

Short abstract
Human disease genes differ significantly from their rodent orthologs with respect to their overall levels of conservation and their rates of evolutionary change. Rodent orthologs of human trinucleotide repeat-expansion disease genes were also found to contain substantially fewer such repeats.
Abstract
Background
Model organisms have contributed substantially to our understanding of the etiology of human disease as well as having assisted with the development of new treatment modalities. The availability of the human, mouse and, most recently, the rat genome sequences now permit the comprehensive investigation of the rodent orthologs of genes associated with human disease. Here, we investigate whether human disease genes differ significantly from their rodent orthologs with respect to their overall levels of conservation and their rates of evolutionary change.
Results
Human disease genes are unevenly distributed among human chromosomes and are highly represented (99.5%) among human-rodent ortholog sets. Differences are revealed in evolutionary conservation and selection between different categories of human disease genes. Although selection appears not to have greatly discriminated between disease and non-disease genes, synonymous substitution rates are significantly higher for disease genes. In neurological and malformation syndrome disease systems, associated genes have evolved slowly whereas genes of the immune, hematological and pulmonary disease systems have changed more rapidly. Amino-acid substitutions associated with human inherited disease occur at sites that are more highly conserved than the average; nevertheless, 15 substituting amino acids associated with human disease were identified as wild-type amino acids in the rat. Rodent orthologs of human trinucleotide repeat-expansion disease genes were found to contain substantially fewer of such repeats. Six human genes that share the same characteristics as triplet repeat-expansion disease-associated genes were identified; although four of these genes are expressed in the brain, none is currently known to be associated with disease.
Conclusions
Most human disease genes have been retained in rodent genomes. Synonymous nucleotide substitutions occur at a higher rate in disease genes, a finding that may reflect increased mutation rates in the chromosomal regions in which disease genes are found. Rodent orthologs associated with neurological function exhibit the greatest evolutionary conservation; this suggests that rodent models of human neurological disease are likely to most faithfully represent human disease processes. However, with regard to neurological triplet repeat expansion-associated human disease genes, the contraction, relative to human, of rodent trinucleotide repeats suggests that rodent loci may not achieve a 'critical repeat threshold' necessary to undergo spontaneous pathological repeat expansions. The identification of six genes in this study that have multiple characteristics associated with repeat expansion-disease genes raises the possibility that not all human loci capable of facilitating neurological disease by repeat expansion have as yet been identified.
Background
Human gene mutations resulting in specific disease phenotypes were first reported in the scientific literature over 50 years ago [1,2]. Since then, protein and nucleotide sequence changes associated with human disease have accumulated at a rapid rate. A large body of literature has appeared on human disease-associated mutations, normal sequence variation, and alterations that acquire pathological significance when combined with other deleterious alleles or second-site mutations. With this information compiled into organized databases [3,4], it is now possible to conduct large-scale, comprehensive analyses of human disease genes. Such studies acquire additional discriminatory power with the availability of multiple genome sequences from model organisms, as comparative studies can provide novel evolutionary insights into the selective relevance of genetic changes. In the present study, we have used a collection of nearly 1,200 human disease gene sequences to perform a large-scale analysis of gene and sequence conservation.
Investigation of evolutionary rates among large sets of genes has become feasible with the availability of the genome sequences of human, mouse and rat [5-8]. The degree of selective pressure to which genes have been subjected is reflected by the ratio of KA, the number of non-synonymous substitutions per non-synonymous site, to KS, the number of synonymous substitutions per synonymous site [9]. Hurst and Smith [10] have compared these ratios for 'essential' mammalian genes (that is, those that are lethal or infertile in genetic knock-out experiments) to those for genes that produce a viable and fertile phenotype when subject to genetic knock-out (non-essential genes). Using a sample size of 67 essential genes and 108 non-essential genes, these authors showed that essential genes manifested significantly lower KA/KS ratios than non-essential genes. Upon further analysis however, they found that immune-system genes, which have high KA/KS values, accounted for much of this effect since these loci were over-represented in the non-essential gene set. Analyses of evolutionary rates must therefore account for rate variation across different tissues.
Using a larger dataset (2,400 human-rodent orthologs and 834 rat-mouse orthologs) and EST information, Duret and Mouchiroud [11] observed that tissue-specific genes, on average, exhibited higher KA/KS ratios than genes expressed in most tissues (so-called 'housekeeping genes'). A more recent study of microarray data confirmed this finding, and demonstrated that much of this effect is explicable in terms of a correlation between a gene's tissue-specificity and the cellular localization of its encoded protein [12].
In this study, we have used genes predicted from the completed mouse, rat and human genomes, and a manually validated set of human disease genes. Our aims were three-fold. Firstly, we sought to determine whether human disease genes are collectively distinguishable, with respect to evolutionary conservation and evolutionary rates, from non-disease genes. Then we investigated whether genes ascribed to different pathophysiological systems exhibit significant differences in evolutionary rates. The results promise to be relevant for the consideration of different types of animal models utilized to investigate the mechanisms of human disease. Finally, we considered the category of human disease genes harboring expansions of trinucleotide repeats. For these genes, a moderate number of repeats is usually compatible with a normal phenotype, whereas further expansions are frequently associated with a neurological disease phenotype. We studied polyglutamine repeats in rat, mouse and human orthologous sequences to obtain an evolutionary perspective on the mechanisms of glutamine-repeat generation.
Results and discussion
Conservation of human disease genes in the rat genome
We considered 1,180 nuclear genes listed in the Human Gene Mutation Database for which missense or nonsense mutations have been reported to be associated with inherited disease [3]. Of these entries, 1,124 were successfully mapped to 1,112 Ensembl human genes (see Materials and methods). Analysis of the distribution of disease genes to human chromosomes (Table 1) indicated that chromosomes 21 and X were particularly enriched in disease genes. The basis for the enrichment of these genes on chromosome 21 is unclear, but the enrichment on the X chromosome is probably due to the obligatory expression of recessive phenotypes in males where recessive mutations are necessarily hemizygous. This unique feature of sex-linked inheritance results in the greater phenotypic expression of recessive alleles in carrier populations thereby facilitating both clinical phenotype identification, and genetic and mutational analysis.
Table 1.
Chromosome distribution of HGMD disease gene set
| Chromosome | Ensembl gene number | Disease genes | Disease gene percentage |
| 1 | 2449 | 115 | 4.70 |
| 2 | 1706 | 81 | 4.75 |
| 3 | 1329 | 57 | 4.29 |
| 4 | 1031 | 39 | 3.78 |
| 5 | 1138 | 56 | 4.92 |
| 6 | 1296 | 44 | 3.40 |
| 7 | 1269 | 51 | 4.02 |
| 8 | 860 | 36 | 4.19 |
| 9 | 1031 | 45 | 4.37 |
| 10 | 986 | 43 | 4.36 |
| 11 | 1568 | 77 | 4.91 |
| 12 | 1207 | 52 | 4.31 |
| 13 | 455 | 25 | 5.50 |
| 14 | 736 | 26 | 3.53 |
| 15 | 797 | 29 | 3.64 |
| 16 | 1084 | 50 | 4.61 |
| 17 | 1348 | 68 | 5.05 |
| 18 | 365 | 15 | 4.11 |
| 19 | 1557 | 55 | 3.53 |
| 20 | 704 | 25 | 3.55 |
| 21 | 192 | 15 | 7.81 |
| 22 | 469 | 25 | 5.33 |
| X | 869 | 83 | 9.55 |
Of these 1,112 Ensembl genes, 844 (76%) were found to have orthologous genes in the rat genome (November 2002 assembly), according to Ensembl [13]. This is a significantly greater proportion of Ensembl 1:1 orthologs than is found for the set of all Ensembl genes (46%). One reason for this difference might be a higher fidelity of predicting human disease genes and their rodent orthologs from their genome sequences, compared with other genes. This would be a consequence of the greater availability of transcript evidence, principally cDNA sequences, for disease genes. Imperfections in gene prediction and genome sequence assembly are the major hindrances to accurate orthology prediction.
We next wished to determine whether any of the 268 remaining human disease genes lacked a rat ortholog, perhaps as a consequence of either pseudogene creation or gene deletion in the rat lineage. Bearing in mind that certain regions of the rat genome sequence remain incomplete, that gene and orthology predictions are inexact, and that additional sources of sequence information are available, each of the remaining 268 human genes were aligned against the rat genome (assembly versions 2.0 and 3.1), EST, cDNA and protein sequences using BLAT [14] and BLAST [15]. Using the methods employed, we were able to assign orthology even if gene-duplication events had occurred within the human or rat lineages. Detailed inspection of alignments indicated that only six human disease genes appear to have no orthologous counterparts among available rat sequences.
Of the six missing orthologs, three with known function were found to be present in mouse sequences: orthologs of human genes HLXB9 (homeobox gene HB9), SGSH (N-sulfoglucosamine sulfohydrolase) and GP6 (glycoprotein VI, platelet) with LocusLink identifiers 3110, 6448 and 51206, respectively. Hence, these genes might yet be found in the portion of the rat genome that still remains to be sequenced. Two of the three genes missing from both mouse and rat appear to have become pseudogenes relatively recently given that there are known hamster orthologs [16,17]. These include cholesteryl ester transfer protein (CETP), which is associated with the deficiency in CETP activity described in rat and mouse [16] and Fuc-TIII (FUT3), an α-(1-3)-fucosyltransferase involved in the synthesis of milk oligosaccharides [18]. A third gene, KAL1 (encoding the Kallman syndrome, or anosmin-1, protein) is entirely absent from the sequenced portions of the rat and mouse genomes. However, rodent Kal-1 genes may yet be found in the pseudoautosomal regions of their genomes [19]. Kal-1 is present in Caenorhabditis elegans [20], amphibians, fish and birds, and its rat and mouse orthologs have been reported to be detectable using an antibody to the human KAL1 gene product [19].
Thus, of the 1,112 human disease genes examined, evidence that all are represented as functional genes in the rat genome was found, except for the six genes discussed above. Clearly, the set of genes identified as being associated with inherited disease in humans is highly conserved in the rat genome.
Mapping human disease mutations to rat genes
We compared sequence variants that result in human inherited disease with amino-acid substitutions that have accumulated since the common ancestor of rat and human. In all, 12,549 missense mutations were mapped to codons in the pairwise alignments of Ensembl human and rat 1:1 orthologs. As expected, the majority (89.6%) of these sites contain the same amino acid in both human and rat wild-type sequences. This exceeds the 82.2% of all sites that are identical for all 1:1 ortholog pairs [8], indicating that such sites are subject to a greater degree of purifying selection. Of the remaining 10.4% of sites, 4.6% were unable to be aligned with precision, while 4.9% exhibited amino-acid substitutions in the rat ortholog that differed from the human disease missense mutations.
The remaining 104 sites each contained an amino acid that is present for both the human disease-associated variant and the rat wild-type sequence (see Additional data file 1). We considered whether these instances might represent genetic variations in human for which associations with disease were erroneously noted, were more tenuous or were unvalidated. Detailed examination of the literature revealed that there was compelling experimental evidence for a direct causal relationship between the reported sequence variant and human disease for only 15 of these sites (Table 2). Of these 15, the rodent sequence is not likely to be in doubt in eight cases because the mouse sequence is identical to the rat at this site. If the establishment of the genotype-phenotype relationship is valid in these cases, then the rat may not represent an appropriate model for studying the human disease processes associated with these variants. On the other hand, the results may represent opportunities to define alternative pathways or differences in protein structure that could be utilized in therapeutic intervention. As noted by Gao and Zhang [21], the most likely explanation for fixation of human disease-associated mutations in the mouse is the presence of other compensatory changes suggesting that potentially interesting structural or functional insights may be revealed by these cases.
Table 2.
Instances where a substituting amino acid in a human disease mutation is identical to the wild-type sequence of the rat genome
| Ensembl reference | LocusLink identifier | Codon start | Nucleotide change | Amino acid change | Disease |
| ENSP00000318731 | 355 | 225 | ACA-AAA | Thr-Lys | Autoimmune lymphoproliferative syndrome |
| ENSP00000260947 | 580 | 295 | AAT-AGT | Asn-Ser | Breast cancer |
| ENSP00000256993 | 4607 | 59 | cACA-GCA | Thr-Ala*§ | Cardiomyopathy, hypertrophic |
| ENSP00000253496 | 2161 | 398 | CGG-CAG | Arg-Gln | Factor XII deficiency |
| ENSP00000326824 | 114548 | 198 | cGTG-ATG | Val-Met* | Familial cold autoinflammatory syndrome |
| ENSP00000324427 | 4653 | 445 | GCA-GTA | Ala-Val | Glaucoma 1, open angle |
| ENSP00000322421 | 3039 | 68 | AACg-AAA | Asn-Lys | Hemoglobin variant |
| ENSP00000298599 | 5979 | 251 | cGAG-AAG | Glu-Lys*§ | Hirschsprung disease |
| ENSP00000298599 | 5979 | 654 | tGCC-ACC | Ala-Thr | Hirschsprung disease |
| ENSP00000291550 | 875 | 354 | gGTG-ATG | Val-Met* | Homocystinuria |
| ENSP00000250087 | 23746 | 302 | CGC-CTC | Arg-Leu* | Leber congenital amaurosis IV |
| ENSP00000273783 | 8893 | 113 | CGC-CAC | Arg-His* | Leukoencephalopathy with vanishing white matter |
| ENSP00000294717 | 24 | 1898 | CGC-CAC | Arg-His* | Macular degeneration, age related |
| ENSP00000310389 | 6622 | 53 | gGCA-ACA | Ala-Thr*§ | Parkinson disease |
| ENSP00000233139 | 6716 | 227 | CGA-CAA | Arg-Gln | Steroid-5 alpha-reductase deficiency |
*Cases where the mutation is also identical to the wild-type mouse sequence. §These three cases were previously reported as being present in wild-type mouse sequences [7]).
Nucleotide substitution rates
We sought to investigate whether synonymous and non-synonymous nucleotide substitution rates differ between disease genes and other genes. We calculated the KA/KS ratio for each human and rat 1:1 ortholog pair. By dividing ortholog pairs into a set that contained known human disease genes, and a set that contained genes not known to be associated with disease, we were able to compare their KA/KS distributions. Only a marginally significant difference was found between these two distributions using a Kolmogorov-Smirnov test (P = 0.035) (Figure 1a). This implies that selective pressures have been applied relatively uniformly between these two gene classes. However, a highly significant difference (P = 9.4 × 10-8) was observed between the KS distribution of ortholog pairs containing human disease genes, and the KS distribution of pairs not containing disease genes (Figure 1b). A smaller difference (P = 3.9 × 10-4) was found between the KA distributions (Figure 1c).
Figure 1.
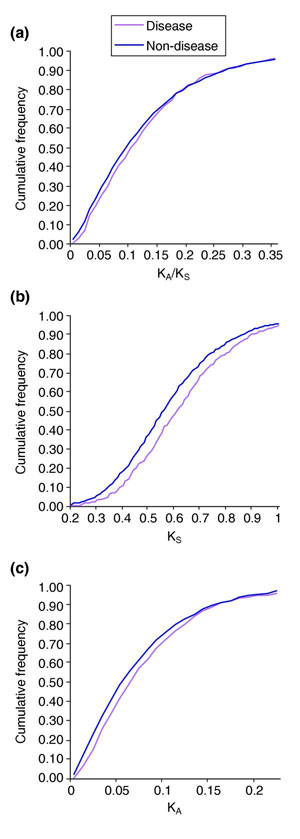
KA/KS, KS, and KA distributions of ortholog pairs for disease versus non-disease genes. (a) The KA/KS ratio, (b) KS, the number of synonymous substitutions per synonymous site, and (c) KA, the number of non-synonymous substitutions per non-synonymous sites, were calculated for 1:1 human:rat orthologs for a set containing human genes associated with disease and a set containing human genes not known to be associated with disease.
We considered whether the highly significant difference between the two KS distributions arose from a small number of outlier disease genes associated with high KS values. However this appears not to be the case since removal of the top 10% of data points in both datasets did not reduce the divergence of the two distributions (data not shown).
Recently, Smith and Eyre-Walker [22] calculated evolutionary rates for rat-human orthologs of 387 human disease genes and 2,024 non-disease genes, taken from the Duret and Mouchiroud [11] and Jimenez-Sanchez et al. [23] datasets, respectively. They noted that KA/KS and KS values were significantly elevated for disease genes compared with non-disease genes. Although the findings relating to KS are fully supported by our study, our results indicate only a modest difference between KA/KS distributions of disease, and non-disease, genes (human-mouse: P = 0.044; human-rat: P = 0.032), rather than the 24% difference (P < 0.0001) reported by Smith and Eyre-Walker [22]. We attribute this difference to the variation that can arise from sampling error when smaller gene sets are employed.
One interpretation of the findings reported here is that substitutions at non-synonymous sites have indirectly affected silent substitution rates [11,24]. However, such an effect is unlikely to be highly pronounced since the significance of the KS distributions' difference was several orders of magnitude higher than that of the KA/KS distributions. The finding rather suggests that human disease gene sequences, and their rat orthologs, have mutated faster than their non-disease counterparts. If so, then it would appear that disease genes differ from other genes in one respect: they are more frequently encoded in hypermutable genomic regions. One possibility that could account for an elevated mutation rate is if the disease gene set were to contain a disproportionately lower number of genes expressed in germ cells. This is because mutations in such genes might be expected to be more frequently repaired by transcription-coupled repair [25,26]. Another possibility is that disease genes are more prevalent in genomic regions that suffer elevated mutation rates for other, as yet unknown, reasons. Certainly, neutral rates have been found to vary significantly between distinct genomic regions [27].
Human disease genes and pathophysiology-based disease systems
A sufficient number of human disease genes have now been characterized in adequate detail to permit grouping them by disease system categories for large-scale analysis. We therefore categorized 1,178 human disease genes according to which organ or pathophysiological system the disease best fitted with respect to a specific pathological variant (Additional data file 2). For example, adenosine-deaminase deficiency is caused by ADA gene mutations that reduce or eliminate enzyme function; these alterations result in frequently fatal severe combined immunodeficiency owing to the toxicity of the accumulating substrates, adenosine and 2'-deoxyadenosine. Considering both the nature of the mutation and the gene it alters, as well as the impact of the resulting disease, this gene is categorized under both metabolic- and immune-disease systems. Among the disease-geneset, 889 genes were categorized into a single disease system whereas 289 genes were categorized into two systems.
Figure 2 delineates the 16 disease categories employed and depicts the number of disease genes in each category. 'Neurological' and 'metabolic' categories together comprised almost 50% of the characterized human disease genes. For some disease systems, few genes directly causing disease have been defined. For example, only 17 genes are currently known to cause pulmonary disease when mutated. Small numbers of genes identified for a given disease system could be a reflection of the complexity of the genetic contribution to disease in that system, particularly for those disease systems requiring large-scale studies to identify genetic factors (for example, [28] for asthma, a pulmonary disease). Thus, it may be that disease systems with few characterized genes may have a greater number of contributory (or modulating) genes associated with complex inheritance patterns, relative to other disease system categories.
Figure 2.
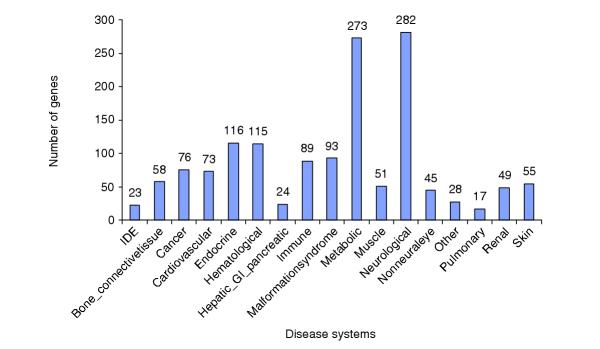
Human disease gene distribution across pathophysiology systems. The horizontal axis represents different disease systems. The vertical axis represents the number of genes present in each disease systems; these numbers are provided at the top of each bar. IDE, insufficient disease evidence.
Selection mechanisms acting on human disease genes in the rat and mouse
Next we investigated whether selection has acted differentially on human disease genes of the 16 disease-system categories. We determined the human-rat and human-mouse distributions of KA/KS ratios for these categories; median KA/KS values are presented in Figure 3. We present median, rather than mean, KA/KS values as these values are not normally distributed. For the same reason we employed the multi-level, non-parametric Wilcoxon-Kruskal-Wallis analysis to identify the significance of differences between categories. We then performed two-level comparisons between each category and the remaining samples. The results of these analyses are shown in Figure 4. To distinguish between disease systems with similar results, two closely scoring systems were tested against each other. For example, when neurological-system genes were compared directly with malformation-syndrome system genes, no significant difference was observed. However, immune-system genes exhibited significantly higher KA/KS values than hematological-system genes (data not shown). Based on these results, we conclude that, on average, immune system disease genes exhibit the highest KA/KS ratios between rat and human, whereas neurological and malformation-syndrome system genes have the lowestKA/KS ratios. Similar results were found for the analysis based on human-mouse orthologs (gray bars in Figures 3 and 4).
Figure 3.
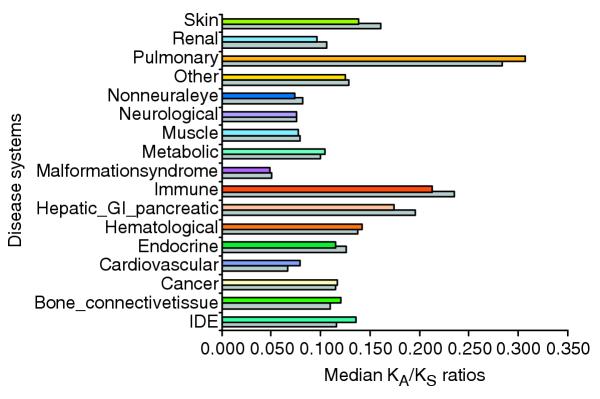
Median KA/KS ratios for rat and mouse orthologs of human disease genes. Median KA/KS values for each disease category are depicted for rat (color) and mouse (grey) demonstrating differences by disease category. The disease categories exhibiting the greatest purifying selection, the neurological and malformation-syndrome disease systems, show the lowest median KA/KS ratios.
Figure 4.
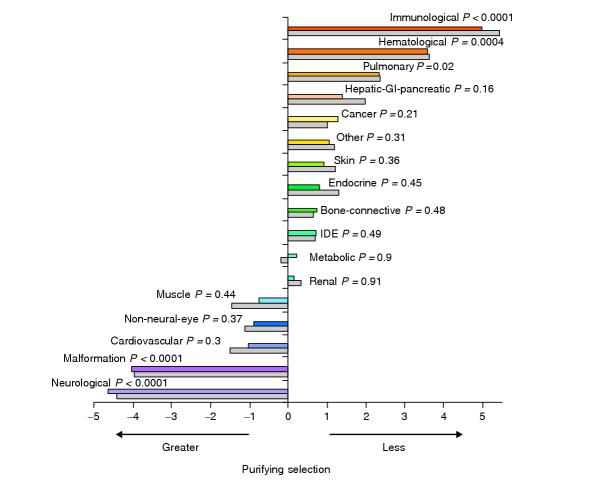
KA/KS differences by disease system. Significance of KA/KS differences determined by Wilcoxon analysis indicate that both rat (color) and mouse (grey) demonstrate significantly strong purifying selection for the orthologs of the neurological and malformation-syndrome disease categories. The immunological, hematological, and pulmonary disease systems demonstrated significantly lower conservation. Disease categories are listed along the vertical axis in the order of standardized score from low to high. The P value is indicated above the bars in the figure; P values of less than 0.05 are considered statistically significant.
We find that significant differences exist between the KA/KS ratio distributions for the different pathophysiological classes. For example, within the neurological-disease system, 95% of the genes were subject to purifying selection (KA/KS < 0.25). This is in contrast to immune-system disease genes where only 65% were found to exhibit such low rates. Thus among all pathophysiological categories, it would appear that the genes of the neurological-disease system have been constrained by purifying selection the most, whilst those of the immune system have been constrained the least. No genes in our study met the strict criterion for positive selection (gene-averaged KA/KS ratios > 1.0), although adaptive evolution is more likely to have occurred at single sites for genes with ratios closer to 1.
In contrast to the findings for KA, KS and KA/KS comparing complete disease and non-disease gene sets, no significant differences in KS were found among different disease systems despite known differences among tissues [12]. The significant differences identified in KA/KS ratios are likely to reflect differences in non-synonymous substitution rates (KA) across different disease systems.
We next investigated whether these variations in KA/KS ratios either arose from associations between physiology and organs or tissues, or were due to intrinsic properties of the human disease gene set under study. We studied two sets of genes: 586 sequences that were retrieved from the Human Proteome Survey Database (HPSD) [29] using gene ontology (GO) terms associated with immune function; and 761 genes retrieved using GO terms with neurological associations. Rat, mouse and human 1:1:1 orthology relationships were available for 200 of these 586 'immune' category genes (Additional data file 3) and for 304 genes of the 761 'neurological' genes (Additional data file 4); orthologs of human disease genes were disallowed from these sets.
We found no significant difference between the human-rat or human-mouse KA/KS distributions of the human 'immune' and human 'immune disease' gene categories (P = 0.0897). We then combined the disease and non-disease immune gene sets and determined the KA/KS values of this larger immune-system gene set compared to a set that included 7,641 non-immune control genes that have 1:1:1 orthologous relationships in mouse, rat and human. This control experiment confirmed our initial findings that the genes involved in the human immune response contain, as a set, fewer members subject to purifying selection than controls not involved in immune function. Utilization of the larger gene set in this analysis reduces the likelihood that results derive from sampling error. We therefore conclude that elevation of KA/KS values for disease genes of the immune system is a general property of immunologically relevant genes, rather than being specific to immune-system disease genes. This conclusion is consistent with findings from studies demonstrating that lymphocyte- or thymus-specific genes evolve relatively rapidly [11,12].
Similar results were obtained for the human neurological-disease genes (data not shown): the significant KA/KS ratio differences evident in Tables 3 and 4 are similar for human genes involved in neurological processes and are not a characteristic restricted to human neurological-disease genes. Equivalent controls comparing genes associated with neurological processes compared to a control group of all non-neural orthologs similarly confirmed that the KA/KS ratio differences are significant but are observed for all neurologically relevant genes.
Table 3.
Conservation index differences by disease system for model organisms
| Disease system | Mouse | Rat | Fish | Fly | Nematode | Yeast |
| IDE | -0.46 | -0.49 | -0.91 | 0.04 | -0.52 | -0.42 |
| Bone_connectivetissue | 1.14 | 0.04 | -0.85 | -1.70 | -2.42 | -0.26 |
| Cancer | -1.01 | -1.42 | -1.26 | -0.85 | -0.56 | -0.01 |
| Cardiovascular | 0.90 | -0.15 | -1.33 | -1.70 | -0.72 | -1.74 |
| Endocrine | -0.96 | -0.30 | 0.19 | -1.34 | -1.81 | -2.26 |
| Hematological | -5.05 | -4.96 | -0.68 | -0.59 | -0.21 | 0.59 |
| Hepatic_GI_pancreatic | -2.41 | -2.67 | -0.07 | -1.01 | -1.05 | -1.36 |
| Immune | -6.93 | -5.52 | -5.81 | -3.42 | -1.52 | 0.05 |
| Malformationsyndrome | 4.17 | 3.06 | 0.09 | -2.88 | -3.67 | -2.55 |
| Metabolic | 2.46 | 2.88 | 4.34 | 5.64 | 7.43 | 2.18 |
| Muscle | 3.24 | 2.24 | 0.87 | -1.17 | -0.66 | 1.42 |
| Neurological | 4.95 | 4.91 | 1.93 | 1.24 | -0.30 | 0.55 |
| Nonneuraleye | 0.46 | 0.43 | 0.04 | 0.58 | -1.00 | -0.07 |
| Other | -0.87 | -0.46 | -1.46 | -0.13 | 0.62 | 2.03 |
| Pulmonary | -3.67 | -3.77 | -0.34 | -0.46 | -3.03 | -1.18 |
| Renal | -1.64 | -1.26 | -0.01 | 0.61 | 0.01 | -0.52 |
| Skin | -1.52 | -0.86 | -0.25 | -0.17 | -0.09 | 0.04 |
Standardized scores from multi-level Wilcoxon analyses are shown. Categories that contribute to the global differences among systems with a sample size greater than 2% of the total are shown in bold, and were also used to generate Figure 6.
Table 4.
Human genes with poly-glutamine repeat tracts expanded in the human lineage and not at present known to be associated with disease
| Gene name | Gene symbol | Ensembl reference | LocusLink identifier |
| CAGH3 transcription factor | TNRC3 | ENSG00000179637 | 10292 |
| DNA polymerase gamma subunit 1 | NFYC | ENSG00000140521* | 4802 |
| Decapping enzyme | DCPIB | ENSG00000151065* | 196513 |
| Nuclear receptor coactivator 3 | NCOA3 | ENSG00000124151 | 8202 |
| Retina-derived POU-domain factor-1 | RPF-1 | ENSG00000106536* | 11281 |
| Retinoic acid induced 1 isoform 2 | RAI2 | ENSG00000108557* | 10742 |
*EST (brain).
From these studies, we conclude that KA/KS ratios differ significantly for genes involved in either neurological or immune processes as compared with the set of all genes examined (See Materials and methods). Although less significant, differences in KA/KS ratio distributions were also identified for three other gene sets: the malformation syndrome, pulmonary and hematological categories. Genes in the neurological and malformation-syndrome systems display, on average, lower KA/KS ratios, whereas genes of the other three categories have, on average, higher KA/KS ratios. Thus, we conclude that the pathophysiological system differences we observe derive from organ, tissue and physiological characteristics rather than arising from properties unique to disease genes and their potential impact on fitness. Although these findings are not specific to genes associated with human disease, they could influence the selection of animal models used to investigate human disease.
Functional-annotation distribution by human disease system
Functional annotations can be examined in large-scale studies through the use of text mining and analysis tools. To this end, we considered whether GO terms [30] and domain terms were over- or under-represented for different disease systems. Results demonstrate that GO terms associated with human disease genes demonstrate significant differences in distribution across pathophysiological systems; for example, annotations associated with cancer (DNA repair, cell proliferation, protein kinase and nucleus) were significantly over-represented in this disease-gene category (see Figure 5). Similarly, membrane protein, G protein-coupled receptors (GPCR) and ion transport protein annotations were more frequently associated with neurological disease genes. Figure 5 depicts over- or under-represented terms identified for each pathophysiological system. Although many of these terms are consistent with our current understanding of disease processes, text mining and analysis methods have the advantage of defining comprehensive search profiles that can be applied to genes of unknown function. From this analysis, we demonstrate that domain, functional and gene-family annotations are non-uniformly distributed across the pathophysiology categories we have utilized and that these annotations closely match what would be expected for current knowledge regarding the unique disease processes that fall within the pathophysiology underlying human disease.
Figure 5.
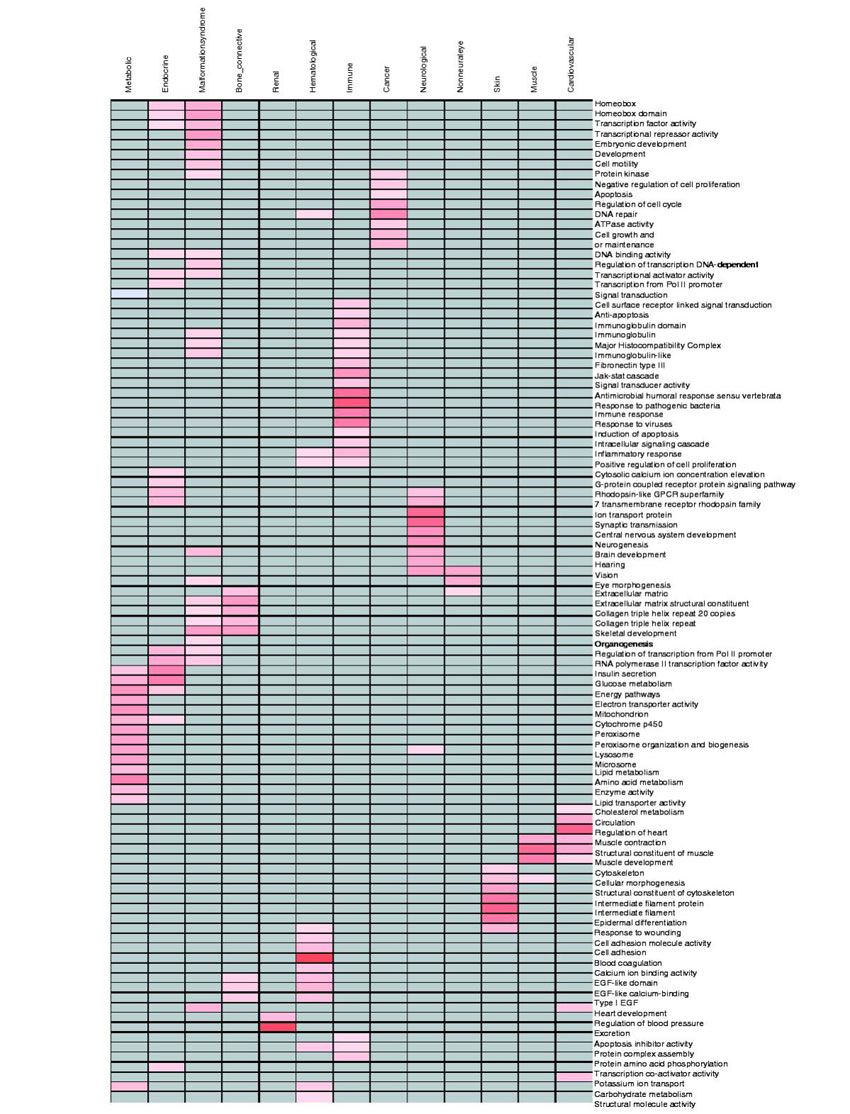
Functional annotation distribution by disease system. Over-representation of gene ontology annotation in different disease systems. Only records with P value ≤0.05 and both expected and observed frequency ≥2%, and with number of records ≥5, are shown. Over-represented functions are labeled red with the color gradient representing the deviation in value. The darker the color, the greater the deviation observed.
We considered subsequently whether the KA/KS differences among pathophysiology system datasets described earlier arose from over- or under-representation of specific domains, functions or evolutionary families. For this we considered GO terms [30] and domain terms. Results indicated that domain names, or their annotations, did not account for differences among median KA/KS values among pathophysiological systems. Thus, although gene-family, domain, and other functional categories are non-uniformly distributed across the pathophysiology groups, their distribution is not the underlying cause of the KA/KS differences we observe.
Conservation of human disease genes in other model organisms
In addition to rodents, other animal models have also been extensively used in the study of human diseases (see review [31]). Given the utility and lower research costs for non-mammalian model organisms, we wished to determine the level of conservation of disease genes in these established models. We thus extended our analysis of rodent orthologs of human disease genes to a broader range of organisms including representative genomes from fish, nematode, fly and yeast.
Conservation metrics were selected for this analysis because comparisons among more distantly-related organisms typically identify multiple substitutions per site, disallowing calculation of KA/KS values from sequence pairs. We defined a conservation index (CI, also known as a score density) as the length-normalized amino-acid similarity between a sequence pair (see Materials and methods). We predicted the number of orthologs and quantiles of CI in each model organism species (Additional data file 5). We then compared CI in different disease systems for each of these organisms. Non-parametric methods were used to calculate the standardized score for each system in each organism similar to those applied in our previous analyses.
With a 16-level Wilcoxon analysis, significant differences between disease systems were identified for each of the species tested. Of the 16 disease systems, five are the main contributors to this difference: immune, hematological, metabolic, neurological and malformation-syndrome. The CI analysis (Table 3, Figure 6) recapitulates the findings from human-rodent KA/KS results. The exception is that metabolic genes appear to be evolving more slowly in invertebrates than in vertebrates.
Figure 6.
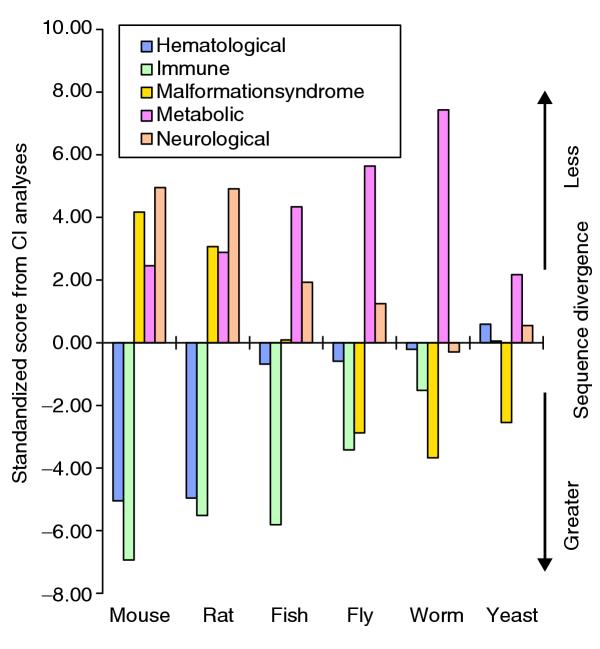
Disease gene system conservation in model organisms. The five disease systems in which significant conservation differences are found (hematological, immune, malformation syndrome, metabolic and neurological) are plotted on the horizontal axis for the six different model organisms (mouse, rat, fish, fly, nematode and yeast). The vertical axis represents standardized score from Wilcoxon analyses for conservation index. The greater the score, the more conserved the disease system.
Thus, for the study of human diseases of immune and hematological systems, primate or human cell models would probably be most suitable. Rodent models are likely to be best suited for studies of genes in neurological, malformation-syndrome and metabolic categories. Neurological and metabolic genes are sufficiently well conserved that fly or fish models are appropriate, whereas the yeast and worm, in general, are perhaps best suited as models of metabolic diseases given the overall lower conservation found for other categories in our study.
These findings parallel previous studies [32,33] that concluded that Drosophila is a good model organism for the study of genes in neurological and metabolic diseases, malformation syndromes and cancer. However, these studies based on 287 human disease genes categorized into 10 pathophysiological systems used the percentage of orthologs present per category to determine significance. By contrast, our study, with a substantially larger disease-gene set and more quantitative analysis, concluded that Drosophila is likely to have more limited utility as a model for the study of human cancer processes.
Amino-acid-repeat expansions associated with human disease
Glutamine expansion is associated with a number of different neurodegenerative disorders. In these diseases, long poly-glutamine tracts result from the expansion of CAG triplets by trinucleotide slippage. To obtain a general picture of poly-glutamine distribution and conservation in mammals, we compared poly-glutamine tracts in human-rat, human-mouse and rat-mouse ortholog pairs. We used aligned sequences to map equivalent (that is, orthologous) repeats and considered tandem repeats either of length 5 or longer, or of length 10 or longer ('very long repeats'). The two rodent species contained a slightly lower number of glutamine repeats than humans (85-88% of the number found in humans), in accord with the generally lower frequency of tandem amino-acid repeats in rodents as compared to humans [34]. For very long glutamine repeats (more than 10 residues), we identified 40 repeats in human and 39 in mouse in the human-mouse comparison; 41 in human and 58 in rat, in the human-rat comparison; and, 83 in rat and 41 in mouse in the rat-mouse comparison. Thus, among very long repeats, an excess in human sequences was not detected and rat sequences contained more repeats than mouse sequences. The number of human glutamine repeats (repeat length 5 or longer) conserved in rat and in mouse was roughly 55% in both cases, slightly higher than the general human repeat conservation level in rodents (46.5% for rat and 52% for mouse).
We next compared glutamine-repeat length differences among orthologous sequences containing very long human glutamine repeats (Figure 7). Human glutamine-expansion-disorder proteins all contain poly-glutamine tracts of 10 or longer in the wild-type protein, except the androgen receptor, which contains a repeat length of 5. In the case of disease-associated genes, except for Machado-Joseph disease protein (MJD), all repeats in the rat and mouse orthologs were less than half the size of the human repeats (that is, those lying below the line in Figure 7), an unexpected finding. Another characteristic of disease-associated genes was that the region encoding the repeat always contained a long CAG tract (repeat length of 8 or longer). In addition, most of the disease-associated genes also contained repeats of other amino-acid types. A group of other genes not known to be associated with disease were found to share these same characteristics (Table 4). Since these genes may also be subject to triplet-repeat expansion in the human/primate lineage, they could be investigated to determine if they are also involved in human disease on the basis of their identification in this study. Of special interest are the four that show EST support for gene expression in the brain (identified by * in Table 4) as these could potentially be associated with neurological disease.
Figure 7.
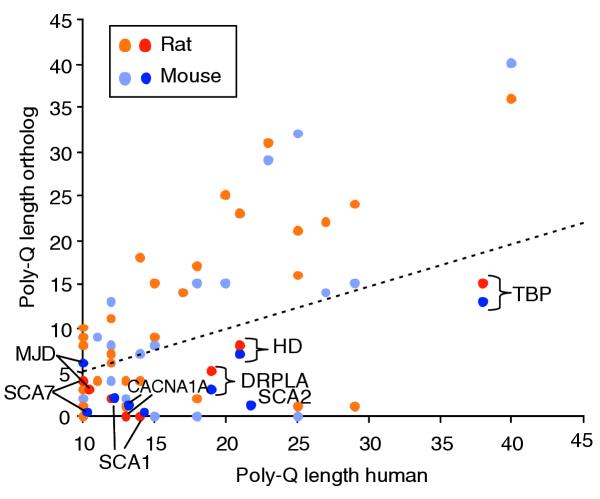
Poly-glutamine repeat length comparison between human-rat and human-mouse orthologous proteins. Comparison of the poly-glutamine length between human-rat orthologous proteins (light orange, dark orange) and human-mouse orthologous proteins (light blue, dark blue). Dark orange and dark blue correspond to repeats in genes associated with repeat-expansion disease in humans: SCA1, spinocerebellar ataxia 1 protein, or ataxin1; SCA2, spinocerebellar ataxia 2 protein; SCA7, spinocerebellar ataxia 7 protein; MJD, Machado-Joseph disease protein, or voltage-dependent calcium channel gamma-1 subunit; CACNA1A, spinocerebellar ataxia 6 protein, or calcium channel alpha 1A subunit isoform 1; DRPLA, dentatorubro-pallidoluysian atrophy protein; HD, Huntington's disease protein, or huntingtin; TBP, TATA binding protein or spinocerebellar ataxia 17 protein. In the case of SCA2 the rat orthologous sequence did not contain the human amino-terminal region, wherein the repeat is located. Points below the diagonal line correspond to a repeat length that is more than double in humans versus rodents.
The comparison of poly-glutamine length in the three mammalian species studied has shown that human disease genes that are associated with glutamine expansion are part of a larger group of genes likely to have experienced repeat expansions in the primate lineage. Examination of CAG and CAA codon repeats in this dataset confirms that lineage-specific glutamine repeats are associated with long CAG tracts whereas those conserved among different lineages tend to be encoded by a mixture of CAG/CAA codons [35]. Comparisons of numbers of very long poly-glutamine repeats also indicate that the rate of glutamine expansion in the rodent lineage may be comparable to that in the human lineage, and that rat sequences may be particularly prone to accumulate long repeats, a desirable feature for transgenic models of triplet-repeat expansion-associated disease. Such models already exist for several such diseases [36-38]. This is in addition to the advantage of using the rat, as opposed to the mouse, as an animal model for the investigation of neurological disorders for in vivo imaging studies because of its larger brain size [38].
Conclusions
Almost all human disease genes have orthologous counterparts in rodent genomes. The set of these disease genes does not differ greatly from the set of other genes with respect to KA/KS ratios although significant differences in synonymous substitution rates (KS) were observed. This suggests that human disease gene sequences and their rat orthologs may have mutated faster (or may have been repaired less efficiently) than their non-disease counterparts. Although the two KS distributions are significantly different, there is considerable overlap between them; the median difference between disease and non-disease distributions (0.05) is significantly smaller than one standard deviation (0.20). This means that the KS value of a particular gene, by itself, is not likely to be a sufficient indicator of whether it is, or is not, associated with disease.
Rodent orthologs of the gene set associated with neurological function exhibit the greatest conservation and are primarily subject to purifying selection. The highest KA/KS ratios were observed for genes that function in the immune system indicating that these genes are under less purifying selective pressure. This finding would be expected if host-pathogen co-evolution drives divergence by pathogen specificity within species. If sequence divergence were to be coupled to functional divergence, then this could suggest that rodent models of human neurological disease are more likely to faithfully represent human disease processes than rodent models of immune disease. Rodent models of human diseases in the immune-, hematological- and pulmonary-system pathophysiological categories should thus be validated particularly carefully before extrapolating from rodent studies to human.
Investigation of repeat-expansion disease genes led to the observation that all rodent homologs of these human disease genes bear shorter poly-glutamine repeat lengths. Furthermore, glutamine repeats in the human disease genes are mostly encoded by long CAG tracts. Rat-mouse-human comparative analysis also identified a number of human genes that, although not known to be associated with disease, share the same repeat characteristics as human disease-associated genes. These genes should be further investigated as potential disease candidates; of special interest are the four for which EST evidence indicates gene expression in the brain. Spontaneous neurological diseases arising through repeat-expansion mutations have not been identified in either rat or mouse laboratory strains or in natural populations. This could be due to ascertainment bias of rare events in rodent colonies or it is also possible that these orthologs fail to achieve a 'critical repeat threshold' required to trigger these mutational mechanisms. With the current successful development of rodent transgenic models using human disease gene constructs, this possibility can now be directly investigated. It will also be instructive to define the normal variation of rodent repeat lengths in natural populations for these genes to determine whether the variation in repeat numbers associated with a normal phenotype parallels that observed for human.
Materials and methods
Validation of disease role and assignment of disease-system annotation
The development of well-curated gene sets is an essential step for genome-scale disease gene analysis. The starting point for the present study was the Human Gene Mutation Database (HGMD) (February 2003 release) [3]. Beginning with 1,178 disease genes in this database, each gene was checked for at least one primary literature reference to confirm that it represented a bona fide gene in which a mutation had manifested an experimentally confirmed disease-association. During the annotation process, genes that did not meet this criterion were placed in an IDE category (insufficient disease evidence) but were not eliminated from the dataset. Thus, all genes that were placed into pathological categories were independently validated for disease association from the literature. Once a gene had passed this validation step, it was placed into one or more categories using the categorization method of Rubin et al. [32] with minor modifications. Thus each gene was assigned to one of the following categories: cancer, cardiovascular, endocrine, hematological, immune, malformation-syndrome, metabolic, neurological, pulmonary, renal or other. However, owing to the larger number of pathological systems represented for the genes categorized in this study, the following categories were added to those used in the Drosophila study [32]: skin, bone-connective tissue, muscle, hepatic-GI-pancreatic, and nonneural eye. Annotation categories were combined where they overlapped functionally (for example, bone-connective tissue and hepatic-GI-pancreatic). Briefly, the annotation method was to read the disease gene entry in the Online Mendelian Inheritance in Man (OMIM) database [4] to see if the pathophysiology category was identified in the synopsis in sufficient depth to make an assignment. This assignment was then confirmed using standard medical texts covering internal medicine, pathology and infectious disease. Annotations were independently determined by at least two individuals and then all genes with discrepant annotations were reviewed by the annotation group. In some cases, two disease-system categories were assigned. For example, mutations in a number of enzyme-encoding genes produce human disease within a narrow pathophysiological area. Thus, both 'metabolic' and the pathophysiology system directly associated with disease would be selected.
Human:rat ortholog pair assignment and KA/KS determination
cDNA 'reference sequences' corresponding to the protein-coding entries in HGMD were mapped to NCBI build 31 of the human genome sequence [39] using BLAT [14] and an alignment identity lower threshold of 95%. HGMD disease entries were assigned Ensembl [13] human gene predictions if the optimal mapping of their cDNA sequences overlapped at least one Ensembl gene exon. Of the 11,522 1:1 rat:human orthologs identified by Ensembl, 11,224 (97.4%) were identified as syntenic with human and are accepted with confidence. The human disease genes in which orthologs were not predicted by Ensembl were individually investigated further using BLAT [14] and BLAST [15]. Additional 1:1 orthology relationships were established or confirmed using rat genome, EST, cDNA and protein sequences on the basis of high amino-acid identity (most more than 80%). Given that the median amino-acid identity among Ensembl's syntenic human-rat orthologs is 88% [8], we are confident that the ortholog assignments utilized in this study are accurate. KA/KS and KS were calculated using the yn00 algorithm [40] implemented in PAML [41] and pairwise alignments of human and rat orthologs, described elsewhere [8].
Ortholog assignment and conservation index determination
Potential orthologs were searched for 1,180 disease genes in the rat, mouse, fish, nematode, fly and yeast genomes using the INPARANOID program [42]. A CI was calculated as the alignment score in bits divided by the alignment length. The number of potential orthologs and quantiles of CI were determined for each species. Although percentage sequence identity could also have been used for this purpose, CI has the advantage of accounting for conservative substitutions. The INPARANOID program utilized BLAST2 to generate alignments and employed the blosom62 amino acid substitution matrix. Species utilized in this study and their on-line sources are: Rattus norvegicus [43], Mus musculus [44], C. elegans [45], Drosophila melanogaster [46], Saccharomyces cerevisiae [47] and Danio rerio [48].
Human disease mutation and rat wild-type genome-sequence comparison
Human and rat ortholog alignments were inspected automatically at positions described by HGMD as human disease mutations. From this, 104 amino-acid sites were found to be identical between the rat sequence and the proposed disease variant in humans. These were investigated further by review of the literature and the relevant HGMD entry. Questionable items (marked as '?' in HGMD), and those for which there was no documented statistical evidence for a causal connection between sequence variation and a clinical phenotype, were excluded, as were entries associated with poor alignment quality.
Text analysis of functional annotation by disease-system categories
Gene Ontology and domain terms were analyzed for over- or under-representation in different disease systems using the CoMet tool within the OmniViz analysis package [49]. This analysis package analyzes associations between terms, categories, clusters and groups by examining the deviation of number of occurrences in a cell from that found in a random distribution; the resulting analyses are then visualized with the CoMet tool which facilitates an overview of correlations among a matrix of variables. Records meeting the criteria of: P value less than or equal to 0.05, expected frequency greater than or equal to 2%, observed frequency greater than or equal to 2%, and record number greater than or equal to 5. For this algorithm, the null hypothesis for a given GO entry would be: 'its frequency in a specific disease system is equal to its frequency in others'. Results are portrayed graphically with over-represented cells labeled in red. A gradient of color hue represents the deviation in value for each category.
Identification of repeat-variation disease gene set
Genes with disease-associated mutations characterized as bearing 'repeat variations' were retrieved from the HGMD database [3]. The dataset included the nine different known CAG-expansion disease genes. Protein and cDNA sequences from humans, rat and mouse were obtained from the Ensembl database [13]. In all, 11,501 human-rat sequence pairs, 12,488 human-mouse sequence pairs and 12,357 rat-mouse sequence pairs were aligned using CLUSTALW [50] and glutamine tandem repeats mapped on the aligned sequences. The length cut-off for considering repeats was five or more glutamine residues in tandem. Conserved repeats between two species were those with a length of 5 or longer in an equivalent position in the two sequences.
Additional data files
The following additional data are available with the online version of this article: the original set of 104 genes where rat wild-type sequence is identical to human disease variant mutation (Additional data file 1), the pathophysiology annotations for human disease genes (Additional data file 2), the list of immune-system genes not identified as disease genes (Additional data file 3), the list of neurological-system genes not identified as disease genes (Additional data file 4) and the potential orthologs of human disease genes identified for each model organism (Additional data file 5).
Supplementary Material
The original set of 104 genes where rat wild-type sequence is identical to human disease variant mutation
The pathophysiology annotations for human disease genes
The list of immune-system genes not identified as disease genes
The list of neurological-system genes not identified as disease genes
The potential orthologs of human disease genes identified for each model organism
Acknowledgments
Acknowledgements
C.P.P., E.E.W. and L.G. are funded by the Medical Research Council UK. H.H., H.W., K.G.W., H.X., K.F. and D.R.S. were funded under grants HG002046 and HG002145 from the National Institutes of Health, USA. M.M.A. acknowledges program Ramón y Cajal and grant BIO2002-04426-C02-01 from the Spanish Ministry of Science and Technology. P.D.S. and D.N.C. acknowledge the support of Celera Genomics, Rockville, MD. We thank the Rat Genome Sequencing Consortium for valuable advice and support during this project.
Contributor Information
Hui Huang, Email: hhuang@genomecorp.com.
Eitan E Winter, Email: eitan.winter@human-anatomy.oxford.ac.uk.
Huajun Wang, Email: huajun@yahoo.com.
Keith G Weinstock, Email: kweinsto@amgen.com.
Heming Xing, Email: Heming.Xing@dds.criver.com.
Leo Goodstadt, Email: Leo.Goodstadt@anat.ox.ac.uk.
Peter D Stenson, Email: stensonpd@Cardiff.ac.uk.
David N Cooper, Email: CooperDN@Cardiff.ac.uk.
Douglas Smith, Email: dsmith@agencourt.com.
M Mar Albà, Email: MAlba@imim.es.
Chris P Ponting, Email: chris.ponting@human-anatomy.oxford.ac.uk.
Kim Fechtel, Email: kfechtel@comcast.net.
References
- Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science. 1949;110:543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- Ingram VM. Gene mutations in human hemoglobin: the chemical difference between normal and sickle cell hemoglobin. Nature. 1957;180:326–328. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD®): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger J, Bocchini C, Valle D, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002;30:52–55. doi: 10.1093/nar/30.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. Genome sequencing of the Brown Norway Rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Hurst LD. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18:486. doi: 10.1016/S0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- Hurst LD, Smith NGC. Do essential genes evolve slowly? Curr Biol. 1999;9:747–750. doi: 10.1016/S0960-9822(99)80334-0. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol. 2000;17:68–74. doi: 10.1093/oxfordjournals.molbev.a026239. [DOI] [PubMed] [Google Scholar]
- Winter EE, Goodstadt L, Ponting CP. Elevated rates of protein secretion, evolution, and disease among tissue-specific genes. Genome Res. 2004;14:54–61. doi: 10.1101/gr.1924004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamp M, Andrews D, Barker D, Bevan P, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, et al. Ensembl 2002: accommodating comparative genomics. Nucleic Acids Res. 2003;31:38–42. doi: 10.1093/nar/gkg083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT-the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. 10.1101/gr.229202. Article published online before March 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi K, Hagi A, Inoue Y. The relationship between plasma high density lipoprotein cholesterol levels and cholesteryl ester transfer protein activity in six species of healthy experimental animals. Biol Pharm Bull. 2001;24:579–581. doi: 10.1248/bpb.24.579. [DOI] [PubMed] [Google Scholar]
- Zhang A, Potvin G, Zaiman A, Chen W, Kumar R, Phillips L, Stanley P. The gain-of-function Chinese hamster ovary mutant LEC11B expresses one of two Chinese hamster FUT6 genes due to the loss of a negative regulatory factor. J Biol Chem. 1999;274:10439–10450. doi: 10.1074/jbc.274.15.10439. [DOI] [PubMed] [Google Scholar]
- Gersten KM, Natsuka S, Trinchera M, Petryniak B, Kelly RJ, Hiraiwa N, Jenkins NA, Gilbert DJ, Copeland NG, Lowe JB. Molecular cloning, expression, chromosomal assignment, and tissue-specific expression of a murine alpha-(1,3)-fucosyltransferase locus corresponding to the human ELAM-1 ligand fucosyl transferase. J Biol Chem. 1995;270:25047–25056. doi: 10.1074/jbc.270.42.25047. [DOI] [PubMed] [Google Scholar]
- Soussi-Yanicostas N, de Castro F, Julliard AK, Perfettini I, Chedotal A, Petit C. Anosmin-1, defective in the X-linked form of Kallman syndrome, promotes axonal branch formation from olfactory bulb output neurons. Cell. 2002;109:217–228. doi: 10.1016/S0092-8674(02)00713-4. [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Di Schiavi E, Hilliard MA, Arbucci S, Ghezzi C, Facciolli A, Coppola G, Ballabio A, Bazzicalupo P. The Kallmann syndrome gene homolog in C. elegans is involved in epidermal morphogenesis and neurite branching. Development. 2002;129:1283–1294. doi: 10.1242/dev.129.5.1283. [DOI] [PubMed] [Google Scholar]
- Gao L, Zhang J. Why are some human disease-associated mutations fixed in mice? Trends Genet. 2003;19:678–681. doi: 10.1016/j.tig.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Smith NGC, Eyre-Walker A. Human disease genes: patterns and predictions. Gene. 2003;318:169–175. doi: 10.1016/S0378-1119(03)00772-8. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- Hess ST, Blake JD, Blake RD. Wide variations in neighbor-dependent substitution rates. J Mol Biol. 1994;236:1022–1033. doi: 10.1016/0022-2836(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Green P, Ewing B, Miller W, Thomas PJ, Green ED, NISC Comparative sequencing Program Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet. 2003;33:514–517. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- Majewski J. Dependence of mutational asymmetry on gene-expression levels in the human genome. Am J Hum Genet. 2003;73:688–692. doi: 10.1086/378134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison R, Roskin KM, Yang S, Diekhans M, Kent WJ, Weber R, Elnitski L, Li J, O'Connor M, Kolbe D, et al. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 2003;13:13–26. doi: 10.1101/gr.844103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RD, Falls K, Simon J, Torrey D, Pandit S, McKenny J, Braunschweiger K, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- BioKnowledge Library http://www.incyte.com/control/researchproducts/insilico/proteome
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harihan IK, Haber DA. Yeast, flies, worms and fish in the study of human disease. N Engl J Med. 2003;348:2457–2463. doi: 10.1056/NEJMon023158. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini ME, Skupski MP, Boguski MS, Hariharan IK. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol. 2000;150:F23–F30. doi: 10.1083/jcb.150.2.F23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albà MM, Guigó R. Comparative analysis of amino-acid repeats in rodents and humans. Genome Res. 2004;14:549–554. doi: 10.1101/gr.1925704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albà MM, Santibáñez-Koref MF, Hancock JM. Conservation of polyglutamine tract size between mouse and human depends on codon interruption. Mol Biol Evol. 1999;16:1641–1644. doi: 10.1093/oxfordjournals.molbev.a026077. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skimmer PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HTL. Ataxin-1 nuclear localization and aggregation: role in poly-glutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/S0092-8674(00)81781-X. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- Van Horsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Bader M, Pabst R, Kobbe P, Krotova J, et al. Transgenic rat model of Huntington disease. Hum Mol Genet. 2003;12:617–624. doi: 10.1093/hmg/12.6.617. [DOI] [PubMed] [Google Scholar]
- NCBI build 31 of the human genome sequence (November 2002) http://hgdownload.cse.ucsc.edu/goldenPath/14nov2002/bigZips/
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- Rattus norvegicus http://hgdownload.cse.ucsc.edu/goldenPath/rnJan2003/bigZips/
- Mus musculus http://hgdownload.cse.ucsc.edu/goldenPath/mmFeb2003/bigZips/
- Caenorhabditis elegans ftp://ftp.wormbase.org/pub/wormbase/archive/wormpep98.tar.gz
- Drosophila melanogaster ftp://ftp.ncbi.nih.gov/refseq/release/invertebrate
- Saccharomyces cerevisiae ftp://ftp.ncbi.nih.gov/refseq/release/fungi
- UniGene - Danio rerio ftp://ftp.ncbi.nih.gov/repository/UniGene/Dr.seq.uniq.gz
- OmniViz http://www.omniviz.com
- Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The original set of 104 genes where rat wild-type sequence is identical to human disease variant mutation
The pathophysiology annotations for human disease genes
The list of immune-system genes not identified as disease genes
The list of neurological-system genes not identified as disease genes
The potential orthologs of human disease genes identified for each model organism