

Short abstract
On the basis of the observation that conserved positions in transcription factor binding sites are often clustered together, a simple extension to the model-based motif discovery method is proposed. Examples with both simulated and real data show that this extension helps discover motifs as the data become noisier or when there is a competing false motif.
Abstract
On the basis of the observation that conserved positions in transcription factor binding sites are often clustered together, we propose a simple extension to the model-based motif discovery methods. We assign position-specific prior distributions to the frequency parameters of the model, penalizing deviations from a specified conservation profile. Examples with both simulated and real data show that this extension helps discover motifs as the data become noisier or when there is a competing false motif.
Background
DNA-binding transcription factors have a crucial role in transcriptional regulation, linking nuclear DNA to the transcriptional regulatory machinery in a sequence-specific manner. Transcription factors generally bind to short, redundant families of sequences. Although experimental methods exist to characterize the sequences bound by a given factor, the systematic enumeration of transcription factor binding sites is greatly aided by computational methods that identify sequences or families of sequences that are enriched in specific collections of regulatory DNA.
Two major strategies exist to discover repeating sequence patterns occurring in both DNA and protein sequences: enumeration and probabilistic sequence modeling. Enumeration strategies rely on word counting to find words that are over-represented [1]. Model-based methods represent the pattern as a matrix, called a motif, consisting of nucleotide base (or amino-acid residue) multinomial probabilities for each position in the pattern and different probabilities for background positions outside the pattern [2,3]. For example, Figure 1 shows the motif representation of the binding sites for the yeast transcription factor Gal4, which regulates the transcription of genes under galactose-rich conditions. The goal of the model-based methods is to estimate the parameters of this model, the position-specific and background multinomial probabilities, and then to determine likely occurrences of the motif by scoring sequence positions according to the estimated motif matrix.
Figure 1.

Binding sites and motif matrix for Gal4. (a) Binding sites obtained from the Promoter database of Saccharomyces cerevisiae (SCPD) [27]. (b) Motif matrix with base frequencies for each of the 17 positions.
Even with weak signals, model-based methods such as MEME [2] and Gibbs Motif Sampler [3] effectively find motifs of variable width and occurrences in DNA and protein sequences. Originally developed to be flexible for finding both protein and DNA patterns, these general motif-discovery algorithms have been enhanced to make them more specific for discovering transcription-factor binding sites [4-8]. Changes include using a higher-order Markov model, genome-wide nucleotide frequencies or a position-specific model for the background distribution [5,7,8] and checking both DNA strands [2,5,6]. Other changes use knowledge about the nature of the interaction between the transcription factor and its binding site. Some transcription factors, like Gal4, bind DNA as homodimers and have palindromic binding sites. The most frequent bases observed at each position, called the consensus, consist of the palindromes CGG and CCG (Figure 1) in the Gal4-binding sites. Several methods have the option to search for palindromic patterns [2,5,9].
Many authors have noted, or showed empirically from structural information on DNA-protein complexes and binding-site examples, that high levels of base conservation at a position correlate with more contacts to the protein [10-14]. For example, Gal4 interacts more closely with the edge positions of the binding site, which is reflected by highly conserved bases in positions 1-3 and 15-17 (Figure 1). This observation has been incorporated into methods for predicting binding sites in new sequences given a motif matrix. The score contribution of the highly conserved positions are upweighted in the scoring functions between the motif matrix and the sequence [10,12]. This has also been incorporated directly to the motif-finding methods. The original fragmentation model of the Gibbs Motif Sampler assigns J positions out of a larger window of motif width W as more important (that is, more conserved), but there is no specification of where they should fall within the W positions.
It has also been observed that highly conserved positions tend to be grouped together within the motif [10,11,13]. This occurs because transcription factors rarely contact only a single base, and not adjacent bases. It follows that the position of high conservation should be clustered within the motif. This grouping has been specified through the use of blocks in BioProspector [5] and earlier in the work of Cardon and Stormo [4]. In BioProspector, the model can be specified for two motif blocks separated by a flexible gap window. The most recent version of the fragmentation model in the Gibbs Motif Sampler includes an option to indirectly specify blocks, by assigning the J positions out of the W to occur at the ends, rather than the middle [8].
Because of the success of these various extensions to the original multinomial motif model, it is widely recognized that making the model more specific improves the detection of real binding sites [2-7]. However, these methods have still maintained their generality so as not to make them specific to particular data or transcription factor. In our approach, we propose another extension to the model that strictly incorporates the observations previously discussed: highly conserved positions within the motif are clustered. For improving motif discovery, we incorporate the ideas behind both the fragmentation model in the Gibbs Motif Sampler and the two-block model of BioProspector, but make use of more restrictive assumptions. The original fragmentation model labels some positions as more important but their location within the motif is not specified. For the two-block model in BioProspector and the newest version of Gibbs Motif Sampler, the positions are clustered but they are not restricted to all be highly conserved. In contrast, we strictly enforce the motif to consist of consecutive highly conserved positions. Our model is still general for different types of binding sites and flexible enough to incorporate the other useful extensions mentioned above, such as palindromicity and alternative background models. In the next section we provide a rationale for our method using empirical data on binding sites.
Rationale
The information content of aligned and experimentally verified binding sites for several transcription factors is shown in Figure 2. A 20 bp flanking region has been included on each side. Peaks in this graph show regions of high base conservation. The shapes of these plots can be described as bimodal, for Gal4-, Abf1- and Crp-binding sites, or unimodal, for Pho4- and PurR-binding sites. These plots reflect the structural constraints discussed above. Positions that have more contacts to the protein are highly conserved and these positions tend to cluster because the protein contacts multiple adjacent bases. Although exceptions exist, the plots of information content for many binding-site motifs look similar to these examples. Therefore, our goal is to search for motifs that are uni- or bimodal.
Figure 2.
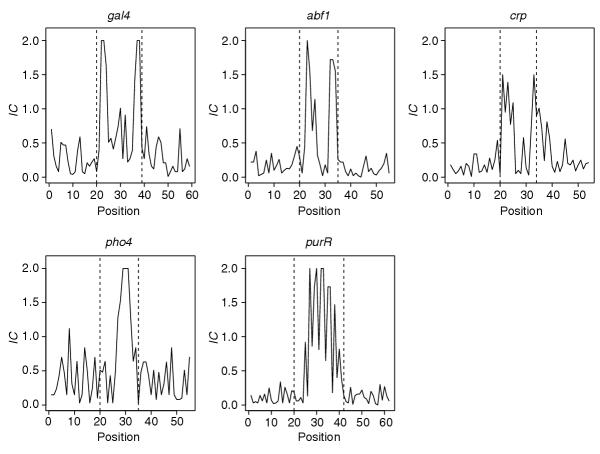
Plots of information content (IC = 2 + Σi pilog2pi) for example motifs. The binding sites have been extended 20 bp on each side and dotted lines mark proposed boundaries of the known sites.
The shapes in these plots can also be coarsely described as blocks of alternating strongly conserved positions and moderately or minimally conserved positions. In our framework, we assign blocks of motif positions a conservation type: strong (regime 1), moderate (regime 2) or low (regime 3). For positions that are specified as strongly conserved, the maximum possible conservation occurs if only one base is observed. Similarly, in the moderately conserved case, perhaps only two bases are conserved, with equal probability or such that their probabilities add to one. The low-conservation case, regime 3, corresponds to three or four bases appearing.
Bimodal motifs can be described as two regime 1 blocks separated by one regime 2 (or regime 3) block. This is illustrated in Figure 3a. For example, the binding site for Gal4 has two sets of three strongly conserved positions separated by a block of 11 positions with relatively low conservation (Figure 2). Other sites, such as those for Pho4 and PurR, are unimodal and have a block of regime 1 positions in the center with a regime 2 block (or regime 3) at either end. This is illustrated in Figure 3b.
Figure 3.
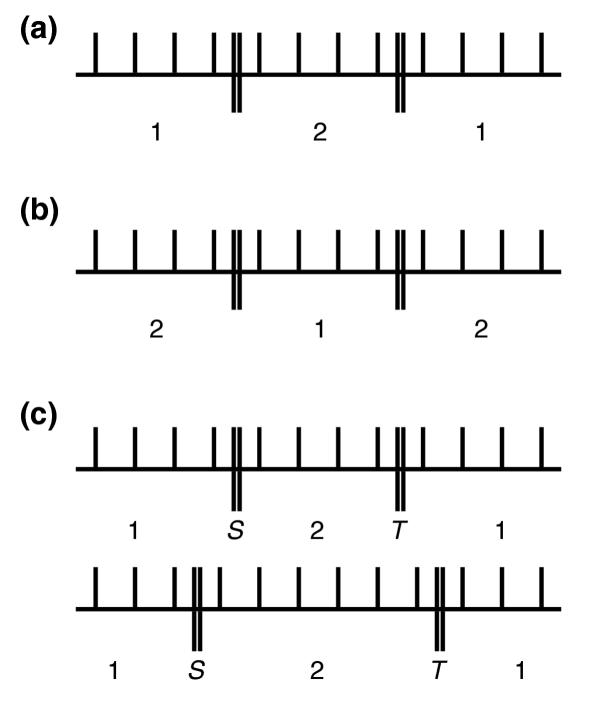
Diagrams illustrating regime blocks and change points. (a) Bimodal information motif. (b) Unimodal information motif. (c) Two different possibilities for a bimodal motif. Vertical lines correspond to positions in the motif and double vertical lines show boundaries between blocks. S and T are the first and second change points, respectively, between blocks.
In our method, we extend the model that was the basis for MEME and Gibbs Motif Sampler. We use the expectation maximization (EM) algorithm, as in Lawrence and Reilly [9], and MEME to estimate the parameters of the model. According to the regime type for each motif position, determined by the blocks, we assign a prior distribution to the multinomial probabilities. This is equivalent to a penalized likelihood method [15]. If a position is assigned as strongly conserved (regime 1), deviation from perfect conservation will be penalized. At each iteration in the algorithm, this translates to upweighting the frequency of the most common base, while downweighting the rest. For the moderately conserved case (regime 2) it translates to upweighting the frequency of the two most common bases, while downweighting the frequencies of the other two. These two situations result in changes that are easy to implement in the original EM algorithm of Lawrence and Reilly.
Results
Basic model and algorithm
We now elaborate on the theory behind our method. Let 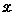 denote the collection of N sequences we examine. Each sequence Xi, i = 1,...,N, consists of Li bases,
denote the collection of N sequences we examine. Each sequence Xi, i = 1,...,N, consists of Li bases,
![]()
Xik is the nucleotide base at position k in sequence i. To simplify notation, all sequences are set to the same length, Li = L, but there is no difficulty in changing back to the more general case. In this paper, we assume that in each sequence there is an occurrence of a conserved pattern of width W, referred to as a motif. This assumption will be relaxed in the future to allow for any number of occurrences: 0, 1 or more than one. Positions in the motif are labeled w, w = 1,...,W. The start position for the motif in each sequence, mi, occurs in the range 1,...,L - W + 1. The alignment A of the motifs refers to the set of mi. Finally, the set of bases ranges from j = 1,...,J, where J = 4 for nucleotide bases.
Lawrence and Reilly [9] use multinomials to model the sequences given the alignment. The work in Stormo et al. [16] appears to be one of the first uses of this approach. They assume that bases in sequence positions that are not in the motif (background positions) are independent and identically distributed according to a multinomial distribution. Bases in positions that are in the motif are independent but non-identically distributed according to a motif position-specific multinomial distribution. Sequences and positions are assumed to be independent. The background multinomial parameters are denoted by p0 = {p01,...,p04} and the motif position-specific multinomial parameters are denoted by pw = {pw1,...,pw4} for w = 1,...,W. The set of all multinomial parameters in the model is  .
.
In practice, the motif start positions are not known a priori. By expanding the previous parameterization, Lawrence and Reilly introduced a random variable for the start position to the model for each sequence. The vector
![]()
contains the alignment information, where Yik = 1 at the start position k = mi and 0 elsewhere. The sum constraint
![]()
corresponds to the one motif occurrence per sequence model. The set of all Yik will be denoted 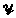 . The prior distribution on is g and following Lawrence and Reilly, we assume g is the uniform distribution along the sequence.
. The prior distribution on is g and following Lawrence and Reilly, we assume g is the uniform distribution along the sequence.
To obtain the maximum likelihood estimates for the motif parameters , the marginal likelihood LX() must be maximized. This is a sum over all possible start positions and is difficult to maximize directly. There are several different approaches for estimating the model parameters. Lawrence and Reilly [9] and Bailey and Elkan [2] use the EM algorithm [17], while Liu et al. [3] use the Gibbs sampler [18,19]. The EM algorithm is guaranteed to reach a maximum, but depending on the initial starting points it may get trapped by local maxima. Alternatively, the Gibbs sampler is a stochastic algorithm, which has the ability to escape local maxima, but there are no guarantees for reaching a maximal solution. Furthermore, there is no clear benchmark for determining the stopping time for Markov chain Monte Carlo methods such as the Gibbs sampler [20]. Following Lawrence and Reilly, we also use EM to obtain the maximum likelihood estimates, but we will discuss alternatives later. The EM algorithm is a two-stage procedure and the steps from Lawrence and Reilly are outlined below for the basic model at the r + 1 iteration. The complete derivations are given in [21].
E-step
The unobserved start position variable Yik is replaced by the probability that it is a start site for a motif, given the current values of the parameters and the data,
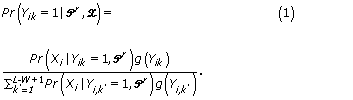
The term Pr(Xi |Yik = 1,  ) is a product of multinomials.
) is a product of multinomials.
M-step
The background multinomial probabilities are updated,
![]()
where 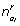 is the expected number of base j in the background after the rth iteration. Similarly, the parameter estimates are updated for each motif position w,
is the expected number of base j in the background after the rth iteration. Similarly, the parameter estimates are updated for each motif position w,
![]()
where 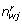 is now the expected number of base j at that position after the rth iteration,
is now the expected number of base j at that position after the rth iteration,
![]()
The parameter estimates at each step are based on the occurrences of bases at each position, weighted by the posterior probabilities of the positions being in a motif, which were calculated in the E-step.
Model with priors
Below, we discuss the details of our extensions to this model and outline the corresponding EM algorithm. For each position, we assume a prior distribution on the multinomial parameters to capture the type of base conservation patterns observed for real binding sites in Figure 2.
Blocks
As discussed above, the bi- and unimodal shape of the information content for motifs can be described as a block of moderately conserved positions separated by two blocks of strongly conserved positions or vice versa. The concept of blocks has been used before [4,5], but we also enforce a specific conservation pattern within the block. The multinomial parameters at each position are assigned a prior distribution according to the block regime specification.
Blocks of motif positions will be assigned a conservation type: strong, moderate or low. Let Iw be the conservation type for motif position w,

The regime 3 case is roughly equivalent to the background distribution for the positions not in the binding site, therefore, we will not consider regime 3 and focus the discussion on regimes 1 and 2. For Pho4, a unimodal motif with W = 10, we assign I = {2,2,2,1,1,1,1,2,2,2}. For Gal4, a bimodal motif with W = 17, we assign I = {1,1,1,2,2,2,2,2,2,2,2,2,2,2,1,1,1}. Depending on whether Iw = 1 or 2, a different prior distribution will be assigned to position w. In the following section we will elaborate on the two different forms of the prior.
Hereinafter, to specify the regime types for a motif I, we will use abbreviated notation. For example, [2(3), 1(4), 2(3)] is equivalent to I = {2,2,2,1,1,1,1,2,2,2}. In this notation, the number in bold indicates the type of regime (1 or 2) for each of the three blocks and the number in parenthesis indicates the width of the block.
Prior distribution
Let f(pw) be the prior on the multinomial probabilities for position w. For f, Liu et al. [3], among others, use the Dirichlet distribution, the conjugate prior of the multinomial. In these methods, the same Dirichlet distribution can be used for each motif position or the Dirichlet parameters at each position can be set by using previous knowledge about the relative base frequencies at the different positions [8]. In contrast, we use a prior distribution that is position specific, depending on the block regime specification, and that is independent of base composition. This prior distribution captures a certain overall base conservation without indicating the base identities.
Because we are ignoring base identity, it would be necessary to use a mixture of Dirichlet distributions for the prior at each position. To obtain the many parameters for the Dirichlet mixtures, we must then train on a relatively small set of example binding-site motifs. To avoid this estimation, we consider two other possibilities for f, the double exponential or normal distribution and qualitatively assign the parameters. Using the double exponential or normal distribution for the prior corresponds to using a certain type of penalty in the likelihood. In these two cases, the penalty function takes the form of the L1 or L2 norm respectively after taking the logarithm. For the double exponential case (L1),
![]()
while for the normal case (L2),
![]()
subject to the constraints
![]()
and 0 ≤ pj ≤ 1 for all j. The L1 and L2 penalty forms are similar to the penalties used for shrinkage in lasso and ridge regression respectively [22,23].
The prior distribution has two parameters, λ and δ. The strength of the prior on the model is determined by λ, where λ ≥ 0. The contribution of the prior to the likelihood increases as λ increases. When λ = 0, the model simplifies to the original model without priors. We assign values for the parameter δ depending on the regime assigned to position w. Below, we discuss the possible values of δ for regime 1 and 2. The w notation is dropped for simplicity.
Regime 1
For positions that are specified as strongly conserved (Iw = 1), the maximum conservation occurs if only one base is possible. That is, for some base j, pj = 1, while for all j' ≠ j, 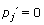 . Thus, the prior can be set as a penalty against deviations from this conservation. For ordered j, such that p(1) ≥ p(2) ≥ p(3) ≥ p(4),
. Thus, the prior can be set as a penalty against deviations from this conservation. For ordered j, such that p(1) ≥ p(2) ≥ p(3) ≥ p(4),
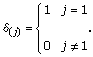
Regime 2a
Similarly, in the moderately conserved case (Iw = 2), perhaps only two bases are conserved, with equal probability. Then,
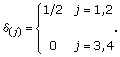
Regime 2b
This previous constraint is somewhat arbitrary. It could very well be that the frequencies for the two bases are  and
and  . A more general variant would be to constrain the sum of the probabilities of the two bases to 1. Now, for L1, the right side of Equation (4) is,
. A more general variant would be to constrain the sum of the probabilities of the two bases to 1. Now, for L1, the right side of Equation (4) is,
![]()
Note, however, that regime 1 is nested in this model (that is, a position that has a small penalty value under regime 1 will also have a small penalty value under regime 2b). The results using simulated data show that the nested nature of the regimes compromises the effectiveness of the method in certain situations.
The constant in Equations (4) to (6) is the log of the normalizing factor for f. The space of p is limited to the 4-d simplex, with the following order constraints: p(1) ≥ p(2) ≥ p(3) ≥ p(4). Because of these complicated constraints, there is no closed form solution for the normalizing factor. However, it does not depend on and is dropped in the derivations of the EM algorithm.
As described previously, we are specifying a model that will bias the search for uni- or bimodal motifs. We look for the motif that maximizes the likelihood of the data given this model. Equations (4) to (6) are the essential component of our method and work as a penalty in the log likelihood. If a potential motif does not follow the indicated shape, it will not score as well, in terms of the log likelihood, as another candidate motif that does follow the shape. More specifically, if a position is specified as regime 1, then δ is set to the value discussed above. If the base frequencies at that position, p, deviate from δ, then the values for Equation (4) will be large and negative and, therefore, reduce the log likelihood.
Algorithm
Assigning a prior distribution on the multinomial parameters for each position only alters the EM algorithm slightly. For the E-step, the update formula for  is the same as in the case without priors. For the M-step, the updates for the background multinomial parameters p0 are the same as with the basic model. The updates for the motif positions pw take on different forms depending on Iw and the functional form of f and are listed in Figure 4.
is the same as in the case without priors. For the M-step, the updates for the background multinomial parameters p0 are the same as with the basic model. The updates for the motif positions pw take on different forms depending on Iw and the functional form of f and are listed in Figure 4.
Figure 4.

Update formulae for motif parameters. Updates in M-step depend on Iw (regime 1 or 2) and the functional form of f (L1 or L2). For position w after the rth iteration, is the expected number of base j at the wth motif position. For ease of notation, the superscript r and subscript w are dropped. The bases, j, are ordered such that n(1) ≥ n(2) ≥ n(3) ≥ n(4).
For the L1 prior and regime 1, using the positive root for γ in Equation (8), the  are rescaled versions of the original maximum likelihood estimates. The base that occurs more frequently is upweighted relative to the other bases. For regime 2, in Equation (10), using the positive root for γ, the top two occurring bases are upweighted relative to the other two bases. If λ = 0, as in the original model, γ = N and the equal the original weighted frequencies fromEquation (3). We do not derive regime 2a, where the top two bases have equal probability,
are rescaled versions of the original maximum likelihood estimates. The base that occurs more frequently is upweighted relative to the other bases. For regime 2, in Equation (10), using the positive root for γ, the top two occurring bases are upweighted relative to the other two bases. If λ = 0, as in the original model, γ = N and the equal the original weighted frequencies fromEquation (3). We do not derive regime 2a, where the top two bases have equal probability,
![]()
for the L1 prior. We cannot safely assume
![]()
to ignore the absolute values and to obtain a closed form solution as above. In this case we will need to directly maximize over a four-dimensional nonlinear equation with constraints, for each position. To simplify the updates, we only use regime 2b with L1.
For the L2 prior, there is no simple closed form solution for γ. Nevertheless, the problem of determining the one-dimensional γ is still a reduction in the complexity of the original maximization in four dimensions of a nonlinear equation with constraints. To solve for γ in R, we use the uniroot function based on the algorithm in Brent [24]. For L2, we do not derive regime 2b because no simplifications are possible as in regime 1 and regime 2a. The penalty (p(1)+p(2)-1)2 in regime 2b causes dependencies between p(1) and p(2) that cannot be factored out into a simple form. Thus, to simplify the updates, we only use regime 2a with L1.
In summary, by including a prior distribution on the multinomial parameters, only the M-step changes. For either type of prior, L1 or L2, there is a closed form solution for the parameter updates depending on the coefficient γ. This coefficient, called the Lagrange multiplier, ensures that the constraint Σj pj = 1 is satisfied. For L1, γ is a unique positive solution to a quadratic equation, while for L2, γ is a unique positive solution to a monotone decreasing nonlinear equation. Thus, there is either an explicit solution or one that can be obtained quickly. For L1 and L2, we use the two different variations of regime 2, 2b and 2a respectively. This is necessary so that in the M-step there is a closed-form solution or an optimization in one dimension. If we do not use these variations, we cannot avoid more costly computations in higher dimensions.
Model with change points
In the previous section, the locations of the blocks were designated in advance. In many situations, the borders between blocks will not be known a priori. We will now expand the current model parameterization to include unobserved random variables for the borders between blocks, referred to as change points. For example, the diagrams in Figure 3c depict two different possibilities for a bimodal motif.
Let S and T denote the first and second change points, respectively, between blocks where -1 ≤ S <T ≤ W. The values of S and T determine each Iw,
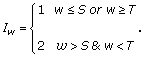
For example, in Gal4 where I = {1,1,1,2,2,2,2, 2,2,2,2,2,2,2,1,1,1}, S = 3 and T = 15. This characterization also applies to the unimodal type of sites, but the previous designations for Iw = 1 and Iw = 2 should be reversed. To include the case where all Iw = 2, the lower range of values for S extends to -1 and the range of T extends to W.
It may not be known which choices for S and T are preferable. Therefore, when S and T are not known, we introduce a random variable cst, -1 ≤ s <t ≤ W. It is an indicator for the two change points, where
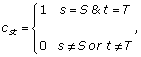
and
![]()
The variable cst determines Iw for all w and as a result, it determines which prior is assigned to each pw. Let 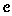 denote the collection cst. There are
denote the collection cst. There are 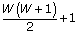 unique cs,t.
unique cs,t.
In practice, W is usually between 6 and 20, which translates into 22 to 211 different cst. We also specify h, the prior distribution on . The ratios of the lengths of the three blocks to W are assumed to follow a Dirichlet distribution. The possible lengths are not continuous but increment by discrete positions, therefore, we use a discretized form of the Dirichlet for h. Change points have also been used to model heterogeneity in base composition along a sequence [25]. In this context, both the locations and the number of change points are random variables.
Algorithm
Now, in the E-step, besides the term , we also need to compute the posterior probability of cst given the current values of the parameters and the data,
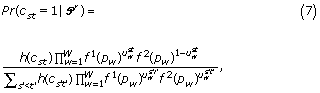
where f1 and f2 are the prior distributions for regime 1 and 2 positions respectively and  indicates that regime 1 is associated with motif position w given the change points s and t (cst = 1). For the M-step, the updates for the background multinomial parameters p0 are the same as with the basic model. The updates for the motif position parameters, pw, take on different forms depending on the functional form of f and are listed in Figure 5.
indicates that regime 1 is associated with motif position w given the change points s and t (cst = 1). For the M-step, the updates for the background multinomial parameters p0 are the same as with the basic model. The updates for the motif position parameters, pw, take on different forms depending on the functional form of f and are listed in Figure 5.
Figure 5.

Update formulae for motif parameters using model with change points. Updates in M-step depend on the functional form of f (L1 or L2). See details in Figure 4. See [35] for solutions to the cubic equation.
Given the data and the current values of the parameter, the term d in Figure 5 is the posterior probability that Iw = 1 for that position, while the term e is the posterior probability that Iw = 2. In the updates for both forms of the priors, when e → 0, and therefore d → 1, then 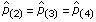 , analogous to the regime 1 estimates in Equations (8) and (12). Alternately, if d → 0, and therefore e → 1, then
, analogous to the regime 1 estimates in Equations (8) and (12). Alternately, if d → 0, and therefore e → 1, then 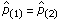 , equivalent to the regime 2 estimates in Equations (10) and (13). As in the previous model, for both types of priors, there is a closed form solution to the parameter updates depending on the Lagrange multiplier γ. For L1, γ is a unique positive solution to a cubic equation, while for L2, γ is a unique positive solution to a monotone decreasing nonlinear equation.
, equivalent to the regime 2 estimates in Equations (10) and (13). As in the previous model, for both types of priors, there is a closed form solution to the parameter updates depending on the Lagrange multiplier γ. For L1, γ is a unique positive solution to a cubic equation, while for L2, γ is a unique positive solution to a monotone decreasing nonlinear equation.
Fixed and variable change point model
Hereinafter, the two versions of our model will be referred to as the fixed change point model, from the section 'Model with priors', and the variable change point model, from the section 'Model with change points'. In Figure 6, we list the steps in the algorithm for estimating the parameters in the two cases. This algorithm has been implemented in the statistical software R [26] for evaluation purposes. Currently, a working version of the algorithm in C is also being completed to increase the speed of the program.
Figure 6.

Expectation maximization algorithm. Differences between the fixed and variable change point model are labeled (F) and (V) respectively.
Our method relies on the most basic motif model introduced in Lawrence and Reilly. This original model has many limiting assumptions, which have been addressed by more recent work in MEME and the Gibbs Motif Sampler. We have not incorporated the more recent adaptations, such as variable motif width, multiple motif occurrences per sequence and non-uniform distribution for g, so that we focus our attention on the conservation trends across motif positions. Nevertheless, because we are using the basic framework that is common to all the model-based methods, we can incorporate these approaches into our method as well.
In practice, this method should be used as follows. First, a set of upstream sequences from co-regulated genes is selected as input for the algorithm. Next, information about the structure of the proposed transcription factor involved in the regulation of these genes can be used to specify the motif width W and whether the search should be for a uni- or bimodal motif. For example, binding sites for helix-turn-helix homeodomain proteins generally have a core of four or five highly conserved bases flanked on either side by another one or two partially conserved bases. In this case a unimodal specification would be input into the algorithm. Otherwise, if more detailed information is known, then the vector I can be specified completely. The width usually ranges from 6 to 20 positions. From the examples we observed, unimodal motifs tend to be shorter (W = 8-10) than bimodal motifs, (W = 12-17).
Examination of transcription factor-DNA complexes suggests that factors within the same broad structural class bind DNA in a similar manner. Although the structures of many transcription factors have not been solved, sequence homology or other means may indicate that a transcription factor may be a member of a particular structural class of factors. This information can be used to select between a uni- or bimodal specification.
Simulations
First, we use simulation methods to compare the different prior functions (L1 or L2) and possible regime specifications in the fixed and variable change point models. We also use the simulations to evaluate the performance of our method with data that consist of both a real motif and a competing false motif. For evaluation purposes, we focus on the simple model of one motif occurrence per sequence with fixed overall width.
For both the simulations and the real data in the following section, we focused on binding sites in S. cerevisiae and E. coli for two reasons. There are many verified binding sites in databases specific to these organisms and the simple model of one occurrence per sequence is a reasonable assumption for both organisms.
For each of the five test sets in Figure 2, which we designate as gal4, abf1, crp, pho4 and purR, we inserted the experimentally verified binding sites for these examples in simulated sequences. We also permuted the positions of the binding sites and inserted a permuted version of the site in each generated sequence. More details about the simulation procedure, starting points and evaluation of the final results are described in Additional data file 1.
Ignoring the background positions, the permuted motif should have an equal chance of being discovered as the real motif because it has the same likelihood, but the information content across positions will not look like characteristic transcription factor binding sites.
We repeated the simulation procedure described in Additional data file 1 with 100 datasets for the five different transcription factor binding site examples. To display the results for increasing λ, we plot the percentage of correctly identified real motifs and the percentage of correctly identified permuted motifs, averaged over the 100 simulated datasets. We refer to this graph as a λ plot.
Recall that λ is a parameter in the prior distribution. It controls the contribution of the prior to the model. The larger λ is, the more deviations from the specified regime types are penalized. When λ = 0, this is equivalent to the original model, where both the real and permuted motifs are equally likely to be discovered. When the algorithm discovers either of the two motifs, the lines in the λ plots should add to 100%. If the lines do not add to 100%, then the algorithm does not find either but instead finds a spurious motif.
Fixed change points
First, we explore the situation where the borders between regimes, the change points, are fixed. For example, in Gal4, the binding site is 17 bp long and the specified regime types for each position in order are: [1(3), 2(11), 1(3)]. We compared the results between L1 and L2, in addition to whether including or not including the regime 2 penalty is beneficial. We find that using the L1 prior without the regime 2 penalty performs the best (that is, detects more real motifs for most λ values). The results for this model are displayed in Figure 7.
Figure 7.
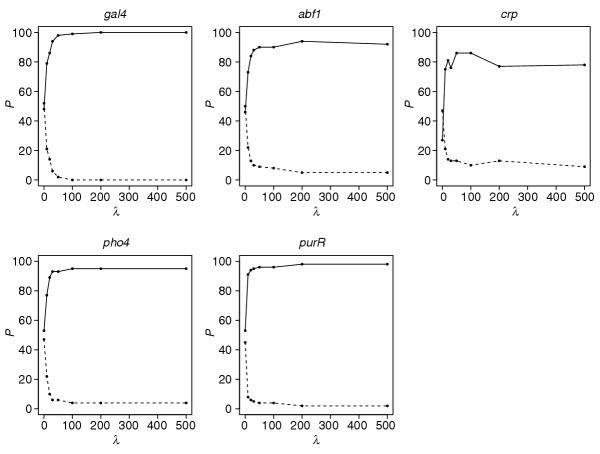
λP plots for the fixed change point model using the L1 prior and no penalty for regime 2. In all λP plots, the solid line shows the percentage of simulated datasets where at least 50% of the real sites were correctly identified. The dashed line shows the percentage of simulated datasets where at least 50% of the permuted sites were identified. The regime type specifications are [1(3), 2(11), 1(3)] for gal4, [1(4), 2(5), 1(3)] for abf1, [1(5), 2(6), 1(5)] for crp, [2(3), 1(4), 2(3)] for pho4 and [2(5), 1(10), 2(5)] for purR.
The parameter λ plays an important role in the performance of the algorithm. As expected, the motifs have about equal probability of being detected at λ = 0. Any variations are likely to be due to random noise for the 100 simulation trials. As λ increases, the real motif is preferred over the permuted one, and the improvement levels off or begins to decrease around λ = 100. In all but one example, this model discovers the real site in 95 to 100% of the datasets. The only exception is the motif for Crp, a relatively weak motif, where the percentage is roughly 90%.
In general, the L1 norm is a stronger penalty because Σj | pj - δj | ≥ Σj (pj - δj)2 for Σj δj = 1, 1 ≥ δj ≥ 0,∀j and Σj pj = 1, 1 ≥ pj ≥ 0, ∀j. Our results also indicate that most of the signal is driven by regime 1. There is little improvement or a negative effect when including regime 2 (data not shown). In addition, we also explored the effect of misspecifying the regimes by one position. For example, the regimes for Gal4 were specified as [1(4), 2(9), 1(4)] instead of [1(3), 2(11), 1(3)]. For all the test sets, this caused a 10-25% drop in the percentage of datasets where the real motif was discovered. Trying different specifications or, equivalently, altering the change points can be done systematically by using the variable change point model (see below).
Variable change points
In Figure 7, the change points between regimes are specified in advance. This information is not always available, so we outlined the algorithm for the situation where the change points are unobservable. For this method, it is only necessary to specify whether the motif is uni- or bimodal. For the variable change point method, we evaluated the results for both L1 and L2. A penalty for regime 2 is necessary, otherwise, the likelihood is maximized when all positions are labeled as regime 2.
In this extension, L2 performs better than L1 for almost all test examples. The nested nature of the L1 regime 2 penalty causes the likelihood to be maximized when all positions are labeled as regime 2. Figure 8 displays the results for L2. The real motif is preferred over the permuted motif as λ increases and reaches a maximum percentage in the range 50-100. Not surprisingly, the algorithm performs worse than when the change points are known a priori (Figure 7).
Figure 8.
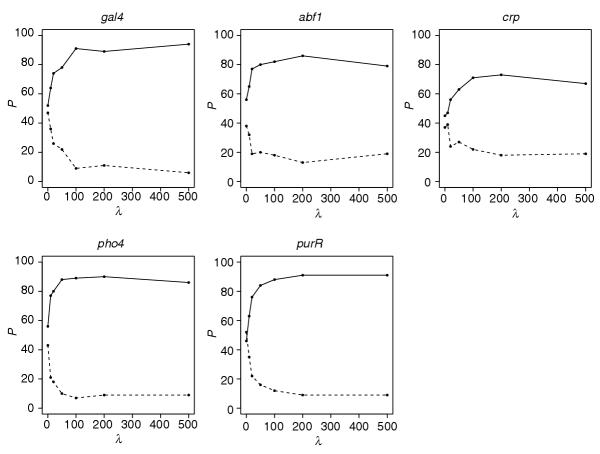
λP plots for the variable change point model using the L2 prior. In all λP plots, the solid line shows the percentage of simulated datasets where at least 50% of the real sites were correctly identified. The dashed line shows the percentage of simulated datasets where at least 50% of the permuted sites were identified. We use the following model specifications: W = 17 and bimodal for gal4; W = 12 and bimodal for abf1; W = 16 and bimodal for crp; W = 10 and unimodal for crp; and W = 20 and unimodal for purR.
Despite the overall drop in performance, we still observe a 70-80% increase in the percentage of datasets where the real motif is discovered. These results are similar to those obtained with the fixed change point model when we misspecified the regimes. The algorithm performs the best with Gal4, identifying the real motif almost 95% of the time, and performs the worst with Crp, only identifying the real motif 75% of the time. These results should not be compared directly with the L1 fixed change point model (Figure 7), but with the L2 fixed change point model, which performs slightly worse than L1 (data not shown).
In summary, when the positions of high, moderate or low conservation in a proposed motif are known a priori, the double exponential prior in our model performed the best at improving the detection of real motif versus decoy motifs in the simulated data. Otherwise, when it is only known that the motif has uni- or bimodal information content, then the normal prior performed the best in improving the detection of the real motifs versus the decoys.
Real data
To assess the performance of our method with real data instead of simulated data, we explored sets of genomic data containing experimentally verified binding sites. Using a specified length, we extracted the genomic sequences containing the binding sites from databases. As we extracted longer and longer sequences, the size of the data increased, adding more noise to the problem, but the number of binding sites, the signal, stayed the same. In particular, we explored two issues with this data: first, the effect of the number of starting points on the algorithm; and second, the effect of λ on the ability of the algorithm to detect the known binding sites in the data.
As previously discussed, we use the EM algorithm to find the motif that maximizes the likelihood of the data given the model. Because of the many local maxima in the likelihood function, the number and type of starting points used for the EM algorithm is a critical issue. It is beyond the scope of this paper to make a rigorous comparison of different procedures for obtaining starting points and to determine the optimal number of starting points. We discuss several examples which show that by increasing the number of starting points, the performance of the method improves. In the light of these results, the number of starting points is selected to be very large. We then explore the effect of including prior knowledge about the positional information content of the motif for the detection of the real sites.
Data
We looked for bimodal and unimodal motif examples that had a relatively weak signal and contained at least 10 sites that were found in the regulatory region of at least 10 different genes. The formal definition of a weak signal is discussed in the following section. Briefly, motifs that are no longer discovered as the size of the data is increased (that is, the noise level grows) are defined as having a weak signal, but motifs that are still detected in the noisier data are defined as having a strong signal.
We examined the five test sets used in the simulations and examples in the SCPD and DPInteract databases [27,28]. Overall, we found four examples of transcription-factor-binding sites that satisfied our criteria, those for Abf1, Crp, Rap1 and Reb1, which we designate as abf1, crp, rap1, and reb1. In Table 1, we summarize the information on these test sets. There were six sets (cpxR, gal4, lexA, repcar1, pho4, purR) that did not satisfy our criteria, which were used as a training set for fitting the parameters of our prior distribution (See Materials and methods for details).
Table 1.
Summary of test sets
| Dataset | Organism | N | Unimodal/Bimodal | W |
| crp | E. coli | 17 | Bi | 16 |
| rap1 | S. cerevisiae | 15 | Uni | 13 |
| reb1 | S. cerevisiae | 14 | Uni | 13 |
| abf1 | S. cerevisiae | 18 | Bi | 12 |
Columns list the dataset name, its source organism, the number of sequences (N), whether it has a uni- or bimodal motif, and the motif width (W).
The motif example crp is of particular interest for several reasons. It has a relatively weak signal that can be observed in Figure 2. Even in the simulated data, with length 100, the crp sites are more difficult to detect than the other test sites (Figure 7). Furthermore, this example has been used as a test set for several other motif-finding methods and is considered the 'gold standard' in this literature [2,5,9].
Using the real data, we evaluated the performance of our method as the background noise level grew. We obtained the sequences that contained the sites starting at length L = 100 bases and then incremented by 100 bases to create new datasets. We first determined the location of the site (or multiple sites) for each sequence and elongated the sequence on each side by a random amount of flanking sequence to obtain the desired overall length L. Although promoter regions are rarely longer than 500 bp for E. coli and 800 bp for S. cerevisiae, we still include larger datasets to address the more general problem of finding a weak signal within long stretches of genomic sequence. For more details about the data and evaluation of the final results see Additional data file 1.
Starting points
In this exercise, we used the MEME starting-point selection procedure. In the MEME approach, each subsequence in the data, of the prespecified motif width, is converted into a motif matrix and used as a starting point for one step of the EM algorithm. After that one step, the motif matrix that has the highest likelihood for the data, given the model, is used as the single starting point for the EM algorithm and the algorithm runs until convergence. See Bailey and Elkan [2] for more details about this procedure.
Effect of the number of starting points
We ran three variations of the method to evaluate the effect of the number of starting points. First, we compared the popular software MEME with our method. Although there are differences in implementation heuristics between MEME and our method, we can still use the MEME results as a benchmark. For each dataset starting at L = 100 and incrementing by 100 bases, we first ran MEME with the following options: -nmotifs 1 -dna -mod oops -brief -noshorten -b 0 -adj none. These options were chosen so as to match our method as closely as possible.
Next, we also ran our algorithm with λ = 0 according to the MEME starting-point procedure described above. The λ = 0 option for our method essentially ignores the prior knowledge and should be equivalent to the MEME model.
Finally, we increased the number of starting points for our algorithm by taking the top 100 selected by the MEME procedure (that is, the 100 that have the highest likelihood after one step of the EM algorithm). We ran our algorithm with λ = 0 for each of these starting points until convergence. The motif with the highest final likelihood from the 100 starting points was selected as the final motif. There is no option to increase the number of starting points with MEME, but similar results between MEME and our method with λ = 0 for one starting point indicate that the results from running MEME and our method with λ = 0 for 100 starting points will also be similar.
In Table 2, we list the results for each L in the crp test set (the results for the other test sets can be found in Additional data file 2). The length was extended such that the motif is no longer found in the previous four lengths for any of the three runs described above. We also repeated this experiment on the four other datasets that had enough sequences (cpxR, lexA, repcar1, purR). These datasets were declared as having strong signals because we could extend the length up to 2,000 and MEME was still able to discover the correct motif. Therefore, we use these four and the other two that did not have enough sequences (pho4 and gal4) as our training set (see Materials and methods).
Table 2.
Percentage of correctly identified sites for crp using different methods, varying number of starting points and different dataset lengths L
| Method | Number of starting points | L | |||||||
| 100 | 200 | 300 | 400 | 500 | 600 | 700 | 800 | ||
| MEME | 1 | 88 | 76 | 65 | 0 | 0 | 0 | 0 | 0 |
| λ = 0 | 1 | 71 | 76 | 65 | 0 | 0 | 0 | 0 | 0 |
| λ = 0 | 100 | 88 | 76 | 65 | 65 | 0 | 6* | 0 | 0 |
The rows display the results for each method. First row: MEME software with the options -nmotifs 1 -dna -mod oops -brief -noshorten -b 0 -adj none. Second and third rows: the variable change point algorithm with normal prior, λ = 0 and motif width and motif specification (uni- or bimodal) listed in Table 1. The number of starting points in the algorithm was set at 1 or 100. Starting points were selected according to the MEME procedure as described in the text. The entry labeled with an asterisk corresponds to a trial in which the correct motif was not found, but one or two sites were correctly predicted by chance, with a spurious motif.
For all four sets, the results of MEME and our method (λ = 0) with one starting point are similar. Although the exact percentage of predicted sites at each L is not the same for the two methods, they both fail to find the correct motif at the same length. This indicates that the λ = 0 implementation of our algorithm is comparable to MEME, based on the options mentioned above. Overall, for the different test sets, neither method finds the correct motif if L is extended beyond 300 for crp, 700 for rap1, 600 for abf1 and 500 for reb1.
The second and third rows in Table 2 illustrate that the number of starting points affects the discovery of the motif. Except for abf1, by using 100 starting points, our method with λ = 0 is able to find the correct motif in longer lengths. For crp and reb1, the maximum length where the motif is discovered, denoted by L*, is increased by 100 bp, while for rap1, the final length is increased by 700 bp.
These results indicate that up to a certain length, increasing the number of starting points gives the algorithm an advantage for discovering weak motifs as the noise level increases. It is advantageous to use many different starting points because the likelihood surface is high-dimensional with many local maxima. However, having too many starting points compromises the speed of the method. In summary, these results show that for shorter lengths, MEME can be improved by altering its implementation. In the next section, we will show that for longer lengths more starting points do not help and that the changes to the model we propose further improve the method.
Effect of λ
We also used the real test sets to explore the effect of our prior, which is controlled by λ, on the algorithm's performance as L increases. So that the number of starting points is not a confounding factor for interpreting the results, we chose the top 2L starting points selected by the MEME procedure for each dataset of length L. For rap1 we used an alternative starting-point selection procedure, which is discussed in Additional data file 1. The number 2L was arbitrarily chosen so that it was sufficiently large and dependent on the size of the data, which the simulations indicate is an important factor.
We focused on the more challenging datasets to determine whether increasing λ improves the detection of the real sites. We started at the last length, L*, in which the motif is discovered with 100 starting points: for crp, L* = 400; for rap1, L* = 1,400; for abf1, L* = 600; and for reb1, L* = 500. For the four test sets, Table 3 lists results using λ = 0, 10, 20, 30, 50, 100, 200 and 500 for length L ≥ L*. The number of starting points was 2L for each L dataset and the same starting points were used for each λ. The algorithm was run with the variable change point model, normal prior with motif width and specification listed in Table 1. Each entry of the table is the percentage of correctly identified sites from the final motif with the highest likelihood for that particular dataset.
Table 3.
Percentage of correctly identified sites for different values of λ and length L
| L | |||||
| crp | λ | 400 | 500 | 600 | 700 |
| 0 | 47 | 0 | 0 | 0 | |
| 10 | 53 | 47 | 0 | 0 | |
| 20 | 53 | 47 | 0 | 0 | |
| 30 | 53 | 47 | 47 | 0 | |
| 50 | 0 | 0 | 0 | 0 | |
| 100 | 0 | 0 | 6* | 6* | |
| 200 | 0 | 0 | 0 | 0 | |
| 500 | 0 | 0 | 0 | 0 | |
| L | |||||
| abf1 | λ | 600 | 700 | 800 | 900 |
| 0 | 56 | 0 | 0 | 0 | |
| 10 | 56 | 0 | 0 | 0 | |
| 20 | 56 | 56 | 50 | 0 | |
| 30 | 56 | 56 | 50 | 0 | |
| 50 | 50 | 0 | 0 | 0 | |
| 100 | 0 | 0 | 0 | 0 | |
| 200 | 0 | 0 | 0 | 0 | |
| 500 | 0 | 0 | 0 | 0 | |
| L | |||||
| rap1 | λ | 1,400 | 1,500 | 1,600 | 1,700 |
| 0 | 80 | 0 | 0 | 0 | |
| 10 | 80 | 0 | 0 | 0 | |
| 20 | 80 | 0 | 0 | 0 | |
| 30 | 73 | 80 | 67 | 0 | |
| 50 | 73 | 80 | 67 | 0 | |
| 100 | 73 | 0 | 67 | 0 | |
| 200 | 73 | 73 | 67 | 0 | |
| 500 | 67 | 60 | 67 | 0 | |
| L | |||||
| reb1 | λ | 500 | 600 | 700 | |
| 0 | 86 | 0 | 0 | ||
| 10 | 86 | 64 | 0 | ||
| 20 | 79 | 64 | 0 | ||
| 30 | 0 | 0 | 0 | ||
| 50 | 0 | 7* | 0 | ||
| 100 | 0 | 7* | 0 | ||
| 200 | 0 | 0 | 0 | ||
| 500 | 0 | 0 | 0 | ||
Rows show the results with the specified λ value and columns correspond to different values of L. The variable change point model and normal prior is used with motif width and motif specification (uni- or bimodal) listed in Table 1. For each dataset L, the top 2L starting points were used according to the MEME starting point selection procedure. For the rap1 test set, a different set of random starting points was used for each L. Entries labeled with an asterisk correspond to a trial in which the correct motif was not found, but one or two sites were correctly predicted by chance, with a spurious motif.
At the maximum value where the motif is detected with 100 starting points, L = L*, either the same or more sites are identified as λ increases from 0 for each test set. For large values of λ, which results in the prior information dominating the likelihood, the performance drops for most of the test sets. This behavior at large λ is not surprising because perfect conservation at a site is strictly enforced, which is not reflective of these relatively weak signals.
Looking across columns, as expected, the ability to detect the real sites drops as L increases. Adding more starting points does not help with these larger lengths. For example, at L = 500 in crp, even with 1,000 MEME starting points, no sites are identified with λ = 0. The limiting factor does not seem to be the number of starting points in these larger datasets.
As we include the prior information by increasing λ, the motif is detected in many cases for L >L*. The maximum length where we discover the motif is increased by 200 bp for crp, abf1 and rap1 and 100 bp for reb1. In all cases, λ in the range 10-30 is best. The simulations (Figures 7,8) also show that the most drastic improvement in performance appears in this range.
Comparison with BioProspector and Gibbs Motif Sampler
Our method is based on the observation that highly conserved positions tend to be grouped together within the motif. Comparing our method with MEME is unfair because MEME does not use information of this type to search for motifs. However, the software BioProspector and Gibbs Motif Sampler have options for specifying blocks but do not have as restrictive assumptions as our model. With our test sets, we also ran these two methods to evaluate how our algorithm compared to these alternative approaches. See Additional data file 1 for the options used in both software.
BioProspector
For BioProspector, there are two main options: a one-block or a two-block motif. These options are analogous to our uni- and bimodal models. The user specifies the width of the blocks and, for the two-block motif, a flexible gap that separates the two blocks. Their gap is analogous to the middle block in our bimodal motif model, but they allow flexibility in its width.
Table 4 lists the percentage of correctly identified sites for the different runs on the four test sets. Except for reb1, we found that BioProspector is sensitive to the choice of block and gap widths. For the two bimodal examples, the motif was found for the larger datasets, L >L*, only if the gap was specified correctly. When the gap was allowed to vary, the motif was only found for L* + 100. For the unimodal example rap1, BioProspector found the motif for L >L*, only for block width equal to 7. For reb1, BioProspector found the motif in the larger-length datasets regardless of the specified block width.
Table 4.
Percentage of correctly identified sites using different options in BioProspector
| L | ||||||
| crp | Options | 400 | 500 | 600 | 700 | 800 |
| 5:6:5 | 47 | 53 | 65 | 41 | 0 | |
| 4:6:4 | 29 | 41 | 24 | 29 | 0 | |
| 6:6:6 | 41 | 53 | 0 | 0 | 6* | |
| 5:(5-6):5 | 41 | 35 | 0 | 6* | 0 | |
| 5:(6-7):5 | 47 | 0 | 0 | 0 | 0 | |
| 5:(5-7):5 | 47 | 0 | 0 | 0 | 0 | |
| L | ||||||
| abf1 | Options | 600 | 700 | 800 | 900 | |
| 4:5:4 | 56 | 44 | 50 | 33 | ||
| 5:5:4 | 39 | 0 | 0 | 6* | ||
| 5:5:5 | 28 | 0 | 0 | 6* | ||
| 4:(4-5):4 | 78 | 56 | 0 | 0 | ||
| 4:(5-6):4 | 33 | 39 | 0 | 0 | ||
| 4:(4-6):4 | 50 | 44 | 0 | 0 | ||
| L | ||||||
| rap1 | Options | 1,400 | 1,500 | 1,600 | 1,700 | 1,800 |
| W = 5 | 0 | 0 | 0 | 0 | 0 | |
| W = 6 | 0 | 0 | 0 | 0 | 0 | |
| W = 7 | 60 | 60 | 47 | 57 | 47 | |
| W = 8 | 73 | 0 | 0 | 0 | 0 | |
| L | ||||||
| reb1 | Options | 500 | 600 | 700 | ||
| W = 5 | 79 | 71 | 71 | |||
| W = 6 | 71 | 71 | 86 | |||
| W = 7 | 79 | 86 | 79 | |||
| W = 8 | 93 | 71 | 79 | |||
The bimodal examples (crp, abf1) use three options: first block width, gap length and second block width. The values for these options are separated by colons in the table. For example, 5:6:5 corresponds to two blocks of five positions separated by a gap of six positions. The gap ranges are denoted by a dash. For the unimodal motifs (rap1, reb1), there is only one option for the block width, denoted by W. Entries labeled with an asterisk correspond to a trial in which the correct motif was not found, but one or two sites were correctly predicted by chance, with a spurious motif.
Gibbs Motif Sampler
Gibbs Motif Sampler was less successful than BioProspector at discovering the motifs in the test sets. Full results are described in Additional data file 2. In summary, the real motif was never discovered for rap1 and abf1, even at L*. The original fragmentation model was able to find the motif for crp with W = 20 and 24, but only up to L = 500. For the reb1 motif, several different combinations were adequate for its discovery up to length 700.
Discussion
Our results strongly suggest that prior knowledge of specific and general positional constraints on information (conservation) in a transcription-factor binding site greatly helps in its discovery. However, is such knowledge generally available? We believe it is. There are many applications where binding sites for a particular factor are being sought - for example, when the targets of a particular factor have been identified by chromatin immunoprecipitation [29]. The structural class of factors can generally be inferred from homology, and the information profile in turn inferred from related factors. Our method can then be used, allowing only small variations on the constraints obtained from the inferred profile. Where the identity of the factor or factors is not available, a general constraint - allowing for uni- or bimodal motifs of various sizes - can be used and will still be useful because it greatly narrows the space of possible motifs and will therefore improve the specificity of the method.
Below, we discuss several issues regarding the model and the implementation of the algorithm. The original intent of the analysis with real data was to observe the effect of using the prior distribution we proposed. As a byproduct of this analysis, we found that the likelihood surface has many local maxima and that, consequently, the starting points have a critical role. We found that to improve the detection of the correct motifs, the number of starting points should be increased with larger data. These observations suggest that the model-based methods using the EM algorithm can be improved simply by using more starting points or by looking into alternative starting-point procedures. However, there is a limit for this improvement. For the very long lengths, we found that increasing the number of starting points proportionally was no better than using only a fixed number of 100.
For the data with very long lengths, where adding more starting points was not effective, we found that including the prior also improves the performance of the basic model. The extent of improvement varied across the datasets, mostly because of the noise level. It was more drastic in the simulated data, which was much less noisy. Depending on the strength of the motif signal, in all results, a λ value in the range 10 to 50 was adequate. We suggest running the algorithm for a few cases of λ to see if the results are consistent. λ controls the contribution of the prior to the model. If we optimize the likelihood over λ directly, λ would approach zero because the likelihood is maximized when there is no penalty (λ = 0). Thus, in future work, we plan to perform cross-validation trials for determining good values for λ in advance.
Although BioProspector and the variants of Gibbs Motif Sampler were developed to account for motifs with block structure, they do not impose the same restrictions as our method. We tried different values for the options in these programs to reproduce our model specification but obtained different results. However, we used default algorithm settings for both programs, and there could be improvement in the results if options, such as the stopping time for the Markov chain, are altered. It is beyond the scope of the paper to systematically explore such options and optimize the results of these methods.
From the options we selected, BioProspector performed better than Gibbs Motif Sampler. On comparing all three methods, the results from BioProspector and our method are the most similar. This is not surprising, because both rely on explicit block structure within the motif. The specification of blocks in Gibbs Motif Sampler is more indirect. Even with the fragmentation from center option, which tries to force the important positions into blocks at the edges, with the bimodal examples, Gibbs Motif Sampler tends to find motifs where the blocks are contiguous, equivalent to a unimodal motif.
The results show that neither our method nor BioProspector is clearly superior in all cases. Depending on the prior information about the overall width and blocks of the expected motif, one method may be better than the other for different data because they rely on different assumptions. BioProspector performed better than our method when the gap and/or block widths were specified correctly. However, these results relied on more information than we use in our variable change point model. The individual block widths in our method are not specified and the prior on the change point positions is trained by other examples. For a more balanced comparison, the results of BioProspector with a variable gap and different block widths should be evaluated. In that case, our method performed better than BioProspector for all test sets except reb1.
Besides variations in the model assumptions, BioProspector and Gibbs Motif Sampler also differ from our method because both use the Gibbs sampler to obtain the maximum a posteriori estimates for and . The Gibbs sampler, a Markov chain Monte Carlo method, is a stochastic algorithm, where and are sampled iteratively according to their full conditional distributions. The Markov sequence generated by repeatedly sampling from 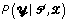 and
and 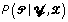 should converge to the joint stationary distribution of and ,
should converge to the joint stationary distribution of and , 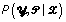 . In contrast, we use the EM algorithm to obtain the maximum a posteriori estimates of the multinomial parameters,
. In contrast, we use the EM algorithm to obtain the maximum a posteriori estimates of the multinomial parameters,  , and use those to calculate
, and use those to calculate 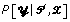 .
.
Although in theory, one starting point for the Gibbs sampler should be adequate, the results on the real data with BioProspector and the Gibbs Motif Sampler suggest that this algorithm may also be affected by the underlying problem that the likelihood function for the data has many local maxima. The developers of these programs recognize this issue and the default option is to use 40 starting points in both BioProspector and Gibbs Motif Sampler. It is possible that using this relatively small number of starting points relies too much on the theoretical argument that the Markov chain generated by the Gibbs sampler samples from the entire parameter space. However, we also ran these methods with 200 starting points and the results were similar to those with the default value of 40.
Advantages and disadvantages of using the Gibbs sampler or the EM algorithm have been addressed in the literature [17-20,30]. In the future, we plan to explore the use of the Gibbs sampler with our model and compare its performance and run time with the EM algorithm. However, the results from both BioProspector and Gibbs Motif Sampler do not indicate that there will be a drastic improvement in performance.
Conclusions
In summary, to improve the discovery of regulatory motifs, we altered the underlying model used in motif-discovery methods. We assigned a prior distribution to the base frequency parameters to capture the uni- or bimodal shapes observed in the information content plots of real binding site examples. Our methods are motivated by structural constraints in protein-DNA complexes and empirical data on binding sites, as observed in Figure 2. We found that building the information content patterns of the motif into the model was advantageous for discovering motifs when the data become noisier or when there is a competing false motif.
Our goal was to alter the original model to improve performance, but to do so in a manner such that the algorithm for parameter estimation did not increase in computational complexity. Therefore, we did not use values of information content directly. Although it is a useful measure to summarize sequence data, information content has an inconvenient functional form. Instead, we focused on a qualitative definition of conservation at a position as a proxy for information content. Another important consideration in the development of this method was to keep it general for different types of transcription factor binding sites. This algorithm can search for one of two major types of motifs, which have either uni- or bimodal information content shape. More specific information content shapes can also be specified through the 'profile' extension of our method [21].
We used the EM algorithm to estimate the parameters and our new model resulted in relatively minor changes to the original EM algorithm in Lawrence and Reilly (see Figure 6). The two forms of our model, fixed and variable change point, required at most one extra update in the E-step: the calculation of the posterior probability of in the variable change point model. For the updates in the M-step, the two different forms of the prior we considered resulted in a closed form solution or an optimization in one dimension. Overall, the changes we proposed in the model result in only a few extra calculations at each iteration of the algorithm. Furthermore, because we used the basic model framework we can relax assumptions in our model (such as one motif occurrence per sequence; OOPS) as has been done with other methods and incorporate other useful extensions, such as palindromicity and alternative background models.
Materials and methods
Data
The S. cerevisiae sites from abf1, gal4, pho4, rap1, reb1 and repressor of car1 (repcar1) were obtained from the promoter database of Saccharomyces cerevisiae (SPCD) [27]. The E. coli sites from purR, cpxR and lexA were obtained from the DPInteract database [28] and the sites from crp were obtained from Berg and von Hippel [31] and Lawrence and Reilly [9]. Several sites were discarded from SCPD because they were either duplicates, unalignable with all other sites or their location in the upstream or downstream region could not be located with the specified ORF or gene label.
For all the real datasets, we used the ORF or gene label to obtain the upstream, or occasional downstream, sequence containing the sites from RSA tools [32]. For crp, two sequences containing sites, colE, a plasmid, and pbr322, a cloning vector, were obtained from the Entrez Nucleotides Database [33] with accession numbers NC_001371 and J01749 respectively. For these two sequences, an 800-bp region was selected so that the binding sites(s) were centrally located. The Crp-binding site labeled 'cat' in the references was not found. Overall there are 17 sequences containing 21 transcription-factor binding sites for crp, 14 upstream and one downstream sequences containing 19 sites for rap1, 14 upstream sequences containing 17 sites for reb1 and 20 upstream sequences containing 23 sites for abf1. The sites for gal4, pho4, cpxR, lexA, repcar1 and purR were used as training sets to estimate the parameters for the prior (see below). The first two sets (gal4 and pho4) consisted of sites that were contained in less than 10 sequences and the last four sets had a strong signal as defined in Results.
Parameters for prior distributions
There are three prior distributions in the two models: g for the location of the motif start site, f for the multinomial parameters at each position in both the fixed and variable change point models, and h for the change point pairs in the variable change point model. We set g as the uniform distribution along each sequence from 1 to L - W + 1, which is common practice in many methods. We explain our selection for the parameters of f in Results. Finally, for h, we use the training set (consisting of four bimodal and two unimodal motifs) to fit the parameters. Recall that h is the distribution on the ratios of the lengths of the three regime blocks to W. These ratios are assumed to follow a discretized form of the Dirichlet distribution. Using the training sets, we used a methods of moment estimator to fit the parameters of the Dirichlet distribution. The estimates are (2.5,5,2.5) showing that the middle block tends to be twice as large as the two end blocks.
Availability
The ANSI C source code of our algorithm, TFEM (Transcription Factor Expectation Maximization), will be made available at [34].
Additional data files
The following files are available with the online version of this paper: a pdf file giving a detailed account of the data for both the simulations and real data analysis, methods for selecting starting points, evaluation diagnostics and a discussion of the options used in BioProspector and Gibbs Motif Sampler (Additional data file 1); and a pdf file giving additional results for the sections 'Effect of the number of starting points' and 'Gibbs Motif Sampler' (Additional data file 2).
Supplementary Material
A pdf file giving a detailed account of the data for both the simulations and real data analysis, methods for selecting starting points, evaluation diagnostics and a discussion of the options used in BioProspector and Gibbs Motif Sampler
A pdf file giving additional results for the sections 'Effect of the number of starting points' and 'Gibbs Motif Sampler'
Acknowledgments
Acknowledgements
We thank Derek Chiang, Alan Moses, Sündüz Keleş, Mark van der Laan, Eric Xing and Dick Karp for valuable conversations and helpful comments. We also thank Mark Richards for converting the R test version of our algorithm to C. This work was partially supported by a National Science Foundation Graduate Research Fellowship (to K.J.K.) and by NIH grant R01 HG002779-02 (to M.B.E.). M.B.E is a Pew Scholar in the Biomedical Sciences.
Contributor Information
Katherina J Kechris, Email: kechris@genome.ucsf.edu.
Erik van Zwet, Email: evanzwet@math.leidenuniv.nl.
Peter J Bickel, Email: bickel@stat.berkeley.edu.
Michael B Eisen, Email: mbeisen@lbl.gov.
References
- Brazma A, Jonassen I, Eidhammer I, Gilbert D. Approaches to the automatic discovery of patterns in biosequences. J Comput Biol. 1998;5:279–305. doi: 10.1089/cmb.1998.5.279. [DOI] [PubMed] [Google Scholar]
- Bailey T, Elkan C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Machine Learning. 1995;21:51–83. doi: 10.1023/A:1022617714621. [DOI] [Google Scholar]
- Liu J, Neuwald A, Lawrence C. Bayesian models for multiple local sequence alignment and Gibbs sampling strategies. J Am Stat Assoc. 1995;90:1156–1170. [Google Scholar]
- Cardon L, Stormo GD. Expectation maximization algorithm for identifying protein-binding sites with variable lengths from unaligned DNA fragments. J Mol Biol. 1992;223:159–170. doi: 10.1016/0022-2836(92)90723-w. [DOI] [PubMed] [Google Scholar]
- Liu X, Brutlag D, Liu J. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput. 2001:127–138. [PubMed] [Google Scholar]
- Roth F, Hughes P, Estep J, Church G. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Thijs G, Marchal K, Lescot M, Rombauts S, Moor BD, Rouzé P, Moreau Y. A Gibbs sampling method to detect overrepresented motifs in upstream regions of coexpressed genes. J Comput Biol. 2002;9:447–464. doi: 10.1089/10665270252935566. [DOI] [PubMed] [Google Scholar]
- Thompson W, Rouchka E, Lawrence C. Gibbs Recursive Sampler: finding transcription factor binding sites. Nucleic Acids Res. 2003;31:3580–3585. doi: 10.1093/nar/gkg608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C, Reilly A. An expectation maximization algorithm for identification and characterization of common sites in unaligned biopolymer sequences. Proteins. 1990;7:41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- Frech K, Herrmann G, Werner T. Computer-assisted prediction, classification, and delimitation of protein binding sites in nucleic acids. Nucleic Acids Res. 1993;21:1655–1664. doi: 10.1093/nar/21.7.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny L, Gelfand M. Structural analysis of conserved base pairs in protein-DNA complexes. Nucleic Acids Res. 2002;30:1704–1711. doi: 10.1093/nar/30.7.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Stormo GD. Consensus patterns in DNA. Methods Enzymol. 1990;183:211–221. doi: 10.1016/0076-6879(90)83015-2. [DOI] [PubMed] [Google Scholar]
- Green P. On use of the EM algorithm for penalized likelihood estimation. J Roy Stat Soc Ser B. 1990;52:443–452. [Google Scholar]
- Stormo G, Schneider T, Gold L, Ehrenfeucht A. Use of the 'Perceptron' algorithm to distinguish translational initiation sites in Escherichia coli. Nucleic Acids Res. 1982;10:2997–3011. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster A, Laird N, Rubin D. Maximum likelihood from incomplete data via the EM algorithm. J Roy Stat Soc Ser B. 1977;39:1–38. [Google Scholar]
- Gelfand A, Smith A. Sampling-based approaches to calculating marginal densities. J Am Stat Assoc. 1990;85:398–409. [Google Scholar]
- Geman S, Geman D. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans Pattern Anal Mach Intell. 1984;6:721–741. doi: 10.1109/tpami.1984.4767596. [DOI] [PubMed] [Google Scholar]
- Smith A, Roberts G. Bayesian computation via the Gibbs sampler and related Markov Chain Monte Carlo methods. J Roy Stat Soc Ser B. 1993;55:3–23. [Google Scholar]
- Kechris K. PhD thesis, University of California, Berkeley, Department of Statistics; 2003. Statistical Methods for Discovering Features in Molecular Sequences. [Google Scholar]
- Draper N, Nostrand RV. Ridge regression and James-Stein estimation: review and comments. Technometrics. 1979;21:451–466. [Google Scholar]
- Tibshirani R. Regression shrinkage and selection via the lasso. J Roy Stat Soc Ser B. 1996;58:267–288. [Google Scholar]
- Brent R. Algorithms for Minimization without Derivatives. Englewood Cliffs, NJ: Prentice-Hall; 1973. [Google Scholar]
- Liu J, Lawrence C. Bayesian inference on biopolymer models. Bioinformatics. 1999;15:38–52. doi: 10.1093/bioinformatics/15.1.38. [DOI] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. A language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- SCPD http://cgsigma.cshl.org/jian
- DPInteract http://arep.med.harvard.edu/dpinteract
- Iyer V, Horak C, Scarfe C, Botstein D, Snyder M, Brown P. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Diebolt J, Ip E. Markov Chain Monte Carlo in Practice. Boca Raton, FL: Chapman and Hall; 1998. A stochastic EM algorithm for approximating the maximum likelihood estimate. [Google Scholar]
- Berg O, von Hippel P. Selection of DNA binding sites by regulatory proteins. II. The binding specificity of cyclic AMP receptor protein to recognition sites. . J Mol Biol. 1988;200:709–723. doi: 10.1016/0022-2836(88)90482-2. [DOI] [PubMed] [Google Scholar]
- Regulatory Sequence Analysis Tools http://rsat.ulb.ac.be/rsat/
- Entrez Nucleotide http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide&itool=toolbar
- Eisen Lab http://rana.lbl.gov
- Spanier J, Oldham K. Atlas of Functions. Washington DC: Hemisphere; 1987. The Cubic Function x3+ax2+bx+c and Higher Polynomials. [Google Scholar]
- Latchman DS. In Eukaryotic Transcription Factors. 3. San Diego CA: Academic Press; 1998. Methods for Studying Transcription. [Google Scholar]
- Ripley B. Stochastic Simulation. New York: Wiley; 1987. [Google Scholar]
- Rosen K, Michaels J, Gross J, Grossman J, Shier D, editor. In Handbook of Discrete and Combinatorial Mathematics. New York: CRC Press; 2000. Sets, Relations, and Functions. [Google Scholar]
- Tatusov R, Altschul S, Koonin E. Detection of conserved segments in proteins: iterative scanning of sequence databases with alignment blocks. Proc Natl Acad Sci USA. 1994;91:12091–12095. doi: 10.1073/pnas.91.25.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A pdf file giving a detailed account of the data for both the simulations and real data analysis, methods for selecting starting points, evaluation diagnostics and a discussion of the options used in BioProspector and Gibbs Motif Sampler
A pdf file giving additional results for the sections 'Effect of the number of starting points' and 'Gibbs Motif Sampler'