

Abstract
Her3 is a member of the human epidermal growth factor receptor (EGFR) tyrosine kinase family, and it is often either overexpressed or deregulated in many types of human cancer. Her3 has not been the subject of small-molecule inhibitor development because it is a pseudokinase and does not possess appreciable kinase activity. We recently reported on the development of the first selective irreversible Her3 ligand (TX1-85-1) that forms a covalent bond with cysteine 721 which is unique to Her3 among all kinases. We also developed a bi-functional compound (TX2-121-1) containing a hydrophobic adamantane moiety and the same warhead of TX1-85-1 that is capable of inhibiting Her3-dependent signaling and growth. Here we report on the structure-based medicinal chemistry effort that resulted in the discovery of these two compounds.
Keywords: Her3, Pseudokinase, Hydrophobic tagging, Cancer, Pyrazolopyrimidine
Graphical Abstract
Her3 is a member of the human epidermal growth factor receptor (EGFR) tyrosine kinase family along with Her1 (EGFR), Her2 and Her4. EGFR family-dependent signaling frequently becomes deregulated in human cancers due to either receptor/ligand over-expression or oncogenic mutations that result in constitutive activation of the kinase domain.1,2 Extensive efforts to target activated mutants of EGFR and Her2 for the treatment of patients with non-small cell lung cancers (NSCLCs) and breast cancer has resulted in development of several FDA-approved drugs such as Gefitinib, Erlotinib, Lapatinib, and Afatinib. In stark contrast, Her3 has not been the intentional target of small-molecule inhibitor discovery efforts because Her3 has been historically classified as a ‘pseudokinase’ because: (1) early biochemical assays showed that Her3 was not capable of catalyzing auto-phosphorylation and phosphorylation of substrates,3,4 (2) Asp 813 of EGFR, a conserved residue acting as a catalytic base for the transfer of the phosphate group, is replaced with the catalytically ineffective Asn 815 in Her3, and (3) substitution of His 740 in Her3 for Glu 738 of EGFR prevents Her3 from forming a key salt bridge that is important for maintaining an active kinase conformation.5,6 A recent report suggests that Her3 may possess very weak kinase activity,6 although it remains unclear whether this activity is required for Her3-dependent signaling. Despite questions regarding its kinase activity, Her3 is well documented as an essential hetero-dimerization partner with EGFR, Her2, and c-Met.1,2,7 The Her3 kinase domain serves as an activator of a heterodimer5,8 resulting in phosphorylation of tyrosine residues in the C-terminus of the heterodimer followed by eventual activation of the PI3K/Akt signaling network.1,2 This crucial hetero-dimerization of Her3 has been suggested as a molecular basis of the acquired resistance in a subset of NSCLCs and breast cancer.7,9 These findings, in addition to the fact that Her3 is over-expressed and deregulated in several human cancers,9-13 have inspired the development of antibody-based antagonists such as Pertuzimab which target the ligand binding domain of Her3.14-17
Here we describe our efforts to develop Her3-targeting small molecules that can down-regulate Her3-dependent signaling. We hypothesized that ATP-competitive Her3 ligands may exhibit pharmacology either because they can induce a conformation of Her3 that results in non-productive heterodimerization or because they block the low but requisite kinase activity of Her3.6 Our strategy to target Her3 was to develop ligands that would be capable of forming a covalent bond with Cys721 which is uniquely present in Her3 relative to all other kinases in the kinome. Cysteine 721 of Her3 is situated on the ‘roof’ of the ATP-binding pocket, located approximately 3.4 Å above the adenine ring of ATP. Kinome-wide sequence alignments demonstrate that this cysteine residue is unique to Her3.18 This suggests that a suitably designed covalent Her3 inhibitor could be exceptionally selective through forming a new covalent bond. One of the common strategies to develop irreversible kinase inhibitors is to employ structure-based design to modify reversible inhibitors with a reactive electrophile.18 When we initiated this work there were no reported Her3 ligands so we developed an ATP-competitive ligand binding assay employing the fluorescence resonance energy transfer (FRET) based LanthaScreen™ Eu methodology19 and screened our in-house library composed of approximately 1500 known ATP-competitive kinase inhibitors. The most potent Her3 binders identified from the screening assay were KIN001-111,20 dasatinib,21 bosutinib,22 and KIN001-51,23,24 all of which possessed IC50 of below 100 nM against Her3 in the binding assay (Fig. 1). We selected KIN001-51 as our starting point to develop covalent Her3 binders because of its molecular simplicity and potential for facile modification to synthesize analogs. Molecular docking studies using the Her3 crystal structure (PDB ID: 3KEX) indicated that the phenyl ring immediately adjacent to the pyrrolopyrimidine ring of KIN001-51 is located close to Cys 721. Modeling results suggested that installation of an electrophile at the meta position of the phenyl ring might provide the correct trajectory for forming a covalent bond with Cys 721 (Fig. 2). Based on these docking study results, SML4-82-1 (1a) possessing a meta-acryl amide, was synthesized as our first covalent Her3 binder. SML4-82-1 was prepared by iodination of 4-amino-7H-pyrrolo[2,3-d]pyrimidine followed by N-alkylation with bromocyclopentane (Scheme 1). The 4-phenoxyphenyl group was installed by a Suzuki coupling using 3-nitro-4-fluorophenylboronic acid and subsequent nucleophilic aryl substitution reaction with phenol. The protection of the exoamine with di-Boc group, reduction of the nitro group, selective formation of an acryl amide followed by the final deprotection of the di-Boc group provided the target compound. The IC50 of SML4-82-1 was 286 nM in the fixed time-point binding assay. In order to determine whether SML4-82-1 could covalently modify Her3, we incubated purified Her3 with SML4-82-1 at a molar ratio of 1:10 (Her3/SML4-82-1) for 3 hours at ambient temperature. Electrospray ionization mass spectrometry detected a single adduct between Her3 and SML4-82-1, although complete labeling was not achieved under the conditions examined (Fig. S1A). Several other incubation conditions were tested to improve the labeling efficiency without success. We attributed this, in part, to the intrinsic instability of purified Her3 protein which prevented incubations longer than 3 hours or elevation of the temperature above 25 ºC. Despite the incomplete labeling, proteolytic digestion and analysis of the resulting peptides by LC/MS demonstrated the exclusive covalent labeling of Cys 721 (Fig. S1B). These combined results demonstrate that SML4-82-1 is a potent, covalent Her3 binder.
Figure 1.
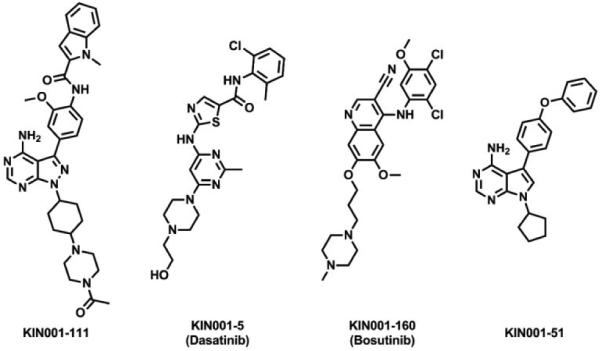
The four most potent Her3 binders obtained from high-throughput screening of our in-house kinase inhibitor library employing a FRET-based protein binding assay.
Figure 2.
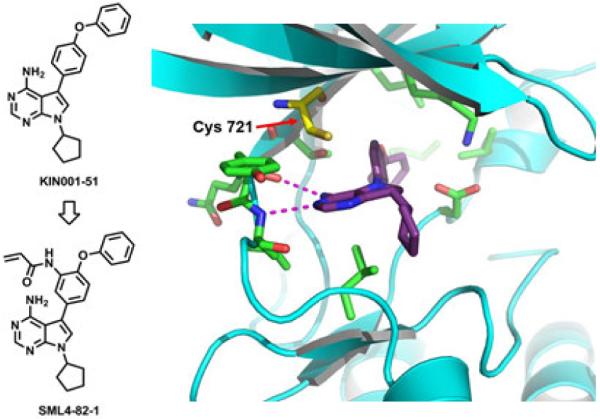
A docking study of KIN001-51 against a Her3 crystal structure (PDB ID: 3KEX) suggested that SML4-82-1, the meta-acryl amide, might be capable of forming a covalent bond with Cys 721 of Her3.
Scheme 1.
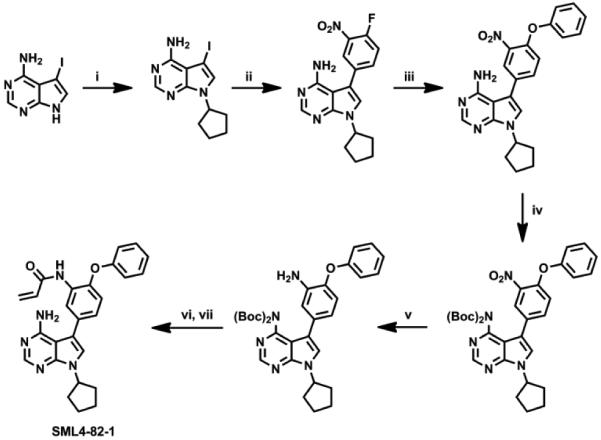
Synthesis of SML4-82-1. Reagents and Conditions: (i) bromocyclopentane, K2CO3, DMF, 100 °C, 28%; (ii) 3-nitro-4-fluorophenylboronic acid, PdCl2(PPh3)2, 1 M Na2CO3, dioxane, 100 °C, 42%; (iii) phenol, K2CO3, DMF, 80 °C, 95%; (iv) (Boc)2O, DMAP, DCM, rt, 70%; (v) Raney Ni, H2, MeOH, rt; (vi) acryloyl chloride, DIPEA, DCM, 0 °C; (vii) TFA, DCM, rt, 61% for 3 steps.
Despite SML4-82-1 being a covalent binder, based on our fixed 3 hour binding assay, its IC50 is approximately 3-fold less potent than the reversible, non-acrylamide modified KIN001-51 (IC50 = 85 nM). One possible interpretation is that Her3 labeling is incomplete after 3 hours in agreement with the labeling efficiency determined by mass spectrometry. We therefore hypothesized that optimization of the electrophile or the trajectory of the acrylamide moiety would improve the potency of this compound. Therefore, we screened several different electrophiles varying the length and the reactivity to identify ones that could more rapidly modify Her3. The α-chloroacetamide 1b and the methylene acrylamide 1c were synthesized analogously to SML4-82-1. The α,β-unsaturated sulfone 1e, methyl ester 1f, and ketone 1g were synthesized by a Suzuki coupling reaction with 4-fluoro-3-formylphenylboronic acid followed by Horner–Wadsworth–Emmons reaction with corresponding phosphonate esters respectively. Unfortunately, none of these alternative electrophiles resulted in more efficient labeling of Her3 relative to the acryl amide (Table 1). We next sought to alter the dihedral angle between the pyrrole core and phenyl substituent by introducing ortho nitrogen. We observed that switching from the pyrrolopyrimidine core to the pyrazolopyrimidine (1a vs 1d) increased the potency by approximately 2 fold (Table 1). Considering the most potent hit KIN001-111 also contains the pyrazolopyrimidine core, we decided to synthesize acryl amides of the pyrazolopyrimidine for the further development of potent, covalent Her3 binders.
Table 1.
Effects of electrophiles and heterocyclic cores on potency of compounds.
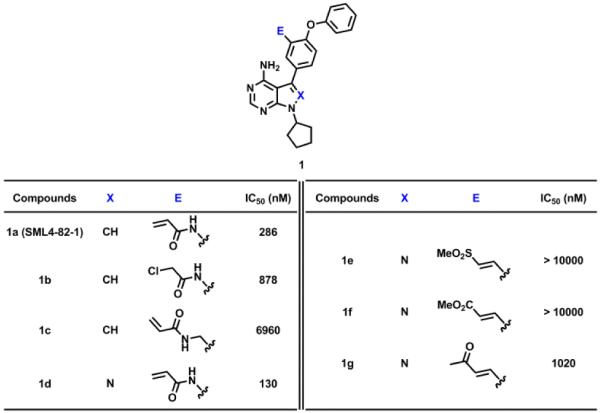
|
To further improve the labeling efficiency of the compounds, we employed structure-guided hybridization of our lead compounds with alternative Her3 scaffolds as this has previously proven to be a productive approach.25,26 We envisioned that incorporation of the solubility enhancing tail of the most potent hit KIN001-111 to 1d might provide more potent covalent Her3 binders, which led us to synthesize TX1-85-1 (Fig. 3). The synthesis of TX1-85-1 commenced with installation of the solubilizing tail to 3-iodo-4-aminopyrazolo[3,4-d]pyrimidine through a 3-step sequence: Mitsunobu reaction with 1,4-dioxaspiro[4.5]decan-8-ol, deprotection of the ketal group, and reductive amination with 1-acetylpiperazine (Scheme 2). The synthesis of TX1-85-1 was completed using the same route as we described in the synthesis of SML4-82-1. TX1-85-1 possessed a substantially improved IC50 of 23 nM in our FRET-based binding assay, approximately 6 times more potent than 1d. Electrospray ionization mass spectrometry unambiguously revealed covalent modification of Her3 by TX1-85-1, although the labeling was still not complete after the 3 hour incubation (Fig. S1C).
Figure 3.

Design of TX1-85-1 by structure-guided hybridization between 1d and KIN001-111. TX1-85-1 is composed of the covalent bond forming moiety of 1d (square) and the solubilizing tail of KIN001-111 (oval).
Scheme 2.
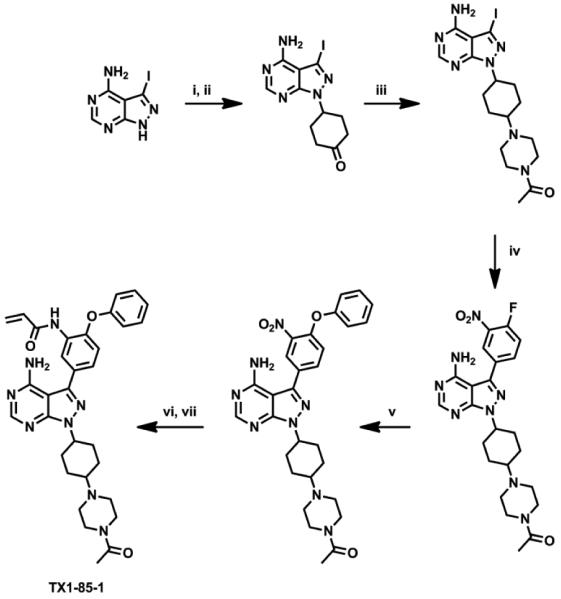
Synthesis of TX1-85-1. Reagents and Conditions: (i) 1, 4-dioxaspiro[4.5]decan-8-ol, Ph3P, DEAD, THF, rt; (ii) acetone, 5 M HCl, 45% for 2 steps; (iii) 1-acetylpiperazine, Na(OAc)3BH, ClCH2CH2Cl, rt, 62%; (iv) 3-nitro-4-fluorophenylboronic acid, PdCl2(PPh3)2, K2CO3, dioxane/H2O (3:1), 100 °C, 75%; (v) phenol, K2CO3, DMF, 80 °C, 90%; (vi) Raney Ni, H2, MeOH, rt; (vii) acryloyl chloride, THF, NaHCO3, 0 °C, 35% over 2 steps.
We investigated the specificity of TX1-85-1 against the entire kinome. A live cell chemical proteomics experiment using the KiNativ approach27-29 demonstrated that TX1-85-1 potently binds to Her3 as well as to Lyn, Her2 and several other Src family kinases at 2 µM (Table S1). We next evaluated the ability of TX1-85-1 to inhibit Her3-dependent signaling and cell proliferation. However, as reported previously,30 TX1-85-1 showed an EC50 of greater than 10 µM in cancer cell lines validated as Her3 dependent for growth by siRNA-mediated depletion of Her3.30 Moreover, there was no inhibition of Akt phosphorylation, an important downstream effector of Her3 at a concentration of 5 µM. These results indicate that despite successful intracellular target engagement of Her3 by TX1-85-1,30 the compound is not capable of inhibiting Her3-dependent growth and signaling in the lines we tested. Collectively, although we cannot exclude the possibility of residual Her3 activity following inhibitor treatment, these data suggest that inhibition of low kinase activity of Her3 with ATP-competitive, covalent Her3 binders may not be an effective strategy to inhibit Her3-dependent functions.
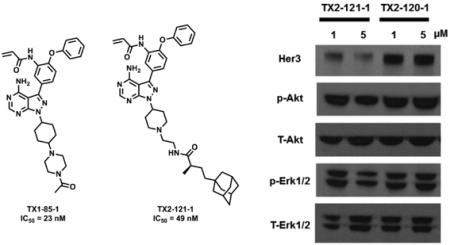
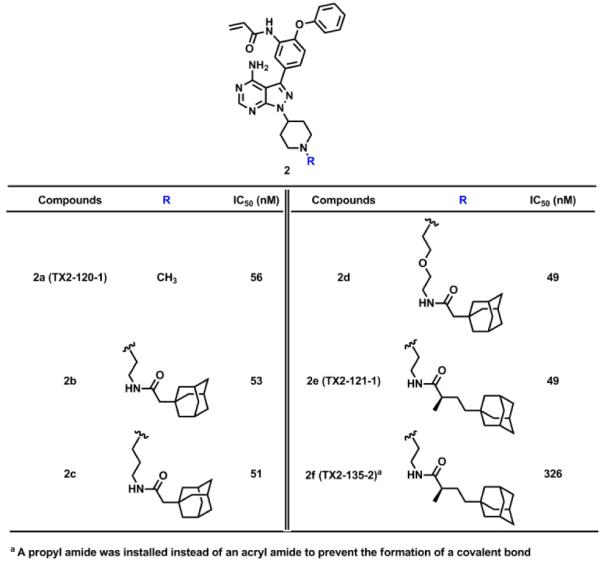
Her3 requires hetero-dimerization with either EGFR, Her2, and c-Met to initiate Her3 cellular signaling. Despite its low kinase activity, the Her3 kinase domain has proven to serve as an “activator” within the asymmetric heterodimer resulting in Her3-dependent signaling.5,8 We hypothesized that perturbation of Her3 dimer may impair the activating function of Her3 and eventually inhibit Her3-dependent functions. One approach we envisioned was targeted, selective Her3 degradation by using our selective, covalent Her3 binders as a foothold. Recent reports have shown that covalent modification of target proteins with hydrophobic groups can result in proteasome-mediated degradation.31-33 A detailed understanding of this phenomenon has not been fully achieved but presumably relies on mimicry of a partially unfolded state for the target protein by adding a hydrophobic tag which turns on the protein homeostasis machinery degrading the target protein by the proteasome.31 We modified our covalent Her3 binders with dense hydrophobic adamantine tags, which have been used previously for induction of proteasomal degradation with a minimal increase of molecular weight.32 One of these bivalent conjugates TX2-121-1 (2e) was prepared by N-alkylation of 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine with tert-butyl 4-bromopiperidine-1-carboxylate followed by deprotection of Boc group to furnish a free piperidine (Scheme 3). Another N-alkylation with N-boc-2-bromoethylamine, subsequent installation of the 4-phenoxyphenol group, and deprotection of Boc group provided a free primary amine. The adamantane moiety was introduced by an EDC-mediated amide coupling with (R)-4-((3R,5R,7R)-adamantan-1-yl)-2-methylbutanoic acid,32 and the final formation of an acryl amide completed the synthesis of TX2-121-1. The other four compounds were prepared in an analogous fashion. The FRET-based binding assay indicated that these adamantane conjugates still retain the ability of binding to Her3, showing IC50s of most conjugates were close to 50 nM (Table 2). We also performed a live cell chemical proteomics experiment using the KiNativ approach for TX2-121-1, which proved again that TX2-121-1 is a selective binder of Her3 at 2 µM (Table S1). Next, we tested the feasibility of the degradation of Her3 with 4 adamantane conjugates and TX2-120-1 (2a) (a negative control possessing no adamantane group) that possessed an IC50 of approximately 50 nM. Treatment of PC9 GR4 cells with 4 conjugates at concentrations of 1 and 5 µM for 12 hours induced partial degradation of Her3 (Fig. 4). The degree of Her3 degradation was dependent on both the type and length of the linker, and the best potency was seen with TX2-121-1 which we advanced for further investigation of the effects of the adamantane conjugates on Her3-dependent cell growth and signaling. To examine the requirement of the covalent bond formation, we replaced the reactive acryl amide group with the non-reactive propyl amide to produce a negative control (TX2-135-2 (2f)) that maintained the binding ability to Her3 showing an IC50 of 326 nM (Table 2).
Scheme 3.
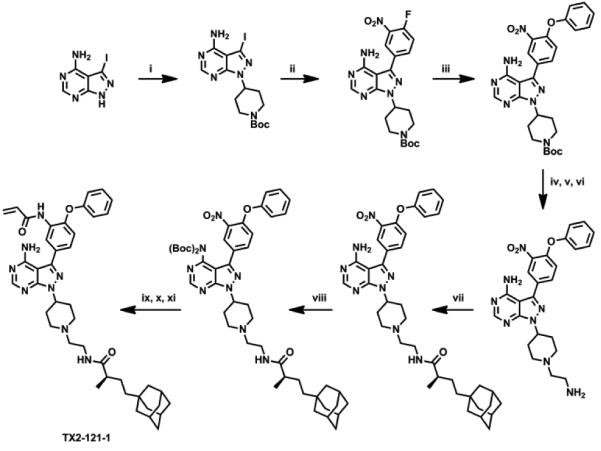
Synthesis of TX2-121-1. Reagents and Conditions: (i) tert-butyl 4-bromopiperidine-1-carboxylate, K2CO3, DMF, 64%; (ii) 3-nitro-4-fluorophenylboronic acid, PdCl2(PPh3)2, K2CO3, dioxane/H2O (3:1), 100 °C, 75%; (iii) phenol, K2CO3, DMF, 80 °C, 90%; (vi) 1 M HCl, THF, rt, 95%; (v) N-boc-2-bromoethylamine, K2CO3, DMF, 80 °C, 81%; (vi) 1 M HCl, THF, rt, 98%; (vii) (R)-4-((3R,5R,7R)-adamantan-1-yl)-2-methylbutanoic acid, EDCI, DMAP, DIPEA, DCM, 43%; (viii) (Boc)2O, DMAP, DCM, 95%; (ix) Raney Ni, H2, MeOH, rt; (x) acryloyl chloride, THF, NaHCO3, 0 °C; (xi) TFA, DCM, rt, 32% over 3 steps.
Table 2.
Bivalent TX1-85-1-adamantane conjugates to induce the degradation of Her3.
|
Figure 4.
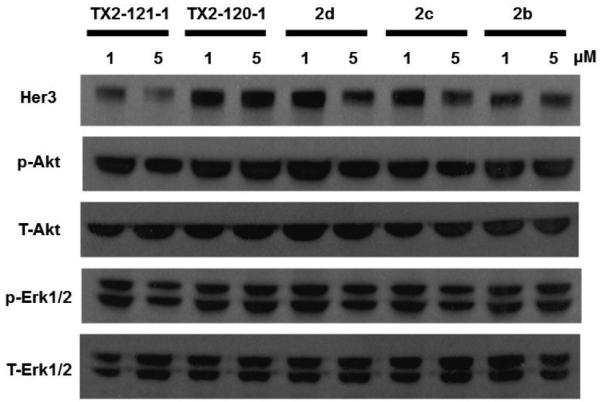
After treatment of PC9 GR4 cell lines with bi-functional conjugates for 12 hours at concentrations of 1 and 5 µM, the degradation of Her3 was most evident with TX2-121-1 at both concentrations.
With TX2-121-1 and its two different negative controls (TX2-135-2 and TX2-120-1) in hand, we investigated and compared the effect of these three compounds on downstream signaling of Her3 by western blot.30 We observed that treatment of TX2-121-1 with serum-starved PC9 GR4 cells for 12 hours at concentrations of 0.5 and 2 µM followed by stimulation with Neuregulin (NRG) attenuated phosphorylation of downstream Her3 effectors (Erk and Akt), while the non-covalent analogs, TX2-135-2 and TX2-120-1, lacking the adamantane group were less effective at inducing Her3 degradation or blocking Her3-dependent signaling.30 Furthermore, an anti-proliferation assay with PC9 GR4 cell lines also demonstrated that TX2-121-1 was approximately 7-fold more potent than TX2-135-2, and TX2-120-1 apparently showed little anti-proliferation effects.30 These combined results emphasize the importance of bivalent conjugates in that both covalent modification of Her3 and the hydrophobic adamantane moiety are required to inhibit Her3-dependent signaling and cell proliferation. Finally, we examined the effect of the partial degradation of Her3 on the hetero-dimerization of Her3, especially with Her2 and c-Met by treating PC9 GR4 cells with TX2-121-1 at 1 µM for 6 hours and washed out unbound compounds. We observed that unlike TX2-135-2, TX2-121-1 reduced the co-precipitation of both Her2 and c-Met with Her3, which indicates that our bivalent conjugates can also perturb hetero-dimerization of Her3.30
To summarize, we have developed the first ATP-competitive, covalent Her3 binders targeting Cys 721 using high-throughput screening followed by structure-based drug design. The most potent covalent Her3 binder, TX1-85-1 was obtained by structure-guided hybridization between the most potent screening hit KIN001-111 and 1d which was evolved from the very first covalent Her3 binder SML4-82-1 by adapting the pyrazolopyrimidine core. The covalent modification of Her3 by TX1-85-1 was unambiguously confirmed by mass spectrometry, and the KiNativ profiling demonstrated that TX1-85-1 is a highly selective Her3 binder. Despite complete intracellular target engagement of Her3 with TX1-85-1, however, TX1-85-1 did not effectively inhibit either Her3-dependent signaling or cell proliferation, suggesting that inhibition of the low kinase activity of Her3 may not be effective for blocking Her3-dependent biological functions. It is well established that hetero-dimerization of Her3 is required to activate Her3-dependent signaling, and perturbation of the hetero-dimerization of Her3 has emerged as an alternative approach to inhibit Her3-dependent signaling. For this purpose, we pursued the hydrophobic tagging approach for the selective degradation of Her3, resulting in the synthesis of the bivalent TX1-85-1-admantane conjugate, TX2-121-1. This bivalent conjugate indeed induced the partial degradation of Her3, attenuated phosphorylation of downstream effectors of Her3, and suppressed the growth of Her3-dependent cell lines. Furthermore, TX2-121-1 perturbed the hetero-dimerization of Her3 with either Her2 or c-Met. Both the covalent bond formation and the adamantane moiety were required for these effects. Although we need to further increase potency of these bivalent conjugates in order to make them clinically meaningful, detailed mechanistic investigation of the protein degradation process will likely enable the optimization of linkers and hydrophobic tags to produce more potent bi-functional conjugates. Her3 has been classified as a ‘pseudokinase’, thus it has been believed ‘undruggable’ by small molecules. Our results provide one of the first successful demonstrations of employing small molecules to target Her3-dependent functions, and we hope to stimulate subsequent research efforts to inhibit Her3-dependent signaling with small molecules.
Supplementary Material
Acknowledgments
This work is supported by the Dana Farber Cancer Institute Lander Fellowship, Claudia Adams Barr Program Award, US National Institutes of Health (NIH) grant AI084140, Cancer Prevention Research Institute of Texas grant R1207, creative/challenging research program of National Research Foundation of Korea NRF-2011-0028676, and NIH grant P01 CA154303.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at
References and notes
- 1.Yarden Y, Sliwkowski MX. Nat. Rev. Mol. Cell Biol. 2001;2:127. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Hynes NE, Lane HA. Nat. Rev. Cancer. 2005;5:341. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 3.Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL. Proc. Natl. Acad. Sci. U.S.A. 1994;91:8132. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sierke SL, Cheng KR, Kim HH, Koland JG. Biochem. J. 1997;322:757. doi: 10.1042/bj3220757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jura N, Shan YB, Cao XX, Shaw DE, Kuriyan J. Proc. Natl. Acad. Sci. U.S.A. 2009;106:21608. doi: 10.1073/pnas.0912101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi FM, Telesco SE, Liu YT, Radhakrishnan R, Lemmon MA. Proc. Natl. Acad. Sci. U.S.A. 2010;107:7692. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale C–M, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA. Science. 2007;316:1039. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 8.Zhang XW, Gureasko J, Shen K, Cole PA, Kuriyan J. Cell. 2006;125:1137. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Nature. 2007;445:437. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baselga J, Swain SM. Nat. Rev. Cancer. 2009;9:463. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 11.Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, Cheng CL, Wang CH, Terng HJ, Kao SF, Chan WK, Li HN, Liu CC, Singh S, Chen WJ, Chen JJ, Yang PC. New Engl. J. Med. 2007;356:11. doi: 10.1056/NEJMoa060096. [DOI] [PubMed] [Google Scholar]
- 12.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, Manning HC, Chang J, Arteaga CL. Proc. Natl. Acad. Sci. U.S.A. 2011;108:5021. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, Moustafa Z, Thomas RK, Greulich H, Schinzel A, Zaghlul S, Batt D, Ettenberg S, Meyerson M, Schoeberl B, Kung AL, Hahn WC, Drapkin R, Livingston DM, Liu JF. Cancer Cell. 2010;17:298. doi: 10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, Totpal K, Wong A, Lee CV, Stawicki S, Clark R, Fields C, Lewis Phillips GD, Prell RA, Danilenko DM, Franke Y, Stephan JP, Hwang J, Wu Y, Bostrom J, Sliwkowski MX, Fuh G, Eigenbrot C. Cancer Cell. 2011;20:472. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, Burenkova O, Pace E, Walton Z, Nie L, Fulgham A, Song Y, Nielsen UB, Engelman JA, Wong KK. Cancer Res. 2010;70:2485. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berlin J, Keedy VL, Jänne PA, Yee L, Rizvi NA, Jin X, Copigneaux C, Hettmann T, Beaupre DM, LoRusso A. 2011 ASCO Annual Meeting; 2011. P. Abstract of Papers. Chicago, IL; American Society of Clinical Oncology: Alexandria, VA. Abstract 3026. [Google Scholar]
- 17.Baselga J, Cortés J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, Clark E, Benyunes MC, Ross G, Swain SM, CLEOPATRA Study Group New Engl. J. Med. 2012;366:109. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Chem. Biol. 2013;20:146. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lebakken CS, Riddle SM, Singh U, Frazee WJ, Eliason HC, Gao Y, Reichling LJ, Marks BD, Vogel KW. J. Biomol. Screen. 2009;14:924. doi: 10.1177/1087057109339207. [DOI] [PubMed] [Google Scholar]
- 20.Burchat A, Borhani DW, Calderwood DJ, Hirst GC, Li B, Stachlewitz RF. Bioorg. Med. Chem. Lett. 2006;16:118. doi: 10.1016/j.bmcl.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 21.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RI, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, de Fex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J, Barrish JC. J. Med. Chem. 2006;49:6819. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- 22.Boschelli DH, Ye F, Wang YD, Dutia M, Johnson SL, Wu B, Miller K, Powell DW, Yaczko D, Young M, Tischler M, Arndt K, Discafani C, Etienne C, Gibbons J, Grod J, Lucas J, Weber JM, Boschelli F. J. Med. Chem. 2001;44:3965. doi: 10.1021/jm0102250. [DOI] [PubMed] [Google Scholar]
- 23.Arnold LD, Calderwood DJ, Dixon RW, Johnston DN, Kamens JS, Munschauer R, Rafferty P, Ratnofsky SE. Bioorg. Med. Chem. Lett. 2000;10:2167. doi: 10.1016/s0960-894x(00)00441-8. [DOI] [PubMed] [Google Scholar]
- 24.Burchat AF, Calderwood DJ, Hirst GC, Holman NJ, Johnston DN, Munschauer R, Rafferty P, Tometzki GB. Bioorg. Med. Chem. Lett. 2000;10:2171. doi: 10.1016/s0960-894x(00)00442-x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Yang PL, Gray NS. Nat. Rev. Cancer. 2009;9:28. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghose AK, Herbertz T, Pippin DA, Salvino JM, Mallamo JP. J. Med. Chem. 2008;51:5149. doi: 10.1021/jm800475y. [DOI] [PubMed] [Google Scholar]
- 27.Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Biochemistry. 2007;46:350. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- 28.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, Kozarich JW. Chem. Biol. 2011;18:699. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cravatt BF, Wright AT, Kozarich JW. Annu. Rev. Biochem. 2008;77:383. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 30.Xie T, Lim SM, Westover KD, Dodge ME, Ercan D, Ficarro SB, Udayakumar D, Gurbani D, Tae HS, Riddle SM, Sim T, Marto JA, Jänne PA, Crews CM, Gray NS. Nat. Chem. Biol. 2014;10:1006. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM. Nat. Chem. Biol. 2011;7:538. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tae HS, Sundberg TB, Neklesa TK, Noblin DJ, Gustafson JL, Roth AG, Raina K, Crews CM. Chembiochem. 2012;13:538. doi: 10.1002/cbic.201100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long MJ, Gollapalli DR, Hedstrom L. Chem. Biol. 2012;19:629. doi: 10.1016/j.chembiol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.