

Abstract
Hair follicles are mammalian skin organs that periodically and stereotypically regenerate from a small pool of stem cells. Hence, hair follicles are a widely studied model for stem cell biology and regeneration. This protocol describes the use of two-photon laser-scanning microscopy (TPLSM) to study hair regeneration within a living, uninjured mouse. TPLSM provides advantages over conventional approaches, including enabling time-resolved imaging of single hair follicle stem cells. Thus, it is possible to capture behaviors including apoptosis, proliferation and migration, and to revisit the same cells for in vivo lineage tracing. In addition, a wide range of fluorescent reporter mouse lines facilitates TPLSM in the skin. This protocol also describes TPLSM laser ablation, which can spatiotemporally manipulate specific cellular populations of the hair follicle or microenvironment to test their regenerative contributions. The preparation time is variable depending on the goals of the experiment, but it generally takes 30–60 min. Imaging time is dependent on the goals of the experiment. Together, these components of TPLSM can be used to develop a comprehensive understanding of hair regeneration during homeostasis and injury.
INTRODUCTION
Background
Stem cells, which are characterized by their ability to self-renew and differentiate into functional specialized cells, are crucial for tissue development, regeneration and disease1. To have a comprehensive and integrated understanding of the role of stem cells in these processes, it is necessary not only to track individual cell behaviors but also to understand these behaviors in the context of the normal physiology of a living tissue. The hair follicle has been established as a powerful model system for stem cell biology. The hair follicle is a self-contained organ with a resident stem cell population that can periodically fully regenerate a mature hair shaft throughout the lifetime of the organism. Furthermore, the process of hair regeneration is both stereotypical and compartmentalized, and therefore all the different aspects of stem cell biology, including self-renewal and differentiation, can be observed and studied within a miniscule area of the skin. We recently developed2 and describe here a novel approach to studying hair follicle regeneration by intravital imaging.
Development of methods to image stem cells in vivo
The conventional approaches available to elucidate the cellular behaviors that are used during hair regeneration are primarily static, and they are only able to provide snapshots of a highly complex process. Recent technological advances have begun to overcome the limitations of conventional histological analysis, enabling the imaging of stem cells in a mammalian system in vivo. These include in vivo imaging of hematopoietic stem cells in the bone marrow3,4 and in vivo imaging of stem cells in the testes5, among others. Despite these pioneering advancements, there was still a need for a system that allowed for the study of dynamic processes in the same structures and cells without causing injury to the mouse/system under study. These challenges were overcome through the use of TPLSM to study stem cells in a noninjurious, noninvasive, highly accessible system: the skin. Until recently, the implementation of live-imaging approaches to look at stem cells in the skin was limited. Uchugonova et al.6 first reported a noninvasive multiphoton tomography procedure for visualizing nestin-labeled skin stem cells. Furthermore, Li et al.7 described a multiphoton intravital imaging protocol for monitoring immune responses in the mouse ear skin. Although these publications have certainly laid the critical groundwork for live imaging in mammalian skin, our approach focuses on elucidating critical aspects of stem cell behavior by monitoring and tracking hair follicle stem cells and testing the tissue requirements for regeneration. Distinct elements of our approach that are highlighted in this protocol include performing revisits for in vivo lineage tracing and laser-ablating specific cell populations.
In vivo imaging of mouse hair follicles by TPLSM
The hair follicle is an ideal model system for live imaging of stem cell dynamics for several important reasons (see Fig. 1 and refs. 8–11):
As the most external organ, the skin provides us with a system that is easily accessible, allowing it to be imaged without causing any injury to the tissue or compromising the health of the animal under study.
As the skin is a solid tissue, imaging revisits can be performed in order to track the same structures and cells over extended periods of time2. Traditionally, in vivo lineage tracing has relied on separate analyses of littermates. In contrast, TPLSM enables in vivo lineage tracing of the same tissues and cells within the same mouse.
The hair follicle undergoes constant regeneration as a result of stem cell activity. Specifically, the hair follicle alternates between periods of quiescence (telogen), growth (anagen) and regression (catagen). Telogen is the period when the hair follicle does not grow. Anagen is the period when the lower part of the hair follicle expands and differentiated lineages that form a new hair shaft are generated by committed progenitors situated at the lower tip in the interphase with the mesenchyme. Finally, catagen is the period of the hair cycle when the lower part of the follicle retracts to restart the quiescent phase of the next hair cycle12–14 (Fig. 1). This cyclical process occurs in a stereotypical and synchronized manner15,16.
Various stem cell populations are located within distinct compartments or ‘niches’ of the hair follicle2,17 (Fig. 1). This compartmentalization enables us to observe multiple populations of stem cells and to compare their contributions to tissue regeneration.
Figure 1.
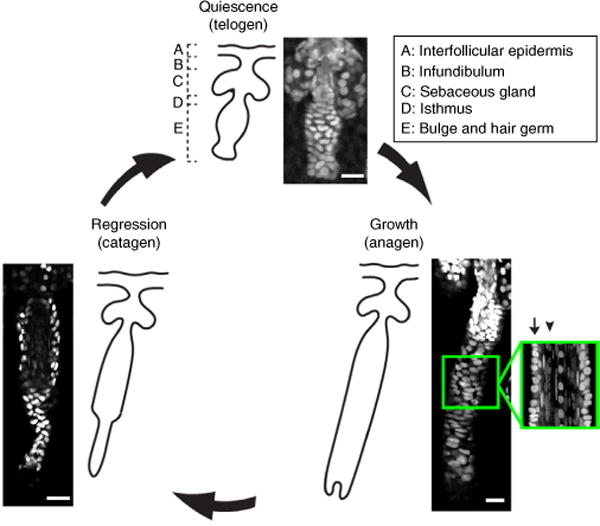
The hair follicle regeneration cycle and anatomy. The hair follicle undergoes cyclical rounds of quiescence (telogen), growth (anagen) and regression (catagen), and consists of heterogeneous compartments (labeled A–E). Bottom right, a magnified optical section of the coronal plane of an anagen hair follicle depicts a high-resolution micrograph of the suprabasal (arrow) and basal (arrowhead) layers of the follicle. All images were taken of a K14H2BGFP mouse using TPLSM. Scale bars, 20 μm. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine and conducted in accordance with the approved animal handling protocol.
An intravital imaging approach in the skin thus provides unique advantages to addressing outstanding questions in the stem cell field given that it enables the noninjurious observation and tracking of stem cell behaviors, such as proliferation and differentiation, to facilitate an understanding of how these behaviors coordinate to maintain a tissue.
Advantages of TPLSM
In traditional epifluorescence, for an excitation event to occur one photon must excite a fluorophore in a single quantum event. By contrast, with two-photon excitation, two lower-energy photons can bring a fluorophore to the same excited state using approximately twice the wavelength required for single-photon excitation. For this to occur, a high flux of excitation photons is required to ensure simultaneous absorption of two photons. This causes a spatial confinement of fluorescence to a small volume defined by a focused laser; in this case it is a mode-locked femtosecond pulsed laser. The key benefits of using photons in the IR spectrum, as is done in TPLSM, are that it substantially decreases phototoxicity and photobleaching, and it enables a greater penetration depth of imaging owing to reduced light scattering. The latter is crucial for the study of skin appendages such as the hair follicle, given that cells in these structures are situated in deeper layers of the tissue.
Our protocol uses a motorized microscope stage with a submicrometer x, y and z axis movement, which is advantageous as it allows easy access to a large sampling area of the skin with high accuracy and reproducibility. For example, a 20× lens, which is most commonly used in TPLSM applications, typically provides a field of view of ~500 μm2. Given that a mouse hair follicle is, at its most minimal state, ~40 μm in diameter and ~100 μm in length, a relatively small number of intact hair follicles can be captured within a single field of view. However, a motorized stage driven by specialized software, which is commonly provided by most microscope manufacturers, can be used to automatically acquire sequential fields of view, and they can be stitched together as tiles of a large mosaic. With this approach, it is possible to image large areas of the skin in a single mouse, which markedly increases the sample number of hair follicles that we can image and allows for the construction of topographical maps of the skin on which the relative location of individual follicles can be established and identified in subsequent experiments. This feature is crucial for implementing lineage tracing in longitudinal studies using the same mice, as explained in detail below.
An added benefit of using TPLSM for studying the skin is second-harmonic generation (SHG) within certain types of highly structured multimerized structures such as collagen, which makes them visible without the need of a specific probe. Type I and II collagen fibers are some well-known proteins that produce SHG. This optical process is not a true autofluorescence, and instead it returns the absorbed energy at half the wavelength of the original excitation source18. SHG is caused by a nonlinear optical effect from intense laser power19. During this process, photons of the same frequency interact with loci that lack a center of symmetry (such as collagen) to combine and generate new photons with twice the energy and, therefore, twice the frequency and half the wavelength of the initial photon. SHG creates emissions that are exactly half the wavelength used for excitation18. This provides a notable advantage, because at a 940-nm wavelength both a GFP signal in the green channel and collagen fibers in the blue channel are visible, thus allowing for simultaneous imaging of two different components. SHG offers valuable information about the state of the collagen-rich extracellular matrix, which constitutes a crucial element of the hair follicle stem cell microenvironment (Fig. 2 and Supplementary Video 1). The specific wavelength for SHG must be chosen carefully for the simultaneous visualization of other tissue components that use true fluorescence.
Figure 2.
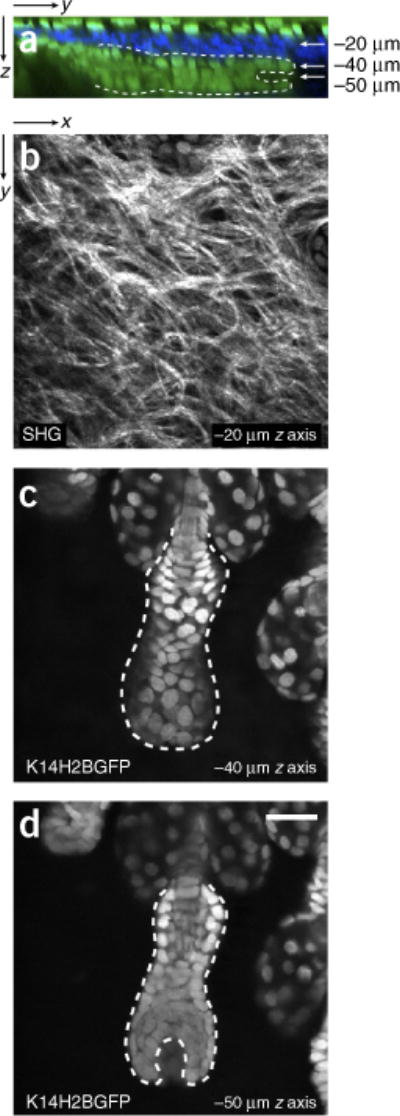
Intravital two-photon imaging of the mouse hair follicle. (a) Reconstructed zy view from a series of serial optical sections in a K14H2BGFP mouse depicting the different epithelial compartments of the skin (in green), including an intact hair follicle in quiescence (telogen). Type I collagen fibers were detected by second harmonic generation (SHG) depicted in blue. (b) Magnified view of type I collagen fibers detected by SHG. (c,d) Two different planes are depicted at the indicated depths where the outer and inner layers of the hair follicle are identifiable by means of nuclear localized signal (K14H2BGFP). The cycling portion of the hair follicle can be seen outlined by the dotted line. See also Supplementary Video 1. Scale bar, 20 μm. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine and conducted in accordance with the approved animal handling protocol.
Experimental design
Challenges of visualizing hair follicles in the mouse skin
Intravital imaging to study the biological basis of hair follicle regeneration relies on two primary objectives: (i) to visualize and resolve the entire cell population that comprises the hair follicle in all stages of the hair cycle and (ii) to follow the behavior and fate of individual stem cells during the entire process of regeneration. This can be accomplished either by time-resolved continuous imaging or by being able to identify and re-image the same hair follicles in separate imaging sessions but on the same animal over an extended period of time. Meeting these objectives can be challenging, and a number of factors need to be taken into account, such as the thickness of the skin, which can affect laser penetration in the deeper layers of the dermis, the density and cycling characteristics of the hair follicles and the stability of the tissue against breathing artifacts.
The skin on the dorsal area of the mouse ear is a region that meets most of these requirements. Owing to the anatomical characteristics of the ear, the skin (both epidermis and underlying dermis) is particularly thin, especially compared with the back skin, which can be two to three times as thick. As a result, hair follicles in the ear skin are situated at a shallow depth within the dermis and at a very steep angle with respect to the plane of the epidermis, and thus can be easily resolved in their entirety by TPLSM. An important consideration is the cycling ability and characteristics of the hair follicles in the mouse ear. The tip of the ear is populated by hair follicles that are more dispersed, have short club hair shafts and are suspended in a prolonged quiescent phase of the hair cycle. In contrast, the part of the dorsal ear skin that is adjacent to the head contains hair follicles that have very similar properties and cycle with similar synchronized kinetics as the hair in the back skin. Finally, the ear offers a great platform for stabilizing this area of the skin and offering protection from breathing artifacts during laser scanning, optical sectioning and time-resolved imaging.
Considerations for optimal skin preparation
To prepare the mouse for imaging, the hair within the area to be imaged must first be removed. This is done first by shaving, and then by applying depilatory cream locally to ensure complete removal of the external hair shafts. This step is crucial not only for overall access to the skin but also because hair shafts substantially scatter light, reducing the overall imaging quality. Although the use of nude mice is an alternative option, we chose not to use these mice in our protocol for reasons of cost, consistency and reproducibility of experimental results using established mouse genetic models with normal hair physiology. When possible, albino mice are always preferred over pigmented mice for imaging, because melanin absorbs large levels of the laser energy delivered to the tissue, generating autofluorescence artifacts and increasing the probability of cell damage after prolonged exposure. Another advantage of using the ear as a preferred area of the skin for imaging is that it has markedly less pigmentation compared with other parts of the body. Importantly, on the basis of our experience, the hair removal processes used in this protocol do not substantially interfere with the fundamental properties of hair follicle stem cell physiology and regeneration.
Ear skin mounting apparatus
To take advantage of these properties of ear skin, we designed and fabricated a custom stage that allows the appropriate mounting and immobilization of the mouse ear skin (Fig. 3). The stage consists of two micromanipulators, with movement on the x, y and z axes, which are used to adjust the relative position of two spatulas. After anesthesia has been administered and the area of the skin has been properly shaved and cleaned, the ear spatula is inserted into the canal of the left ear for mounting (Fig. 3a). An alternative mounting procedure is described by Li et al.7, in which the ear is mounted for imaging using adhesive masking tape atop a conical tube. In our mounting system, a round coverslip is glued on the second spatula, and once it is secured it is pressed gently against the ear skin to achieve stabilization and leveling of the imaging area (Fig. 3b). The angle and orientation of the two micromanipulators have been optimized for mounting and imaging the dorsal skin of the left mouse ear, but the design can be easily modified to accommodate other parts of the skin. This arrangement allows for increased flexibility and it can accommodate appropriate mounting positions for mice of different ages and sizes. The stage also includes a heating plate and mounting provisions for the tubing for inhalable anesthesia.
Figure 3.
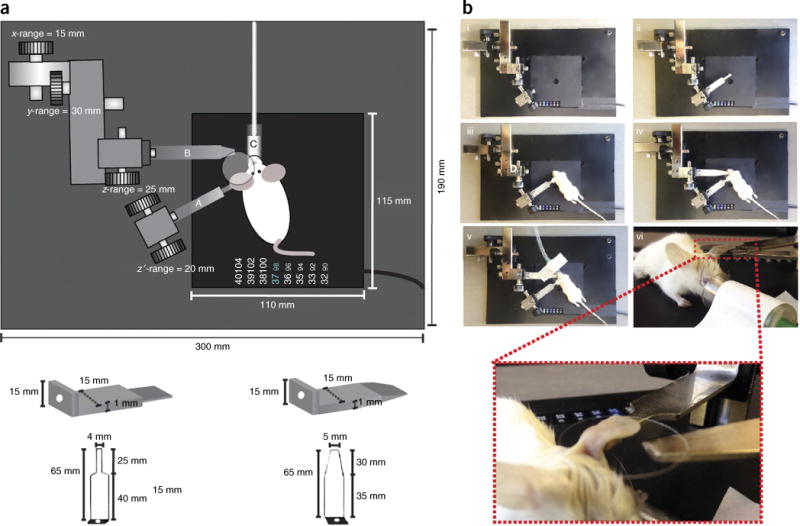
Pictorial and photographic schematics of mounting stage. (a) Bottom left image, spatula used to mount the ear. Bottom right image, spatula used to hold the coverslip. Top, nose cone for inhalable anesthesia. The schematics and dimensions of the stage and spatulas, as well as the ranges of the stage manipulators, are given. (b) Photographic stepwise depiction (i–vi) of stage and mouse preparation. Inset in image vi is a magnified view of a mouse mounted for imaging with the ear lying flat on the mounting spatula and the coverslip pressing gently from the top. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine and conducted in accordance with the approved animal handling protocol.
Fluorescent reporters
Our laboratory takes advantage of a wide range of available fluorescent reporter mouse lines for intravital imaging of the skin (Table 1). Although many different fluorescent lines are suitable for TPLSM, experimenting with a variety of lines revealed one in particular as optimal. This is a transgenic mouse strain in which GFP is fused to the histone H2B under the control of the keratin 14 promoter to label all epithelial nuclei (K14H2BGFP mice, which can obtained by request from the laboratory of E. Fuchs, Rockefeller University)20. The specificity of the keratin 14 promoter allows us to label epithelial cells throughout the hair follicle and the epidermis, and the subcellular nuclear localization of H2B-GFP allows us to distinguish individual cells from their neighbors (Figs. 2 and 4). In addition to facilitating the study of stem cells within their various ‘niches’, TPLSM can be used to observe the effect of epithelial-mesenchymal interactions on hair regeneration. This is achievable through the use of a lymphoid enhancer-binding factor 1 (Lef1)-RFP mouse line that effectively labels mesenchymal cells of the hair follicle, which are located in the dermal papilla21.
TABLE 1.
Reporter mouse lines expressing fluorescent proteins.
| Fluorescent protein | Type | Promoter | Reporter mouse line | Reference |
|---|---|---|---|---|
| H2BGFP | cTg | Keratin-14 (all epithelial cells) | Tg(Hist1H2BJ/GFP) | 20 |
| H2BGFP | cTg | Tre;tetO | pTRE-H2BGFP | 20 |
| RFP | cTg | Lef1 (dermal papilla) | Tg(Lef1RFP) | 21 |
| tdTomato | cKI | CAG | Rosa-CAG-LSL-tdTomato-WPRE | 35 |
| mTmG | cKI | Chicken beta-actin/pCA | RosamTmG | 36 |
Figure 4.
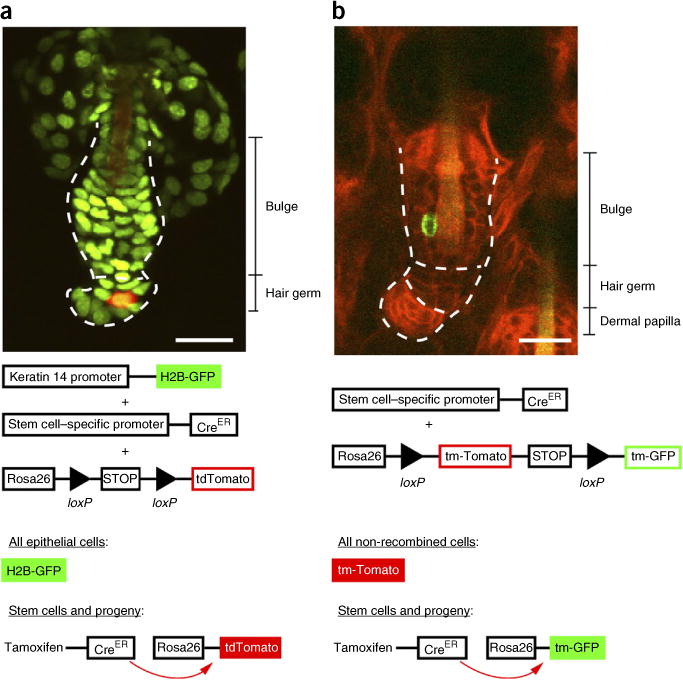
Labeling hair follicle stem cells with inducible Cre reporters. These images demonstrate the diversity of reporters that can be used to target specific cell populations, including stem cells, within the hair follicle. (a) K14H2BGFP;tdTomato;Lgr5CreER hair follicle and, directly below, a corresponding schematic describing the dual fluorescent reporter/Cre recombinase system. Upon Cre recombination, the fluorescent tag switches from nuclear green to cytoplasmic tomato (red). The cycling portion of the follicle is outlined (dotted line) and divided into bulge and hair germ, and all epithelial nuclei are in green. A single tdTomato Cre-recombined epithelial cell is in red. (b) mTmG;K19CreER hair follicle and, directly below, a corresponding schematic explaining the mTmG/Cre recombinase system. The cycling portion of the follicle is outlined (dotted line) and divided into bulge, hair germ and dermal papilla. All epithelial nuclei contain a red membrane label, and the single Cre-recombined cell contains a green membrane label. Both images demonstrate single-cell labeling as a result of low induction of the Cre recombinase system through administration of a low dose of tamoxifen. Scale bars, 20 μm. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine and conducted in accordance with the approved animal handling protocol.
A variety of established reporter lines can be used to achieve the specific labeling of different hair follicle cell populations using the Cre-loxP system22. The Cre-loxP recombinase system acts through Cre-mediated recombination of genes flanked by loxP sites, resulting in the excision of a specific DNA segment23,24. Several transgenic mouse lines exist that can report Cre activity with a fluorescent protein. In this case, the fluorescent reporter is under the control of a ubiquitous promoter but flanked by a loxP-STOP-loxP sequence that inhibits its expression until the Cre is activated, and then excises the stop sequence. The subcellular localization and emission wavelength of existing reporter lines enables the simultaneous visualization of different cell populations, whose specificity is determined by the promoter that controls the expression of Cre recombinase (Fig. 4).
Assessing short-term stem cell behavior in hair regeneration
TPLSM can be used to study real-time behaviors of stem cells using time-resolved imaging. Four-dimensional (4D) data sets can be obtained by optically sectioning the tissue at regular intervals and then by rendering these intervals into a fluid movie by sequentially connecting each time frame of the projected volume. Through controlling the time interval, step size and number of optical sections, critical cellular behaviors can be observed that mediate the process of hair regeneration, including proliferation, migration and cell death (Supplementary Videos 2–4). These dynamic processes are difficult to infer using traditional static histological methods. Despite the advantages of time-resolved imaging, there are still several potential caveats. Fluorescence intensity can gradually decrease as a result of photobleaching during imaging, depending on the laser intensity and dwell time, the number of time points and the type of fluorophore used. Second, any movement of the imaging region owing to the mouse’s breathing or other factors can substantially reduce the quality of imaging data by introducing drift artifacts that are difficult to rectify after acquisition. Furthermore, appropriate measures need to be taken to ensure the proper health and physiology of mice for the duration of an imaging session, with younger mice being generally more vulnerable to negative effects of prolonged sedation.
Assessing long-term cell behavior by in vivo lineage tracing
The Cre recombinase system can be used to target specific cell populations by controlling Cre expression using a cell type–specific promoter25. In order to achieve temporal control of Cre-recombinase activity, the enzyme is fused to the ligand-binding domain of an engineered steroid hormone receptor (estrogen or progesterone, annotated CreER and CrePGR, respectively)26–29. Under normal conditions, the enzyme is sequestered away from the nucleus, but it can be activated with the administration of tamoxifen (CreER) or mifepristone (CrePGR). This treatment releases the Cre recombinase, freeing it to translocate to the nucleus where it binds and splices the loxP sequences, resulting in recombination of the targeted DNA sequence.
Established promoters that target Cre expression in specific stem cell populations within the skin and hair follicle include keratin 14 (all epithelial cells30), keratin 19 (bulge and hair germ31) and Lgr5 (lower bulge and hair germ32; Table 2). The rate of Cre-mediated recombination is regulated through titrating the amount of tamoxifen or mifepristone that is administered to the mouse. This allows for mosaic expression of the Cre reporter within a given population of hair follicle stem cells. These cells and their progeny are labeled irreversibly, and they can be tracked over time using TPLSM. A common approach that our laboratory takes for TPLSM in vivo lineage tracing is to combine three alleles in the same mouse: (i) K14H2BGFP, (ii) an inducible CreER allele under the control of a cell-specific promoter and (iii) a Rosa-loxP-STOP-loxP-tdTomato fluorescent Cre reporter allele (Fig. 4). This approach can result in variable degrees of labeling among follicles within a particular field, which provides the additional benefit of being able to select those follicles whose labeling best suits the purpose of the particular experiment. Our previous work has demonstrated the power of combining genetic lineage tracing with intravital TPLSM for studying hair regeneration by clonally labeling hair follicle stem cells and following their fate to demonstrate how their position influences their contribution to regeneration over time33.
TABLE 2.
Cre mouse lines for targeting specific cell populations.
| Line | Type | Specificity | Reference |
|---|---|---|---|
| K14 Cre | cTg | All undifferentiated epithelial cells | 37 |
| K14CreER | cTg | All undifferentiated epithelial cells | 38 |
| K19CreER | cKI | Bulge stem cells and hair germ | 31 |
| Lgr6CreER | cKI | Isthmus, interfollicular epidermis, infundibulum and occasionally in the bulge and hair germ | 17,39 |
| Lgr5CreERT2 | cKI | Hair germ and bulge | 32 |
| K5-rtTA | cTG | All undifferentiated epithelial cells | 40 |
Revisiting the same hair follicles
Owing to the noninvasive nature of our intravital imaging protocol, the exact same region of the mouse skin can be revisited in separate imaging sessions spaced hours, days or even months apart. This attribute is particularly important because it enables the direct assessment of stem cell fate by monitoring the same stem cells and their lineages throughout the process of hair regeneration, instead of relying on statistical analysis of histological samples from littermates, as is the case in conventional lineage-tracing experiments. For this, it is key to be able to recognize the same region of the skin in subsequent imaging sessions and to establish the identity of individual hair follicles within them. To achieve this, critical topological information needs to be collected in the initial imaging session, including (i) macroscopic features of the general skin region; (ii) number, orientation and clustering of the hair follicles; and (iii) location of individual labeled cells (Fig. 5).
Figure 5.
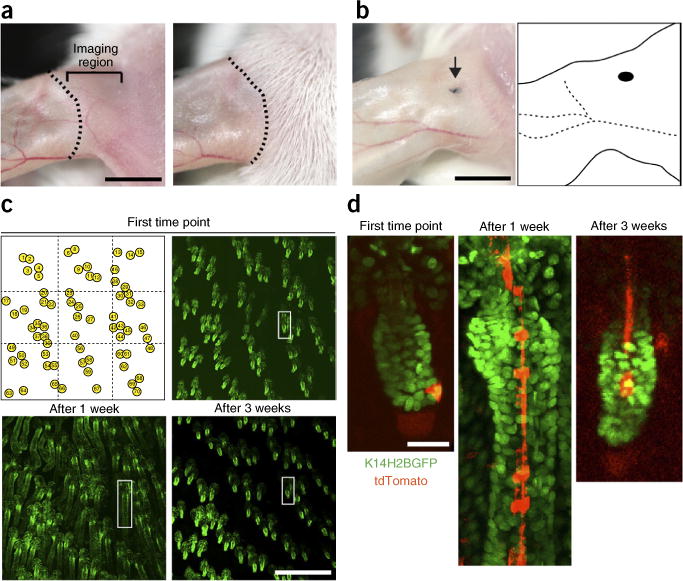
Strategy for repeated imaging of the same skin region. (a) Magnified views of the dorsal ear skin immediately after depilation (left) and 10 d later (right). The dotted line indicates the borders between areas of active hair growth and quiescence. Imaging should be done in the ear region to the right of the dotted line, indicated by a bracket, where hair is actively growing. Scale bar, 5 mm. (b) A punctate tattoo (indicated by the arrow) can be used for creating additional landmarks, which, together with the distinctive pattern of the vasculature network, can be used for navigating back to the same region for subsequent imaging sessions. Scale bar, 5 mm. (c) Digital tiling can be used to identify and revisit the same hair follicles in subsequent imaging sessions. Sequential views of the same skin region, showing hair follicles in different stages of the hair cycle (telogen I, upper right; anagen II, lower left; telogen II, lower right). Scale bar, 0.5 mm. The white box highlights a single follicle magnified in d. Snapshots of a single K14H2BGFP;tdTomato follicle at three different time points during the regeneration cycle, in which a single stem cell is labeled and traced. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine and conducted in accordance with the approved animal handling protocol.
To recognize the same general area of the skin, we rely on inherent topological landmarks of the dorsal ear region, such as the location and branching patterns of the vasculature, which can be easily assessed by visual inspection. In addition, and for greater accuracy, we can introduce new landmarks in the form of a minimal punctate tattoo. The combined information from these landmarks is used in subsequent experiments to mount the same approximate region of the skin under the coverslip. Once the attributes of the general area of the skin have been established, the next step is to identify the same hair follicles in order to follow the fate of labeled stem cells and their progeny as they were imaged in the initial experiment. The number and relative location of individual hair follicles is established in embryonic development and in the absence of injury, and it is maintained throughout the adult life. This allows us to use the relative position and clustering of the hair follicles to put together topographical maps of the entire imaging region.
Typically, the area of the skin that is in direct contact with the coverslip is no larger than 3 mm2. This relatively small area can be fully imaged using a tiling approach. In our case, the entire area is divided into equal imaging fields of 0.5 mm2. Each of these fields is sequentially imaged and optically sectioned. Subsequently, the entire region can be digitally reconstituted using the coordinates of the individual tiles. This can be accomplished manually, as was done during our early work, but it can be particularly labor-intensive. Therefore, we now take advantage of a motorized microscope stage and associated software to perform the tiling function automatically. Most microscope manufacturers have integrated similar functionality into their software packages when a motorized stage is used. Once a digital mosaic of the imaging region is reconstituted, it can be used effectively as a map to identify the relative position of individual hair follicles in subsequent experiments (Fig. 5). Once identified, the hair follicles of interest can be magnified from the same data set to locate singly labeled cells and their direct progeny.
Manipulating stem cell behavior by laser ablation
One of the great attributes of intravital imaging is that it enables the direct visualization of stem cell behavior within their native tissue environment. The next step in elucidating the role and mechanism of the observed stem cell behaviors is to manipulate specific cells, as well as other elements of the tissue microenvironment, in vivo. One way of assessing the role of specific cells in the process of hair regeneration is by ablating them. We use laser-induced cell ablation as a preferred way to eliminate cells because it allows for precise spatiotemporal control of the process. Alternative methods, such as genetic cell ablation based on inducible expression of diphtheria toxin, depend on the specificity of the particular promoter and often show incomplete penetrance. A pulsed Ti: sapphire femtosecond laser such as the one used in TPLSM, combined with a high-numerical-aperture (high-NA) objective lens, allows the concentration of lethal energy into a small focal volume within the tissue. In practice, using laser ablation, we have been able to ablate a very small number of cells within the hair follicle or in the surrounding niche without affecting superficial or other neighboring tissue structures (Fig. 6 and Supplementary Video 5).
Figure 6.
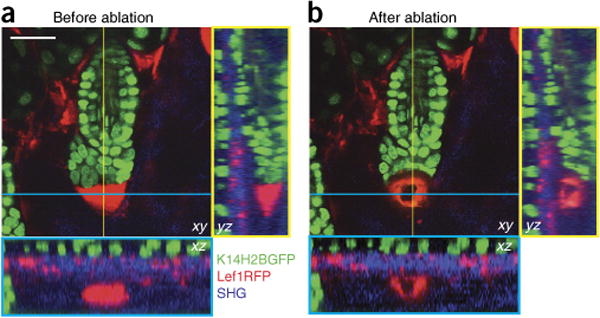
Laser ablation of hair follicle cells. Images are taken of a K14H2BGFP;Lef1RFP mouse. Epithelial nuclei are in green, mesenchymal cells are in red and collagen fibers of the extracellular matrix are in blue. (a) Before ablation, the hair follicle mesenchymal niche (dermal papilla, DP), located immediately below the hair germ, is fully intact, as indicated by a condensed RFP signal. (b) Immediately after ablation of the DP, bright autofluorescence occurs at the site of injury. Yellow- and cyan-bordered panels provide orthogonal views of the indicated y and x axes, respectively. Notice that the tissue surrounding the ablated region is minimally affected. See also Supplementary Video 5. Scale bar, 20 μm. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine and conducted in accordance with the approved animal handling protocol.
To target specific cell populations for laser ablation, an appropriate fluorescent reporter is needed to mark these cells to distinguish them from their surroundings. The highly compartmentalized structure of the hair follicle allows for the precise identification of stem cells, as well as other populations, based on the anatomical features of the organ. After a cell target is identified, laser ablation is carried out by performing a single scan of the selected area, usually between 10 and 50 μm2, at 900-nm wavelength while adjusting the pixel dwell time to achieve optimal results. The lack of a direct and specific readout of cell death can create some ambiguity for assessing the outcome of a laser ablation procedure. Ablation artifacts, such as a characteristic halo of autofluorescence, are often visible immediately after the scan (Fig. 6 and Supplementary Video 5). However, it is often necessary to revisit the ablated region at least 24 h after the procedure to further assess the success of the ablation process by verifying the absence of fluorescence in the targeted region.
MATERIALS
REAGENTS
Mice of interest. For the images found in this protocol, the following fluorescent reporter mouse strains were used: K14H2BGFP (received from the laboratory of Elaine Fuchs at Rockefeller University), mTmG (Jax Stock no. 007576) and tdTomato (Jax Stock, no. 007908). For a comprehensive list of fluorescent and Cre reporter mouse lines used by the laboratory for TPLSM, please see Tables 1 and 2 ! CAUTION Please note that all animal experiments should be performed according to relevant guidelines and regulations. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine and conducted in accordance with the approved animal handling protocol.
Anesthesia
Isoflurane (Henry Schein)
Ketamine (Henry Schein) ! CAUTION Ketamine is a controlled substance. All relevant regulations should be followed.
Xylazine (Henry Schein) ! CAUTION Xylazine is a controlled substance. All relevant regulations should be followed.
Sterile PBS, pH 7.2 (PBS, 1×)
Sterile 10-ml vials (Henry Schein)
Mouse preparation
Depilatory lotion (Nair)
PBS, pH 7.2 (PBS, 1×)
Tattoo ink (optional; Dr. Ph Martin’s Black Star waterproof India ink)
Stye Ophthalmic Ointment 8 ox tube (Del Pharmaceuticals)
Stage preparation
Vacuum grease (Dow Corning, no. 1597418)
Purified water (Milli-Q, Millipore)
EQUIPMENT
Microscope
LaVision TriM Scope II multiphoton laser scanning microscope (LaVision Biotec)
Laser
Ti:sapphire Chameleon Vision II (Coherent)
Objectives
40× (W-LD C-APOCHROMAT, NA 1.1 Zeiss)
20× (W-Plan-APOCHROMAT, NA 1.0 Zeiss)
4× (XL Fluor 4×/340, NA 0.28, Olympus)
Software
ImSpector 400 (LaVision Biotec)
Filters
Olympus filter cube blue excitation, no. U-MNIB3
Olympus filter cube green excitation, no. U-MWIG3
Custom mounting stage
Micromanipulators (Edmund Optics, cat. nos. NT03-682 ×2 and NT36-347 ×2)
Heating pad (CryoLogic, BioTherm SmartStage no. SS12)
Custom-made mounting spatulas
Glass coverslip (22 mm; Electron Microscopy Sciences)
Super glue (Loctite)
Bull’s-eye level (McMaster-Carr)
Grease syringe
Thumb screw (ProTanium)
Mouse preparation
Scale (Ohaus Corporation Model HH120D)
Insulin syringe for intraperitoneal (i.p.) injections (Terumo 27 gauge)
Small, electric hair clipper (Wahl)
Handheld vacuum
Cotton swabs
Adhesive tape
Anesthesia
Wall-mounted anesthesia machine (VetEquip)
Isoflurane vaporizer (VetEquip)
Anesthesia breathing circuit and nose cone (VetEquip)
O2 gas flow regulator for E-cylinders (VetEquip)
O2 tank (E-cylinder; Airgas)
REAGENT SETUP
Injectable anesthetic
For injectable anesthesia, prepare batches of 15 mg/ml ketamine and 1 mg/ml xylazine in PBS (pH 7.2), and store them in sterile 10-ml vials at room temperature (~20–26 °C) for up to 30 d.
EQUIPMENT SETUP
Stage
Ensure that the heating element of the stage is plugged in and that the stage is at 37 °C (as indicated by the temperature indicator strip) (Fig. 3).
Microscope
Turn on the laser, microscope and all imaging components: Objectives, scanning devices, laser sources and detection camera (Fig. 7).
Figure 7.
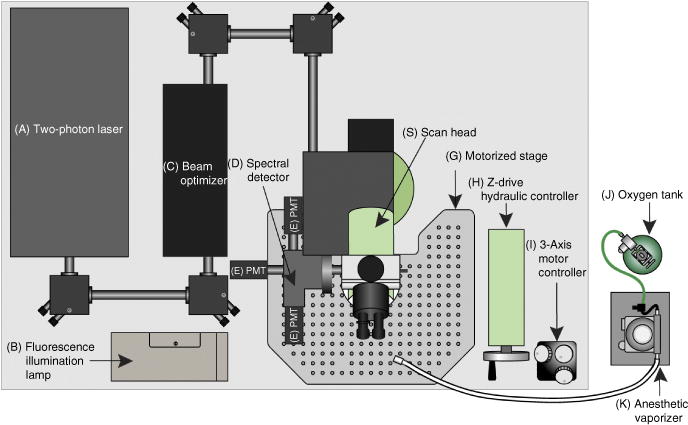
Schematic of the two-photon microscope system used in this protocol. For two-photon imaging, a Ti:sapphire laser generates a beam between 680 and 1,080 nm (A) that is transmitted to the scanner (S) through the beam optimizer (C). Once excitation occurs on the sample, the emission wave is delivered to the spectral detector (D). In the spectral detector, there are two dichroic mirrors (495LP and 560LP) and three filters (450/50, 525/50 and 610/75). Depending on the wavelength, the excitation wave separates into three different PMTs (E). Fluorescence from the sample can be checked visually with wide-field fluorescent illumination (B) before recording. The coordinates of the microscope platform (G) are regulated hydraulically (H) or more precisely with a motorized controller (I). The oxygen tank (J) is connected to the isoflurane vaporizer (K), and the inhalable anesthetic gas is continuously delivered to the mouse through the tubing and a nose cone.
Custom-made mounting spatulas
To construct a custom-made mounting spatula, machine-bend aluminum and cut it to dimensions specified in Figure 3. Drill a 5-mm hole for securing the spatula to the micromanipulator with a thumb screw.
PROCEDURE
Mouse preparation for imaging ● TIMING 15 min
-
1| Weigh the mouse using an electronic scale, and calculate the dose of ketamine (100 mg/kg)/xylazine (10 mg/kg) cocktail to administer. Use a 27-gauge insulin syringe to deliver the anesthetic via i.p. injection. Alternatively, if a second line of isoflurane and oxygen is available in the preparation area, this can be used for the duration of imaging (1% oxygen and ~1–2% isoflurane) to maintain a breathing rate of ~1 breath per second.
! CAUTION All procedures should follow approved institutional and governmental animal protocols.
▲ CRITICAL STEP A state of anesthesia should be verified by the absence of physical (reflex withdrawal after a toe pinch) and physiological responses (change in respiratory or heart rate or change in the character of respiration).
-
2| Once the mouse is fully sedated (~5 min), immediately place it on the stage (heated to 37 °C) and apply eye ointment. Next, use the electric clippers to shave the hair on and surrounding the ear. Use a handheld vacuum cleaner placed just above the mouse to remove shaved hairs from the mouse when you are finished.
▲ CRITICAL STEP The body temperature of a mouse starts to decrease during the anesthesia state. To maintain the proper physiological state of the mouse, ensure that it is placed on the heating pad for the entire duration of preparation and imaging.
-
3| Gently apply depilatory cream (e.g., Nair) onto the shaved area using a cotton tip swab, and let it sit for 2 min. Subsequently, use cotton swabs soaked in PBS (pH 7.2) to gently remove the depilatory cream using repeated rolling strokes toward the tip of the ear.
▲ CRITICAL STEP Strictly adhere to the time indicated, and ensure the complete removal of the depilatory cream after treatment. If the depilatory cream remains on the skin for too long, it can lead to degradation of the cornified layer of the epidermis, which in turn can cause topical inflammation, thus making the area unfit for imaging. Similarly, for the removal of the depilatory cream, always soak the cotton swab in PBS and use gentle strokes to avoid abrasion of the epidermis.
Tattoo (optional) ● TIMING 1 min
-
4| If you wish to mark the area of the mouse ear that is being imaged for later revisiting, a minimal punctate tattoo system can be used (Fig. 5b,c). Dip a 30-gauge needle into a carbon pigment ink solution. After visually checking that a small amount of pigment has been absorbed at the tip, use the needle to singly puncture the skin of the anesthetized mouse, immediately adjacent to the area of the ear that will be imaged.
! CAUTION The tattoo should be used as a reference point when revisiting the same area in subsequent experiments. However, ink pigment absorbs a substantial amount of the laser energy, which can cause image artifacts and in some instances even lead to tissue damage such as re-epithelialization and immune cell infiltration. To avoid adverse effects of the tattoo process on the physiology of the tissue under observation, we recommend that the tattoo be situated outside of the imaging area (at least 500 μm) but adjacent to it, so the same follicles can easily be found during revisits.
▲ CRITICAL STEP The needle should not penetrate the skin more than a quarter of a millimeter below the surface, as the goal is to deposit a minimal amount of pigment immediately below the epidermis and into the dermis. Briefly rinse the skin with PBS using a cotton-tip swab to remove any excess pigment from the surface. Visually inspect the presence of the minimal tattoo deposit inside the dermis.
Mounting the mouse on the imaging stage ● TIMING 5 min
5| Screw the ear-holding spatula onto the micromanipulator (Fig. 3a) and use a bull’s-eye spirit level to make sure that the surface on which the ear will be placed is level. Apply a thin layer of vacuum grease at the tip of the spatula to provide a mild adhesion between the inner surface of the ear and the spatula.
-
6| Gently position the mouse’s ear on the spatula and visually inspect the skin to ensure that it is completely flat.
▲ CRITICAL STEP The mouse’s ear should be attached to the spatula evenly and completely; otherwise, the skin may wrinkle and cause an uneven surface, or it may begin to drift during imaging, both of which can lead to the deterioration of the image quality.
Mounting the coverslip on the skin ● TIMING 5 min
7| Screw the upper spatula (Fig. 3b) into place on the stage, and use a bull’s-eye spirit level to make sure that the surface on which the coverslip will be placed is level. Use the micromanipulator to lower the spatula toward the mounted ear to assess its relative position to the desired skin area.
-
8| Place a round coverslip (22 mm, no. 1 micro coverglass, Electron Microscopy Sciences) on the tip of the upper spatula and secure it in place using a drop of super-glue (Loctite). After the glue has dried, slowly lower the coverslip spatula until it gently comes in contact and presses on the area of the skin to be imaged.
! CAUTION Excessive compression of the skin with the coverslip can have adverse effects on the physiology of the tissue owing to hypoxia or local inflammation. Visually inspect that there is sufficient blood flow in the small vessels and capillaries immediately below the imaging area.
▲ CRITICAL STEP Ensure that the coverslip glues to the spatula in a completely flat manner; otherwise, the resulting unevenness will disturb imaging. The primary purpose of the coverslip is to flatten and minimally compress the area of the skin to be imaged. This action aligns the surface of the epidermis onto a single place and also tilts the hair follicles so that they are positioned almost parallel to the surface of the skin, thus greatly increasing the resolution in the xy imaging plane.
? TROUBLESHOOTING
Setting up the anesthesia nose cone ● TIMING 1 min
-
9| Position the snout and mouth of the mouse fully within the nose cone, and secure the cone to the stage using a piece of tape to ensure that it stays in place for the duration of the imaging session.
! CAUTION Ensure that the entire snout of the mouse is within the cone so that the mouse will remain fully sedated for the duration of imaging.
Connecting and aligning the stage to the imaging platform ● TIMING 10 min
10| Move the stage from the preparation area to the imaging platform (Fig. 7), and then immediately connect the outlet of the heating element to the power supply.
-
11| Connect the tubing from the anesthesia apparatus to the nose cone, and adjust the oxygen tank (Fig. 7j) and vaporizer (Fig. 7k) outlet to deliver 1 liter/min O2 and 1% isoflurane to the mouse for the duration of imaging.
▲ CRITICAL STEP If the breathing of the mouse becomes too heavy, lower the isoflurane by ~0.2% until the breathing returns to normal (~1 breath per second).
? TROUBLESHOOTING
-
12| If a water-immersion lens is used, deposit a meniscus of distilled water on top of the coverslip (~1 ml for a 22-mm-diameter coverslip).
▲ CRITICAL STEP For immersion lenses, a water interphase needs to be maintained between the lens and the coverslip at all times. For short experiments, a self-contained water meniscus is sufficient; however, for long time-lapse recordings, steps need to be taken to replenish the water that is lost by evaporation. A silicone ring or other barrier can be used on the rims of the coverslip to increase the volume of the water meniscus.
? TROUBLESHOOTING
-
13| Place the mounting stage on the microscope motorized platform so that the coverslip is situated directly underneath the objective lens. Use the hydraulic height adjuster to move the microscope platform until contact is established between the objective lens and the water meniscus on the coverslip.
! CAUTION After adjusting the height of the microscope platform to its optimal setting, lock it in place to avoid drift and to ensure stability during imaging.
? TROUBLESHOOTING
-
14| Use wide-field epifluorescence illumination and the appropriate filters to visualize the fluorophores used on the mouse. Adjust the motorized x-, y- and z-plane controller to bring the area of interest into focus.
▲ CRITICAL STEP If the animal was tattooed, the borders of the tattooed area are easy to identify owing to increased contrast.
? TROUBLESHOOTING
15| Pull a lightproof curtain over the microscope to ensure that no light leakage can affect the imaging. Turn off all the lights in the room.
Intravital imaging ● TIMING 30 min–2 h for each mouse
16| For intravital imaging, use a LaVision TriM ScopeII (LaVision Biotec) multiphoton laser-scanning microscope equipped with a Chameleon Vision II (Coherent) Ti-sapphire IR laser. Set up to image with a 20× water-immersion lens (Zeiss W-Plan-APOCHROMAT, NA 1.0, working distance 2.3 mm).
17| Select the image size. For a 20× lens, the typical maximum scanned field area is 0.5 mm2. This area covers ~5–7 hair follicles.
18| Select the pixel density. A higher pixel density will result in an image with greater resolution, but it will require more time to acquire.
19| Select the scanning frequency (~600 Hz). A shorter pixel dwell time will result in a lower quality image, but it requires less time for frame acquisition and it reduces photobleaching.
20| Select the line average. Increased line averages can reduce noise from the background signal and produce a high-quality image, but it will take more time.
21| Choose fluorescence channel detectors and adjust the detector gain values. Our work has used band-pass emission filters of 610/75, 525/50 and 450/50 to receive red, green and blue channel emissions, respectively. Photomultiplier tube (PMT) values should be regulated depending on the intensity of the fluorophores. A high gain produces a strong signal but receives more background noise (a gain value of ~75% of maximum is recommended). PMT values should be regulated depending on the intensity of the fluorophores. The range of gain value is 0–100 and a high gain produces a strong signal, but it receives more background noise (a gain value between 60 and 80 is optimal).
22| Open the laser shutter.
-
23| Select the wavelength of the laser. For two-photon excitation, 940 nm is used for green-yellow fluorophores (e.g., YFP and GFP) and 1,040 nm is used for orange-red fluorophores (RFP, Tomato and mCherry).
! CAUTION Be careful to properly adjust the laser power. If the laser power is too high, it could burn the skin of the mouse. In general, less laser power is needed for short wavelengths (~800–900 nm) because of the higher laser power output. 1–15% power in 940 nm and 5–50% power in 1,040 nm provides a strong but nondamaging signal.
▲ CRITICAL STEP The combination of laser power and PMT gain determines the intensity of the signal. To receive the best quality of imaging, both values should be regulated.
▲ CRITICAL STEP SHG can be used to detect dermal collagen fibers in the skin without the use of specific fluorophores. The collagen is detected by SHG with a broad emission spectrum (~450–550 nm) and excited with a wavelength of double the range (~900–1,100 nm; ref. 18). Type I, II, III and V collagen form fibrils, but type IV forms sheets in basal laminae, and types VI and IX act as links34. In the adult skin, type I collagen is one of the major components of the dermal extracellular matrix, and it can be detected by SHG. However, type IV collagen in the basement membrane is undetectable18.
To place SHG emissions within the blue channel, an excitation wavelength of ~900–1,100 nm is required, which is double that of the destination channel19.
24| Set up the scan range. In this step, the starting point, endpoint and thickness of each optical section (step size) should be determined. Normally, the epidermis is set as the start point and the dermal papillae as the endpoint while using a step size of 2 μm. The number of images within a z-stack is determined on the basis of the scan range and step size. For a typical telogen follicle, the required depth to image the entire follicle is ~50–70 μm. The approximate maximum depth of a follicle during full anagen is 120–150 μm.
-
25| Set up the power increment for the area that you have chosen to image. The deeper the scan, the higher the power needed for full resolution.
? TROUBLESHOOTING
Time-lapse imaging (optional) ● TIMING 1–24 h
-
26| If you are recording time-lapse imaging (Supplementary Videos 2–4), define the time interval and the number of optical sections. The time interval should be longer than the time needed to acquire a single z-stack. In most cases, a time interval >5 min is recommended to prevent excessive tissue damage and photobleaching. In addition, the mouse condition can be checked during that interval. Both the wait time and the number of steps determine the total imaging time.
▲ CRITICAL STEP The time interval must be carefully monitored and balanced. Frequent intervals can make a high-frame movie smoother, but they can also result in decreased fluorescence from frequent exposure.
3D Tiling imaging (optional) ● TIMING 30 min–2 h
-
27| To image an area larger than a single scan field (0.5 mm2), set up the 3D tiling imaging option and adjust the grid size as desired.
▲ CRITICAL STEP In large-area imaging, a slightly tilted coverslip causes the edges of the grid to become unfocused. Ensure that the coverslip is as level as possible.
Laser ablation (optional) ● TIMING 30 min
▲ CRITICAL To eliminate a specific cell population of the skin after imaging, the laser-ablation method can be used (Fig. 6 and Supplementary Video 5). Laser ablation can be carried out as described in this section with the same optics as those used for acquisition.
28| After scanning a full field of view using the image acquisition settings, select a region of interest (ROI). For a small group of 10–20 cells, an area of 10 μm2 is typically selected at the center of this cluster.
29| Select the maximum pixel density (10 pixels per μm).
30| Select the lowest scanning frequency (200 Hz). These settings are aimed to achieve maximum laser dwell time within the ablation area.
31| Inactivate the PMTs or decrease their gain to zero to protect them from the high intensity of the laser pulse.
32| Select the wavelength of the laser. We have achieved the best results using 900 nm, but as laser ablation is mediated through thermal damage the use of a specific wavelength is not critical.
-
33| Set the laser power to 20% and initiate the scan of a single ROI frame.
? TROUBLESHOOTING
34| Switch to image acquisition mode and scan the entire field of view to assess the effect of the ablation scan.
-
35| If necessary, return to laser-ablation mode and repeat Steps 27–32 for the same ROI while increasing the laser power by 5% until the desired effects are achieved.
▲ CRITICAL STEP This protocol uses a pulsed femtosecond IR laser, which, combined with a high-NA objective lens, can deliver a substantial amount of energy to a very limited focal point within the tissue, which can result in the ablation of the cells that are situated within a small radius from this focal point. For a given ROI, the effective thermal damage will extend proportionally at a radius to all directions; therefore, the effective area of ablation is typically larger than the selected ROI. The process of identifying the appropriate dimensions for the ROI to target a selected population of cells requires some experimentation, which should be undertaken in a different area of the skin at the beginning of the ablation experiment. After identifying the appropriate laser power that is sufficient to ablate a given ROI with a single scan, these settings will give reproducible results in equivalent depths within the same tissue. ROIs in greater depths will generally require higher laser intensity to achieve the same effect.
Performing revisits (optional) ● TIMING 30 min–2 h
-
36| To revisit the same area of skin in the same mouse, prepare the mouse as detailed above (Steps 1–4).
! CAUTION Between imaging sessions, ensure that all procedures follow approved institutional and governmental animal protocols in order to maintain the proper health of the mouse. Immediately after imaging, it is recommended that the mouse be allowed to recover on a heating pad (~37–39 °C) before it regains consciousness and is returned to the housing facility. For 48 h after imaging, check to ensure that the mouse is consuming fluids and chow and producing feces and urine.
? TROUBLESHOOTING
-
37| Find the same area by using the tattoo and clusters of follicles as landmarks.
▲ CRITICAL STEP If the mouse skin is in the growing phase of the hair cycle, it is possible that there will be slight changes in hair follicle arrangement; however, the overall formation and patterns of hair follicle clusters will remain the same. Together with the tattoo and hair follicle clusters, this method can be used even when imaging hair follicles at different stages of growth and regression.
-
38| Repeat Steps 1–25 to set up and to image the animal.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
TABLE 3.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 8 | Nuclei in view are not all in focus | The coverslip is not placed evenly on the skin | Adjust the skin as needed using a pair of plastic tweezers. Position it on the spatula so that it is completely flat Adjust the height of coverslip so that it is just lightly touching the skin |
| Change in nuclei shape | Too much pressure from the coverslip | If hypoxic circumstances are prolonged, the nuclei will change shape from an ellipse to a sickle. Lift up the coverslip until blood flow is detected. Wait for ~30 min until the nuclei shapes go back to an ellipse and blood flow can be observed in the veins | |
| Vibration of skin | Insufficient contact with the coverslip | If contact between the skin and the coverslip is incomplete, the skin will vibrate from respiration. Lower the coverslip until the vibration stops and the imaging plane is even | |
| 11 | z-axis shifting | Movement of the mouse | If anesthesia is not suitable (the mouse is breathing heavily or awakening) or if the mouse position is not correct (if the spatula is too high or the mouth cone is tilted), there could be movement of the mouse |
| 12 | Images are dim and out of focus | Water has dried out on the coverslip | In the case of long imaging times, it is easy to dry out the water on the coverslip. Once the water is nearly evaporated, the imaging will dim and start to become unfocused. Add more water during the interval time, or if the interval time is too short, pause the recording and add more water |
| 13 | Images shift | Movement of the stage and/or the platform | If the screws of the spatula of the stage and/or microscope platform are not fastened properly, there could be gradual movement. Fasten all the screws tightly |
| 14 | Images are out of focus | Focusing is set to an area outside of the contact area | If the focusing is outside of the contact area, vibration will be observed. Change the x and y axes and find the appropriate contact region |
| 25 | Different fluorescence intensity between optical sections | Recorded long range of z-stack without adjusting laser power for depth | A deeper optical plane needs more laser power than the top of the skin. The laser should be attenuated through the stack when recording a long z-stack, such that the top of the stack receives more laser power. This option should increase laser power gradually. For example, in a K14H2BGFP mouse, the power settings are as follows: interfollicular epidermis = 2%, bulge = 3% and hair germ = 4% at 940 nm |
| Too much background noise | Laser power is too low | If the laser power is too low, it will not be sufficient to excite the fluorophore, and it will be difficult to distinguish the real signal from the background. Increase the laser power and/or the PMT value to increase the signal | |
| Laser power is too high | Excessive laser power can cause nonspecific autofluorescence in the hair shaft, in melanin and in dust on the epidermis. Decrease the laser power and keep the imaging region as clear as possible. In non-albino mice, try to image a less-melanin-heavy region | ||
| 33 | Target of laser ablation is not eliminated | Insufficient laser power | Insufficient laser power will not eliminate the target and will cause bleached fluorescence. Bleaching and ablation can be distinguished by autofluorescence. Increase the laser power until the target is successfully ablated, which is recognizable by the generation of autofluorescent protein particle aggregates, organized in a halo-like structure |
| Laser ablation causes tissue damage | Excessive laser power | Too much laser power will eliminate not only the target region but also the surrounding area. Start with a low laser power and gradually increase the power until the target area is successfully ablated | |
| 36 | The hair follicles being revisited are not all visible in the imaging area | Grid size is too small | During the growth phase of the hair cycle, the follicles may extend out of the original area selected for the tiling option. Expand the grid size as necessary in the direction in which the follicles are growing to ensure that all follicles of interest are captured |
● TIMING
Steps 1–3, mouse preparation for imaging: 15 min
Step 4, tattoo (optional): 1 min
Steps 5 and 6, mounting the mouse on the imaging stage: 5 min
Steps 7 and 8, mounting the coverslip on the skin: 5 min
Step 9, setting up the anesthesia nose cone: 1 min
Steps 10–15, connecting and aligning the stage to the imaging platform: 10 min Steps 16–25, intravital imaging: 30 min–2 h per mouse
Step 26, time-lapse imaging (optional): 1–24 h per mouse Step 27, 3D tiling imaging (optional): 30 min–2 h per mouse Steps 28–35, laser ablation (optional): 30 min
Steps 36–38, performing revisits (optional): 30 min–2 h per mouse
ANTICIPATED RESULTS
The TPLSM live imaging approach can be used for a wide variety of purposes to not only capture cell behaviors within intact skin but also to manipulate specific compartments of the tissue to probe their in vivo function. 3D imaging generated from optical z-stacks and automated mosaic tiling will provide a complete view of your cells of interest, as well as the surrounding tissue environment (Figs. 2 and 5d; Supplementary Video 1). With the minimal laser power required for a single image acquisition, this process can be iterated over a variety of time scales to generate time-resolved movies. These movies capture several types of cellular behaviors, such as cell migration, proliferation and death. This approach can be extended for revisits, as well as to determine the long-term behavior and fate of single cells. Simultaneous use of multiple cell type–specific fluorescent reports will allow the user to visualize how distinct cell populations coordinate their behaviors in vivo (Supplementary Videos 2–4). Users can also take advantage of the ablative function of the two-photon laser to remove and perturb cells or structures specifically in both space and time. This, in combination with imaging revisits, will allow the user to determine the functional role or requirement of the cells or structures under investigation (Fig. 6 and Supplementary Video 5).
Supplementary Material
Acknowledgments
This work is supported by The New York Stem Cell Foundation and grants to V.G. by the American Cancer Society, grant no. RSG-12-059-02; Yale Spore Grant National Cancer Institute, US National Institutes of Health (NIH) grant no. 1P50CA121974; and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), NIH grant no. 1R01AR063663-01. C.M.P. is supported by the Human Genetics Training Grant (HGTG) through Yale University. S.P. is supported by the James Hudson Brown-Alexander Brown Coxe Postdoctoral Fellowship. K.R.M. is supported by the NIH Predoctoral Program in Cellular and Molecular Biology, grant no. 5T32 GM007223, and is currently a National Science Foundation (NSF) Graduate Research Fellow. A.M.H. is supported by NIAMS Rheumatic Diseases Research Core Centers, grant no. 5 P30 AR053495-07. P.R. is a New York Stem Cell Foundation Druckenmiller Fellow and is supported by the CT Stem Cell Grant 13-SCA-YALE-20. V.G. is a New York Stem Cell Foundation Robertson Investigator. The authors gratefully acknowledge the laboratory of E. Fuchs (Rockefeller University) for providing the K14H2BGFP mice.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS C.M.P., S.P. and P.R. wrote the manuscript and prepared the figures. K.R.M. conducted experiments for figures. M.W., D.G.G. and A.M.H. helped develop and troubleshoot the TPLSM technique. V.G. oversaw and assisted all aspects of the protocol.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Morrison SJ, Spralding AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rompolas P, Greco V. Stem cell dynamics in the hair follicle niche. Sem Cell Dev Biol. 2014:25–26. doi: 10.1016/j.semcdb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lo Celso C, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nat Lett. 2008;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie Y, et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nat Lett. 2008;457:97–101. doi: 10.1038/nature07639. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida S, Sukeno M, Nabeshima Y. A vasculature-associated niche for undifferentiated spermatogonia in the mouse testis. Science. 2007;317:1722–1726. doi: 10.1126/science.1144885. [DOI] [PubMed] [Google Scholar]
- 6.Uchugonova A, Hoffman RM, Weinigel M, Koenig K. Watching stem cells in the skin of living mice noninvasively. Cell Cycle. 2011;10:2017–2020. doi: 10.4161/cc.10.12.15895. [DOI] [PubMed] [Google Scholar]
- 7.Li JL, Goh CC, Keeble JL, Win JS, et al. Intravital multiphoton imaging of immune responses in the mouse ear skin. Nat Protoc. 2012;7:221–234. doi: 10.1038/nprot.2011.438. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs E. Finding one’s niche in the skin. Cell Stem Cell. 2009;4:499–502. doi: 10.1016/j.stem.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuchs E. The tortoise and the hair: slow-cycling cells in the stem cell race. Cell Rev. 2009;137:811–819. doi: 10.1016/j.cell.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotsarelis G. Epithelial stem cells: a folliculocentric view. J Investig Dermatol. 2006;126:1459–1468. doi: 10.1038/sj.jid.5700376. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Clevers H. Coexistence of quiescence and active adult stem cells in mammals. Science. 2010;327:542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cotsarelis G, Sun RR, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329–1337. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 13.Jahoda CA, Horne KA, Oliver RF. Induction of hair growth by implantation of cultured dermal papilla cells. Nature. 1984;311:560–562. doi: 10.1038/311560a0. [DOI] [PubMed] [Google Scholar]
- 14.Muller-Rover S, et al. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Investig Dermatol. 2001;117:3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- 15.Greco V, et al. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell. 2009;4:155–169. doi: 10.1016/j.stem.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plikus MV, et al. Cyclic dermal BMP signaling regulates stem cell activation during hair regeneration. Nat Lett. 2008;451:340–344. doi: 10.1038/nature06457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Page ME, Lomard P, Ng F, Gottgens B, Jensen KB. The epidermis comprises autonomous compartments maintained by distinct stem cell populations. Cell Stem Cell. 2013;13:471–482. doi: 10.1016/j.stem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ. Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc. 2012;7:654–659. doi: 10.1038/nprot.2012.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campagnola PJ, Loew LM. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues, and organisms. Nat Biotechnol. 2003;21:1356–1360. doi: 10.1038/nbt894. [DOI] [PubMed] [Google Scholar]
- 20.Tumbar T, et al. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rendl M, Lewis L, Fuchs E. Molecular dissection of mesenchymal-epithelial interactions in the hair follicle. PLoS Biol. 2005;3:1910–1924. doi: 10.1371/journal.pbio.0030331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwan KM. Conditional alleles in mice: practical considerations for tissue-specific knockouts. Genesis. 2002;32:49–62. doi: 10.1002/gene.10068. [DOI] [PubMed] [Google Scholar]
- 23.Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stemberg N, Hamilton D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J Mol Biol. 1981;150:467–486. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]
- 25.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- 26.Feil R, et al. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci USA. 1996;93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 28.Metzger D, Clifford J, Chiba H, Chambon P. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc Natl Acad Sci USA. 1995;92:6991–6995. doi: 10.1073/pnas.92.15.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res. 1996;24:543–548. doi: 10.1093/nar/24.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Youssef KK, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol. 2010;12:299–305. doi: 10.1038/ncb2031. [DOI] [PubMed] [Google Scholar]
- 31.Means AL, Xu Y, Zhao A, Ray KC, Gu G. A CK19 (CreERT) knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis. 2008;46:318–323. doi: 10.1002/dvg.20397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nat Cell Biol. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 33.Rompolas P, Mesa K, Greco V. Spatial organization within a niche as a determinant of stem-cell fate. Nature. 2013;502:513–518. doi: 10.1038/nature12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox G, et al. 3-dimensional imaging of collagen using second harmonic generation. J Struct Biol. 2003;141:53–62. doi: 10.1016/s1047-8477(02)00576-2. [DOI] [PubMed] [Google Scholar]
- 35.Madisen L, Zwingman TA, Sunkin SM. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 37.Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127:4775–4785. doi: 10.1242/dev.127.22.4775. [DOI] [PubMed] [Google Scholar]
- 38.Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA. 1999;96:8551–8556. doi: 10.1073/pnas.96.15.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snippert H, et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–1389. doi: 10.1126/science.1184733. [DOI] [PubMed] [Google Scholar]
- 40.Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004;64:8804–8807. doi: 10.1158/0008-5472.CAN-04-2623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.