role in the interaction between HER2 and ER in mammogenesis and breast cancer.23,25–28 In ER(+) breast cancer cells, the MAPK pathway induced by Src phosphorylation can further phosphorylate ER and other regulatory factors, such as caveolin-1, which increase the activity of the ER pathway. In addition, abnormal activation of the MAPK pathway may possibly represent the significant mechanism of ER pathway compensatory activation in rBT474 cells.
Conclusion
In order to determine whether inhibition of the ER pathway could prevent the acquired drug resistance induced by lapatinib, MTT and cloning formation assays were used to show that the combined utilization of lapatinib and fulvestrant could inhibit rBT474 cell proliferation, as well as the formation of BT474 clones. Consistent with previous in vitro results, we further confirmed that, in a mouse tumor model, lapatinib and fulvestrant are more efficient in inhibiting tumor growth than either drug alone. Fulvestrant has almost no effect on BT474 tumor growth; therefore, molecular status monitoring should be performed during drug therapy. This would lead small-molecule targeting drugs to a more rational application. All of our results supported that the drug combination against HER2 and ER pathways is effective therapy to prevent lapatinib acquired drug resistance. In an in vivo trial, treatment of HER2-positive xenografts with the combination of lapatinib and a small molecule inhibitor of ER was more effective than either drug alone. ER signaling activation is suggested as a mechanism of lapatinib resistance, indicating that the combination of HER2 and ER inhibition is a therapeutic strategy to prevent and/or overcome lapatinib resistance in HER2-overexpressing breast cancer. Further in vivo or clinical studies are warranted.
Acknowledgments
This project was supported by the National Natural Science Foundation of China (No.51003078), Shanghai Municipal Health Bureau research projects (No.2008133), and the Shanghai Municipal Science and Technology Commission (No.12140902302).
Disclosure
No authors report any conflict of interest.
References
- Higa GM, Abraham J. Lapatinib in the treatment of breast cancer. Expert Rev Anticancer Ther. 2007;7:1183–1192. doi: 10.1586/14737140.7.9.1183. [DOI] [PubMed] [Google Scholar]
- Chan A. Lapatinib - Overview and current role in metastatic breast cancer. Cancer Res Treat. 2006;38:198–200. doi: 10.4143/crt.2006.38.4.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campone M, Juin P, André F, Bachelot T. Resistance to HER2 inhibitors: Is addition better than substitution? Rationale for the hypothetical concept of drug sedimentation. Crit Rev Oncol Hematol. 2011;78:195–205. doi: 10.1016/j.critrevonc.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Wang SE, Narasanna A, Perez-Torres, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Burris HA, 3rd, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23:5305–5313. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- Rexer BN, Ham AJ, Rinehart C, et al. Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene. 2011;30:4163–4174. doi: 10.1038/onc.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CM, Lee IT, Lin CC, Wang CH, Cherng WJ, Hsiao LD. c-Src-dependent MAPKs/AP-1 activation is involved in TNF-alpha-induced matrix metalloproteinase-9 expression in rat heart-derived H9c2 cells. Biochem Pharmacol. 2013;85:1115–1123. doi: 10.1016/j.bcp.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Schlegel A, Wang C, Katzenellenbogen BS, Pestell RG, Lisanti MP. Caveolin-1 potentiates estrogen receptor alpha (ERalpha) signaling. Caveolin-1 drives ligand-independent nuclear translocation and activation of ERalpha. J Biol Chem. 1999;274:33551–33556. doi: 10.1074/jbc.274.47.33551. [DOI] [PubMed] [Google Scholar]
- Long X, Fan M, Nephew KP. Estrogen receptor-alpha-interacting cytokeratins potentiate the antiestrogenic activity of fulvestrant. Cancer Biol Ther. 2010;9:389–396. doi: 10.4161/cbt.9.5.10926. [DOI] [PubMed] [Google Scholar]
- Pommier SJ, Quan GG, Christante D, et al. Characterizing the HER2/neu status and metastatic potential of breast cancer stem/progenitor cells. Ann Surg Oncol. 2010;17:613–623. doi: 10.1245/s10434-009-0730-z. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. (Published erratum appears in 2010; 133–34) [DOI] [PubMed] [Google Scholar]
- Gomez HL, Doval DC, Chavez MA, et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol. 2008;26:2999–3005. doi: 10.1200/JCO.2007.14.0590. [DOI] [PubMed] [Google Scholar]
- Esteva FJ, Yu D, Hung MC, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol. 2010;7:98–107. doi: 10.1038/nrclinonc.2009.216. [DOI] [PubMed] [Google Scholar]
- MacFarlane RJ, Gelmon KA. Lapatinib for breast cancer: A review of the current literature. Expert Opin Drug Saf. 2011;10:109–121. doi: 10.1517/14740338.2011.533168. [DOI] [PubMed] [Google Scholar]
- Eichhorn PJ, Gili M, Scaltriti M, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68:9221–9230. doi: 10.1158/0008-5472.CAN-08-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang B, Chatti K, Qiu H, et al. Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc Natl Acad Sci U S A. 2008;105:12463–12468. doi: 10.1073/pnas.0805009105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Bacus S, Hegde P, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci U S A. 2006;103:7795–7800. doi: 10.1073/pnas.0602468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergina NV, Rausch M, Wang D, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Greger J, Shi H, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009;69:6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- Azuma K, Tsurutani J, Sakai K, et al. Switching addictions between HER2 and FGFR2 in HER2-positive breast tumor cells: FGFR2 as a potential target for salvage after lapatinib failure. Biochem Biophys Res Commun. 2011;407:219–224. doi: 10.1016/j.bbrc.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Bauerfeind I, Elling D, Heinemann V. Lapatinib in the treatment of hormone receptor-positive/ErbB2-positive breast cancer. Breast Care (Basel) 2010;5(Suppl. 1):13–15. doi: 10.1159/000285775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter CA, Perez-Torres M, Rinehart C, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–4919. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- Guo S, Sonenshein GE. Forkhead box transcription factor FOXO3a regulates estrogen receptor alpha expression and is repressed by the Her-2/neu/phosphatidylinositol 3-kinase/Akt signaling pathway. Mol Cell Biol. 2004;24:8681–8690. doi: 10.1128/MCB.24.19.8681-8690.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Lee MY, Han HJ. A potential role for caveolin-1 in estradiol-17beta-induced proliferation of mouse embryonic stem cells: Involvement of Src, PI3K/Akt, and MAPKs pathways. Int J Biochem Cell Biol. 2009;41:659–665. doi: 10.1016/j.biocel.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Jin W, Wu L, Liang K, Liu B, Lu Y, Fan Z. Roles of the PI-3K and MEK pathways in Ras-mediated chemoresistance in breast cancer cells. Br J Cancer. 2003;89:185–191. doi: 10.1038/sj.bjc.6601048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanaskova N, Keshamouni VG, Krueger JS, Schwartz JA, Miller F, Reddy KB. MAP kinase/estrogen receptor cross-talk enhances estrogen-mediated signaling and tumor growth but does not confer tamoxifen resistance. Oncogene. 2002;21:4000–4008. doi: 10.1038/sj.onc.1205506. [DOI] [PubMed] [Google Scholar]
- Todorovic-Rakovic N, Neskovic-Konstantinovic Z, Nikolic-Vukosavljevic D. Cross-talk between ER and HER2 in breast carcinoma. Archive of Oncology. 2006;14:146–150. [Google Scholar]
- Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–376. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]