

Abstract
Maternal smoking during pregnancy repeatedly exposes the developing fetus to nicotine and is linked with attention deficits in offspring. Corticothalamic neurons within layer VI of the medial prefrontal cortex are potential targets in the disruption of attention circuitry by nicotine, a process termed teratogenesis. These prefrontal layer VI neurons would be likely targets because they are developmentally excited and morphologically sculpted by a population of nicotinic acetylcholine receptors (nAChRs) that are sensitive to activation and/or desensitization by nicotine. The maturational effects of these α4β2* nAChRs and their susceptibility to desensitization are both profoundly altered by the incorporation of an α5 subunit, encoded by the chrna5 gene. Here, we investigate nicotine teratogenesis in layer VI neurons of wildtype and α5−/− mice. In vivo chronic nicotine exposure during development significantly modified apical dendrite morphology and nAChR currents, compared with vehicle control. The direction of the changes was dependent on chrna5 genotype. Surprisingly, neurons from wildtype mice treated with in vivo nicotine resembled those from α5−/− mice treated with vehicle, maintaining into adulthood a morphological phenotype characteristic of immature mice together with reduced nAChR currents. In α5−/− mice, however, developmental in vivo nicotine tended to normalize both adult morphology and nAChR currents. These findings suggest that chrna5 genotype can determine the effect of developmental in vivo nicotine on the prefrontal cortex. In wildtype mice, the lasting alterations to the morphology and nAChR activation of prefrontal layer VI neurons are teratogenic changes consistent with the attention deficits observed following developmental nicotine exposure.
Keywords: Medial prefrontal cortex, Nicotinic acetylcholine receptors, In vivo developmental nicotine, Corticothalamic, Electrophysiology
1. Introduction
It is estimated that between 10 and 14 percent of women in North America smoke cigarettes regularly during pregnancy (Osadchy et al., 2009; Tong et al., 2009). Exposure of the developing fetus to cigarette smoke is linked statistically with persistent neurobehavioral consequences including attention deficits that can manifest as Attention Deficit Hyperactivity Disorder (ADHD) (Pauly and Slotkin, 2008; Thakur et al., 2013). Recent experimentation in rodent models suggests that nicotine, a major psychoactive component of cigarette smoke, likely contributes directly to the pathogenesis of these attention deficits (Counotte et al., 2011; Schneider et al., 2011). However, the neurobiological mechanisms by which chronic developmental nicotine exposure disrupts developing attention circuits are not understood, significantly limiting the potential to develop appropriate therapeutic intervention for affected children.
Nicotinic acetylcholine receptors (nAChRs) located on corticothalamic pyramidal neurons within layer VI of the medial prefrontal cortex (mPFC) are uniquely positioned to mediate developmental effects of in vivo nicotine on prefrontal attention circuitry. Stimulation of α4β2* nAChRs on these neurons by the neurotransmitter acetylcholine is critical for optimal attention performance in mature animals (Bailey et al., 2010; Guillem et al., 2011). Signaling at these nAChRs is tightly regulated during development (Bailey et al., 2012; Kassam et al., 2008; Winzer-Serhan and Leslie, 2005), and normal activation of these receptors appears to be required for the morphological maturation of mPFC layer VI neuron apical dendrites from an immature morphological pattern characterized by long apical dendrites that predominantly travel across the width of the mPFC to terminate within cortical layer I, to a mature morphological pattern wherein approximately only half of the apical dendrites terminate within cortical layer I (Bailey et al., 2012). We hypothesize that the larger currents and greater calcium influx of α5 subunit-containing α4β2* nAChRs on mPFC layer VI neurons (Bailey et al., 2010; Tapia et al., 2007) contribute to the cellular mechanisms underlying apical dendritic retraction for a subset of this neuronal population during postnatal maturation (Bailey et al., 2012; Pugh and Berg, 1994). Chronic in vivo nicotine exposure may subvert this developmental role for nAChRs through its ability to desensitize these receptors (Cohen et al., 2005; Pidoplichko et al., 1997; Quick and Lester, 2002), potentially imparting long-term effects on prefrontal attention systems.
To test the hypothesis that developmental in vivo nicotine would change the apical dendrite maturation and nAChR currents of layer VI neurons, we chronically exposed mice to either in vivo nicotine or vehicle control during prenatal and early postnatal development, and examined layer VI neurons in young mice near the end of the treatment period and in adulthood. In these neurons, the nAChR α5 subunit (encoded in mouse by the gene chrna5) appears to normally associate with α4β2* nAChRs, significantly influencing receptor function (Bailey et al., 2010; Kuryatov et al., 2008) and making these receptors resistant to upregulation by in vivo nicotine (Gahring and Rogers, 2010; Mao et al., 2008). In order to determine the contribution of the α5 subunit towards nicotine teratogenesis in this neuronal population, we performed these experiments in wildtype mice and also in mice genetically deleted for chrna5 (α5−/−). We also examined the electrophysiological responses of layer VI neurons to stimulation of nAChRs as well as to desensitization by acute nicotine in brain slice.
In wildtype mice, developmental in vivo nicotine exposure allowed an immature pattern of long apical dendrites to persist into adulthood. In addition, it decreased acute nAChR responses compared with vehicle control both in young and adult mice. In short, developmental in vivo nicotine makes mPFC layer VI neurons of wildtype mice resemble the morphological and electrophysiological phenotypic patterns of those observed in α5−/− mice in previous studies (Bailey et al., 2010, 2012), as well as in the α5−/− mice treated with vehicle control in this current study. These findings suggest that developmental in vivo nicotine acts by desensitizing α5 subunit-containing nAChRs. By contrast, developmental in vivo nicotine exerts a different pattern of changes in layer VI neurons of α5−/− mice, in the opposite direction to its effects in wildtype mice. In summary, in vivo nicotine exposure during development has lasting consequences for the morphology and nAChR activation of medial prefrontal layer VI neurons, and the direction of these effects is determined by chrna5 genotype.
2. Materials and methods
2.1. Experimental animals and developmental in vivo nicotine treatment
Genetically modified mice on a C57BL/6 background had all nAChR α4 subunits labeled with YFP (Nashmi et al., 2007) and were either homozygous wildtype for the nAChR α5 subunit or homozygous null for the nAChR α5 subunit (α5−/−) (Salas et al., 2003). Mice were bred in separate homozygous lines that were less than five generations removed from shared parents that were heterozygous null for the nAChR α5 subunit (α5+/−). The α4-YFP staining was used to verify neuronal localization within layer VI, as previously described (Bailey et al., 2012). Timed-pregnant nulliparous females of each genotype were randomly assigned to receive ad libitum access to food and water containing either 200 μg/mL nicotine hydrogen tartrate (calculated as free base) and 2% (wt/vol) saccharin sodium salt hydrate, pH 7.0, or equimolar tartaric acid and 2% (wt/vol) saccharin sodium salt hydrate, pH 7.0, throughout gestation and postnatal development until offspring weaning on postnatal day (P) 21. Solutions were prepared fresh twice per week. The concentration of nicotine was selected for in vivo treatment based on previous studies examining nicotine teratogenesis in C57BL/6 mice (Heath et al., 2010; Pauly et al., 2004) and because it has been shown to lead to levels of the metabolite cotinine in pregnant mice (Pauly et al., 2004; Sparks and Pauly, 1999) similar to those observed in pregnant women who are heavy smokers (Klebanoff et al., 1998; Kvalvik et al., 2012; Mercelina-Roumans et al., 1996). Mice were separated according to sex at weaning and housed in groups of two to four per cage at an ambient temperature of 22 °C with a 12-h light/dark cycle with lights on at 7:00 A.M. Neurons located within layer VI of the mPFC from male offspring were examined for nAChR currents and morphology while the mice were young (P14-20) and in adulthood (P115-150), as described below. Refer to Fig. 1 for a schematic illustrating the timeline of developmental drug treatment and the ages at which offspring were examined. Five offspring from each experimental group were examined for young mice and four offspring were examined from each experimental group in adulthood. Male mice were randomly sampled from at least three litters per experimental group and age, with the exception of adult male wildtype vehicle control mice, which where sampled from two litters. All data for adult male wildtype vehicle control mice in this study were not statistically different from data generated previously in our laboratory for adult wildtype mice (Bailey et al., 2012). All efforts were made to minimize animal suffering and to limit the number of mice used. Experimental animals were cared for according to the principles and guidelines of the Canadian Council on Animal Care and the experimental protocol was approved by the University of Toronto Animal Care Committee.
Fig. 1.

Schematic illustration showing the timing of developmental chronic in vivo treatments in both genotypes, the ages at which young and adult male offspring were examined, and the measurements that were made from layer VI pyramidal neurons.
2.2. Electrophysiology and neuron morphology
Detailed methods for electrophysiology and neuron morphological analysis have been described previously (Bailey et al., 2012). Briefly, 400 μm thick coronal slices of the mPFC were prepared from offspring at postnatal week 3 (P14-20; hereafter termed “young” mice) and in adulthood (P115-150; hereafter termed “adult” mice) using the appearance of white matter and the corpus callosum as anterior and posterior guides (Gabbott et al., 2005; Paxinos and Franklin, 2001). Slices were cut in 4°C oxygenated sucrose artificial cerebrospinal fluid (ACSF) (254 mM sucrose, 10 mM D-glucose, 24 mM NaHCO3, 2 mM CaCl2, 2 mM MgSO4, 3 mM KCl, 1.25 mM NaH2PO4, pH 7.4) and then recovered for at least two hours in 30°C oxygenated ACSF (composition listed above except 128 mM NaCl was substituted for sucrose). Slices were placed with their posterior sides facing up and analyzed in a modified chamber (Warner Instruments, Hamden, CT, USA) mounted on the stage of an Olympus BX50WI microscope (Olympus, Richmond Hill, ON, Canada), with room temperature oxygenated ACSF flowing over them at a rate of 3–4 mL/min.
Analyzed mPFC neurons were located medial to the white matter of each brain slice, within layer VI of the prelimbic, infralimbic and anterior cingulate cortical regions (Paxinos and Franklin, 2001). The neuron sampling location did not influence any measure in this study. Whole-cell recording of these mPFC layer VI neurons was performed using electrodes containing 120 mM K-gluconate, 5 mM KCl, 2 mM MgCl2, 4 mM K2-ATP, 400 μM Na2-GTP, 10 mM Na2-phosphocreatine, 10 mM HEPES buffer and 0.3% (wt/vol) Neurobiotin Tracer (Vector Laboratories, Burlington, ON, Canada) (adjusted to pH 7.3 with KOH). Recordings were made using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), acquired at 20 kHz and lowpass filtered at 2 kHz using a Digidata 1440A data acquisition system (Molecular Devices), and corrected for the liquid junction potential. Voltage clamp experiments were performed at −75 mV in the presence of 200 nM atropine (to block muscarinic acetylcholine receptors). Each experimental group contained at least 16 neurons in brain slices from at least 4 mice, as illustrated in Table 1.
Table 1.
Neuron electrophysiological properties.
| Wildtype mice
|
α5−/− mice
|
||||
|---|---|---|---|---|---|
| Vehicle control | Developmental in vivo nicotine | Vehicle control | Developmental in vivo nicotine | ||
| Young | Number of neurons | 26 | 24 | 25 | 21 |
| Number of mice | 5 | 5 | 5 | 5 | |
| Membrane potential (mV) | −76.9 ± 0.7 | −76.5 ± 0.5 | −77.3 ± 0.7 | −74.9 ± 1.1 | |
| Input resistance (MΩ) | 164.5 ± 12.7 | 188.1 ± 16.2 | 198.6 ± 22.8 | 177.5 ± 14.9 | |
| Spike amplitude (mV) | 100.8 ± 1.6 | 100.1 ± 1.6 | 99.1 ± 1.6 | 96.6 ± 2.3 | |
| sEPSC frequency (Hz) | 1.0 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | |
| sEPSC amplitude (pA) | 15.4 ± 0.8 | 14.4 ± 0.6 | 13.9 ± 0.8 | 15.4 ± 1.3 | |
| Adult | Number of neurons | 19 | 17 | 16 | 16 |
| Number of mice | 4 | 4 | 4 | 4 | |
| Membrane potential (mV) | −76.8 ± 0.8 | −78.2 ± 0.5 | −78.2 ± 0.5 | −78.1 ± 1.0 | |
| Input resistance (MΩ) | 132.7 ± 11.2 | 143.3 ± 5.3 | 129.6 ± 8.4 | 139.6 ± 10.4 | |
| Spike amplitude (mV) | 97.2 ± 1.3 | 94.2 ± 0.9a | 97.0 ± 1.4 | 92.8 ± 1.5a | |
| sEPSC frequency (Hz) | 0.9 ± 0.2 | 0.8 ± 0.1 | 0.9 ± 0.2 | 0.9 ± 0.1 | |
| sEPSC amplitude (pA) | 17.2 ± 0.4 | 16.7 ± 0.7 | 16.8 ± 0.7 | 17.5 ± 0.7 | |
p = 0.008 for persistent effect of developmental in vivo nicotine treatment to decrease spike amplitude in adulthood (two-way ANOVA).
Upon the completion of electrophysiological recording, slices were fixed overnight in a solution containing 4% (wt/vol) paraformaldehyde in 0.1 M phosphate buffer (pH 7.5) and then probed for α4-YFP* nAChRs according to a previously-reported immunohistochemistry protocol (Bailey et al., 2012). Inclusion of streptavidin-conjugated Alexa Fluor 594 (Life Technologies Inc., Burlington, ON, Canada) in the last step of this protocol allowed for the indirect labeling of both Neurobiotin-filled neurons and α4-YFP* nAChRs using this single fluorophore. Multiphoton imaging of labeled neurons and α4-YFP* nAChRs was performed using a Ti:sapphire laser (Mai Tai, Spectra Physics, Santa Clara, CA, USA) tuned to wavelength 780 nm and an Olympus Fluoview FV1000 microscope with an Olympus XLPlan N 25x, 1.05 NA water-immersion objective. Neurons were traced and analyzed using Neurolucida software (MicroBrightField, Williston, VT, USA). Pyramidal neurons included in subsequent morphology analyses had distinguishable apical dendrites and were fully contained within the fixed brain slice. Labeled neurons having apical dendrites that did not definitively terminate within the brain slice (18 of 100 neurons for young mice and 7 of 69 neurons for adult mice) were not included in the morphology analyses.
2.3. Statistical analysis
All data are presented as mean ± SEM. Contingency analysis of apical dendrite termination layer was performed using the Fisher’s exact test. Morphology and electrophysiology data were analyzed by unpaired Student’s t-test, one-way ANOVA or two-way ANOVA, as indicated. Data are reported as mean values for neurons within each experimental group. All significant effects on neuron morphology were also observed when neuron means per mouse were considered to be independent experimental units (see Supplemental Fig. 1). In addition, estimates of intra- and inter-litter variability did not differ from one another for either morphological or electrophysiological measures. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) and a level of p < 0.05 was required to indicate statistical significance.
3. Results
Our goal in pursuing this study was to assess the long-term consequence of developmental in vivo nicotine on the function and morphology of mPFC layer VI neurons and also to test whether changes arose during or subsequent to the in vivo nicotine exposure. Mice were treated throughout gestation and to weaning with either 200 μg/mL nicotine tartrate or tartaric acid vehicle control in the drinking water, based on the concentration of nicotine required to produce similar concentrations of the metabolite cotinine in pregnant mice (Pauly et al., 2004; Sparks and Pauly, 1999) to that found in heavy smokers while pregnant (Klebanoff et al., 1998; Kvalvik et al., 2012; Mercelina-Roumans et al., 1996). Neurons were labeled for morphological reconstruction and measurements of electrophysiological properties such as nAChR currents were made in young mice or in adulthood. Finally, in order to understand better the effects of developmental in vivo nicotine on nAChRs, we took brain slices from young mice and applied acute nicotine to test desensitization of nAChR currents. Refer to Fig. 1 for a schematic illustration of the developmental drug treatment timeline and the ages at which male offspring were examined. Intrinsic electrophysiological properties for all neurons in this study are reported in Table 1 along with the numbers of mice and individual neurons analyzed.
3.1. Morphological changes exerted by developmental in vivo nicotine in wildtype mice
We first analyzed the long-term effects of developmental in vivo nicotine on mPFC layer VI neuron morphology. In adult wildtype mice that had been treated with vehicle control, half the layer VI neurons displayed a “long” morphology having apical dendrites that terminate within cortical layer I, and half of the layer VI neurons displayed a “short” morphology having apical dendrites that terminate below cortical layer I. The mature pattern and proportion of long:short neurons observed here is similar to that seen in adult wildtype mice in an earlier study (Bailey et al., 2012). This initial, qualitative analysis found that developmental in vivo nicotine had a striking effect on the termination layer for layer VI neuron apical dendrites, shifting the proportion of “long” neurons from 50% (9 of 18 neurons) in vehicle control mice to 87% (13 of 15 neurons) in nicotine-treated mice (Fisher’s exact test, p = 0.03). Example tracings of layer VI neurons representative of these proportions are shown in Fig. 2A.
Fig. 2.

In wildtype mice, developmental in vivo nicotine alters adult morphology and nAChR signaling in medial prefrontal layer VI neurons. A, Representative tracings of adult neurons treated with either vehicle control or nicotine during development from conception to weaning on postnatal (P) day P21. B, Apical dendritic complexity is shown with three-dimensional Sholl analysis of the number of dendrite intersections at concentric spheres of varying distance from the soma for neurons from adult mice from both treatment groups. Developmental in vivo nicotine resulted in more complex distal apical dendrites in adulthood compared with vehicle control (‡p = 0.0002). C, Whole-cell nAChR responses to 1 mM ACh (15 s, in the presence of 200 nM atropine) were significantly affected by age (p = 0.006) and were also significantly reduced by developmental in vivo nicotine (*p = 0.04). D, Example nAChR current recordings from young and adult mice of each experimental group.
We then traced each neuron’s apical dendrite, and found that three-dimensional Sholl analysis confirms an effect of developmental in vivo nicotine to increase the complexity of distal apical dendrites (300 μm from the soma to the terminal) (Fig. 2B, two-way ANOVA, main effect of developmental in vivo nicotine, F(1,620) = 14.01, p = 0.0002). Examination of distal apical neuronal morphology in young mice during the last week of the developmental in vivo nicotine treatment showed that the increased complexity, or apparent lag in morphological maturity, in nicotine-treated mice was already present at this stage of postnatal development (data not shown, two-way ANOVA, main effect of developmental in vivo nicotine, F(1,810) = 7.30, p = 0.007). Interestingly, these increases in distal apical dendritic complexity were not accompanied by changes in overall apical dendrite tree length including all branches along the tree (for adult mice: vehicle control: 1885 ± 134 μm, n = 18, developmental in vivo nicotine: 1960 ± 145 μm, n = 15, Student’s t-test, p = 0.7; for young mice: vehicle control: 2206 ± 83 μm, n = 16, developmental in vivo nicotine: 2342 ± 155 μm, n = 16, p = 0.4).
3.2. Electrophysiological changes exerted by developmental in vivo nicotine in wildtype mice
Since nAChR activation has been implicated in limiting neuronal process length (Lipton et al., 1988; Pugh and Berg, 1994), we had tested whether developmental in vivo nicotine interferes with nAChR signaling in layer VI neurons by measuring whole-cell current responses elicited by 1 mM ACh (15 s application, in the presence of 200 nM atropine) in brain slices from both young and adult mice. Previous work has shown that such nAChR currents are strongly developmentally regulated (Alves et al., 2010; Bailey et al., 2012; Kassam et al., 2008). In wildtype mice, we found not only a normal effect of age (Fig. 2C, two-way ANOVA, main effect of age, F(1,77) = 8.11, p = 0.006), but also a significant effect of developmental in vivo nicotine exposure to decrease nAChR signaling (main effect of developmental in vivo nicotine, F(1,77) = 4.43, p = 0.04). Representative nAChR current recordings from each experimental group in wildtype mice are shown in Fig. 2D.
3.3. Direction of morphological changes exerted by developmental in vivo nicotine depend on α5 subunit genotype
Previous work found that layer VI neurons in adult α5−/− mice are disproportionately of the “long” variety (Bailey et al., 2012), an effect that we have replicated in the initial qualitative analysis of the vehicle control α5−/− mice in this study (71% “long”; 10/14 neurons). Developmental in vivo nicotine did not change the long/short ratio significantly (60% “long”, 9/15 neurons; Fisher’s exact test, p = 0.7). Representative tracings of neurons from α5−/− mice are shown in Fig. 3A.
Fig. 3.

In nicotinic acetylcholine receptor (nAChR) α5 subunit knockout mice (α5−/−), developmental in vivo nicotine alters adult morphology and nAChR signaling in medial prefrontal layer VI neurons. A, Representative tracings of adult neurons treated with either vehicle control or nicotine during development from conception to weaning on postnatal (P) day P21. Note, layer VI neurons from vehicle control α5−/− mice retain long apical dendrites into adulthood (Bailey et al., 2012). B, Three-dimensional Sholl analysis shows the number of dendrite intersections at concentric spheres of varying distance from the soma for neurons from adult mice of both treatment groups. Developmental nicotine exposure resulted in less complex distal apical dendrites in adulthood compared with vehicle control (*p = 0.01). C, Whole-cell nAChR responses to 1 mM ACh (15 s, in the presence of 200 nM atropine) were significantly affected by age (p = 0.0006) and were also significantly increased by developmental in vivo nicotine (‡p = 0.003). D, Example nAChR current recordings from each experimental group of α5−/− mice.
Quantitative, three-dimensional Sholl analysis in adult wild-type mice (from Fig. 2B) and adult α5−/− mice (from Fig. 3B), which were both treated with vehicle control, confirmed previous findings (Bailey et al., 2012) that chrna5 deletion increases layer VI neuron apical dendrite complexity (two-way ANOVA, main effect of genotype, F(1,928 = 3.97, p = 0.047) but was most apparent in the distal region of 300 μm to the terminal (main effect of genotype, F(1,609) = 7.08, p = 0.008). Of note, Sholl analysis within adult α5−/− mice revealed a significant, persisting effect of developmental in vivo nicotine to restore the α5−/− layer VI neurons toward the mature wildtype pattern of less complicated apical dendrites, by decreasing the complexity of their distal apical dendrites (300 μm from the soma to the terminal) compared with the vehicle control α5−/− mice (Fig. 3B, main effect of developmental in vivo nicotine, F(1,546) = 6.73, p = 0.01). This effect was only observed in adult mice and not during the last week of the developmental nicotine treatment (data not shown, two-way ANOVA, main effect of developmental in vivo nicotine, F(1,713) = 0.04, p = 0.8). Again, the change in distal apical dendrite complexity observed for adult α5−/− mice was not accompanied by a change in the overall length of the apical dendrite tree (for adult mice: vehicle control: 2065 ± 119 μm, n = 13, developmental in vivo nicotine: 1877 ± 90 μm, n = 15, Student’s t-test, p = 0.2; for young mice: vehicle control: 2240 ± 129 μm, n = 15, developmental in vivo nicotine: 2198 ± 98 μm, n = 18, p = 0.8). It is important to emphasize that developmental in vivo nicotine yielded the opposite changes to adult distal apical dendrite morphology in wildtype and α5−/− mice.
3.4. Electrophysiological changes exerted by developmental in vivo nicotine depend on α5 genotype
As with wildtype mice, we also observed a normal effect of age to decrease the magnitude of nAChR currents in α5−/− mice in this study (Fig. 3C, main effect of age, F(1,72) = 12.91, p = 0.0006). However, in this genotype group we found the opposite effect of treatment, that developmental in vivo nicotine significantly increased nAChR currents (Fig. 3C, main effect of developmental in vivo nicotine, F(1,72) = 9.39, p = 0.003). It is important to note that this effect of developmental in vivo nicotine in α5−/− mice was again in the opposite direction of that observed in wildtype mice. Moreover, these remarkably different effects of developmental in vivo nicotine across genotypes were observed both in young mice, still receiving in vivo nicotine treatment, and in adult mice, who had not received in vivo nicotine in months. Example nAChR current recordings from each experimental group in α5−/− mice are shown in Fig. 3D.
3.5. Genotype independent effects of developmental in vivo nicotine exposure
The genotype-specific effects of developmental in vivo nicotine observed in this study are limited to the apical dendrite morphology and to nAChR currents. We observed no long-term effects of developmental in vivo nicotine within either genotype on basal dendrite morphology (see Supplemental Fig. 2). Sholl analysis revealed no effect of developmental in vivo nicotine on basal dendrite complexity for adult mice of either wildtype (two-way ANOVA, main effect of developmental in vivo nicotine, p = 0.1) or α5−/− (p = 0.7) genotypes. Moreover, there were no long-term effects of developmental in vivo nicotine exposure within either genotype on total length of basal dendrites, average length of basal dendrite trees, distance of the farthest basal dendrite terminal from the soma, or average distance of basal dendrite terminals from the soma (Student’s t-test, all p > 0.05).
We did identify a genotype-independent effect of developmental in vivo nicotine to decrease average dendrite diameter along the length of all analyzed neurons from adult mice that was significant in apical dendrites (two-way ANOVA, main effect of developmental in vivo nicotine, F(1,56) = 14.94, p = 0.0003) and showed a strong trend towards significance in basal dendrites (two-way ANOVA, main effect of developmental in vivo nicotine, F(1,61) = 3.64, p = 0.06). Average apical dendrite diameters were: wildtype control: 1.25 ± 0.02 μm (n = 18); wildtype developmental in vivo nicotine: 1.17 ± 0.03 μm (n = 15); α5−/− control: 1.25 ± 0.04 (n = 12); α5−/− developmental in vivo nicotine: 1.13 ± 0.01 (n = 15). This effect of developmental in vivo nicotine exposure to decrease apical dendrite diameter was also found when we examined a single point on the proximal apical dendrite: 20 μm from the soma (two-way ANOVA, main effect of developmental in vivo nicotine, F(1,56) = 12.17, p = 0.001). These proximal apical dendrite diameters were: wild-type control: 1.94 ± 0.10 μm (n = 18); wildtype developmental in vivo nicotine: 1.51 ± 0.16 μm (n = 15); α5−/− control: 1.96 ± 0.16 (n = 12); α5−/− developmental in vivo nicotine: 1.51 ± 0.08 (n = 15). Developmental in vivo nicotine exposure did not influence the length of apical dendrite tufts for “long” neurons terminating within cortical layer I (two-way ANOVA, main effect of developmental in vivo nicotine, F(1,28) = 0.34, p = 0.6). Tuft lengths were: wildtype control: 104.3 ± 21.9 μm (n = 6); wildtype developmental in vivo nicotine: 113.1 ± 16.3 μm (n = 12); α5−/− control: 151.7 ± 34.1 (n = 8); α5−/− developmental in vivo nicotine: 112.2 ± 30.6 (n = 6). In addition, developmental in vivo nicotine did not influence the termination layer for apical dendrites of the “short” cells that terminated below cortical layer I, which on average extended to the mid-layer of cortex (53 ± 3% of the total cortical width).
As shown in Table 1, we identified an effect of developmental in vivo nicotine to decrease action potential spike amplitude in adulthood significantly (two-way ANOVA, main effect of developmental in vivo nicotine, F(1,64) = 7.63, p = 0.008). However, we identified no additional effects of developmental in vivo nicotine on other intrinsic electrophysiological properties at either age, including resting membrane potential, input resistance, and spontaneous excitatory postsynaptic potential (sEPSC) frequency and amplitude.
3.6. Interactions between α5 subunit genotype and developmental in vivo nicotine exposure
Chrna5 genotype appears to alter the long-term consequences of developmental in vivo nicotine on mPFC layer VI neurons. We tested this hypothesis by comparing the morphological and electrophysiological effects of developmental in vivo nicotine across genotypes, as illustrated for adult mice in Fig. 4. To permit this comparison, distal apical dendrite complexity was measured by summing the total number of Sholl intersections per neuron within the region of 300 μm from the soma to the dendrite terminal. Two-way analysis in adult mice demonstrated significant interactions between the effects of chrna5 genotype and developmental in vivo nicotine to alter apical dendrite complexity (Fig. 4A, interaction of effects, F(1,57) = 4.63, p = 0.04), and nAChR currents (Fig. 4B, interaction of effects, F(1,64) = 6.91, p = 0.01). Specifically, we observed that the direction of effects following developmental in vivo nicotine is dependent on the presence of the nAChR α5 subunit. Similar analysis in young mice found a significant interaction between the effects of α5 subunit genotype and developmental in vivo nicotine on nAChR currents (data not shown, interaction of effects, F(1,85) = 6.98, p = 0.01). However, since apical dendrite complexity is altered by developmental in vivo nicotine only in wildtype mice at this age (as detailed above), there did not appear to be a morphological interaction in the young mice (data not shown, interaction of effects, F(1,60) = 0.51, p = 0.5).
Fig. 4.

Interactions between nicotinic acetylcholine receptor (nAChR) α5 subunit genotype and developmental in vivo nicotine in medial prefrontal layer VI neurons of adult mice. A, The mean number of Sholl intersections for distal apical dendrites (300 μm from the soma to the terminal in 25 μm increments) was altered in adult mice by developmental in vivo nicotine in a manner that depends on the presence of the α5 nAChR subunit (effect of interaction, p = 0.04). B, Whole-cell nAChR responses to 1 mM ACh (15 s, in the presence of 200 nM atropine) were also altered in adult mice by developmental in vivo nicotine in a manner that depends on the α5 nAChR subunit (effect of interaction, p = 0.01). Data are presented as: wildtype vehicle control (dark blue), wildtype developmental in vivo nicotine (light blue), α5−/− vehicle control (dark red), and α5−/− developmental in vivo nicotine (light red). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.7. Potential mechanism of in vivo nicotine teratogenesis: receptor desensitization
In wildtype mice, developmental in vivo nicotine appeared to have a stronger effect on apical dendrite morphology than it does on the nAChR currents. Specifically, we found that developmental in vivo nicotine led to an aberrant pattern of apical dendritic morphology that resembles that of α5−/− mice, yet the nAChR currents were not reduced to the level seen in α5−/− mice. One potential explanation for this discrepancy is that our acute brain slice recording conditions would permit substantial washout of residual in vivo nicotine. So, instead of seeing the young nAChR currents acutely desensitized in the presence of nicotine, we saw the young nAChR currents after some degree of recovery. We therefore sought to assess the ability of acute nicotine in brain slice to desensitize α4β2* nAChRs in layer VI neurons of young wildtype and α5−/− mice. These desensitization experiments are most relevant to understanding nicotine’s effects in the young mice because they are the ones receiving nicotine or vehicle control in vivo.
To perform this experiment, we measured the nAChR currents in response to 1 mM ACh (15 s application in the presence of 200 nM atropine, as above) before and after slices were exposed to 300 nM nicotine for ten minutes (plus a ten minute washout period to allow holding currents to return to baseline). This concentration of acute nicotine in brain slice is consistent with that observed in the blood of smokers (Henningfield et al., 1993; Rose et al., 2010). Example young nAChR current recordings before and after acute nicotine in brain slice for neurons from each experimental group are shown in Fig. 5A.
Fig. 5.
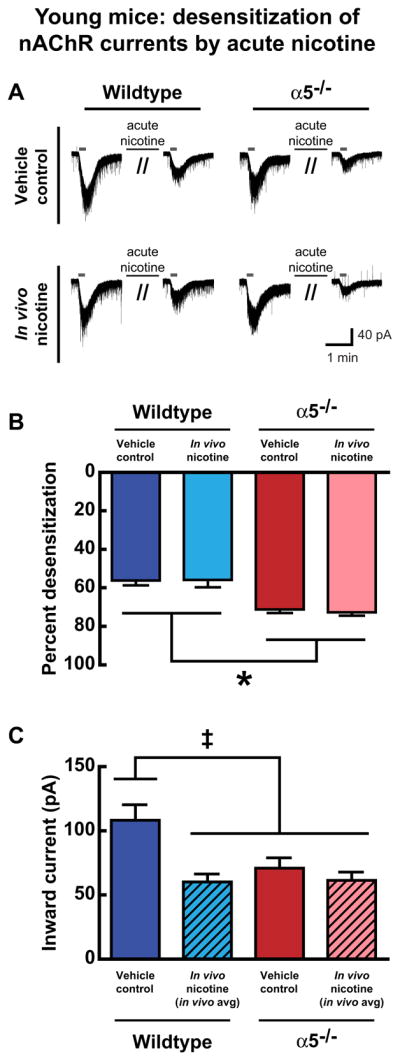
In young mice, the influence of α5 subunit genotype and developmental in vivo nicotine on acute nicotine-induced nAChR desensitization in brain slice. A, Example current recordings of the response to ACh before and after acute nicotine application are shown for one neuron from each experimental group. B, Within individual neurons, nAChR desensitization was calculated by measuring nAChR current responses to 1 mM ACh (15 s, in the presence of atropine) before and after the brain slice was exposed to 300 nM nicotine (10 min). Percent desensitization by acute nicotine exposure was significantly affected by α5 subunit genotype (*p < 0.0001), but not by developmental nicotine exposure (p = 0.8). C, Theoretical in vivo average nAChR currents experienced in mPFC layer VI neurons of mice repeatedly exposed to nicotine during development were calculated by averaging nAChR currents measured before and after receptor desensitization by acute exposure to 300 nM nicotine, and compared with normal, non-desensitized nAChR currents in the vehicle control groups. This approach was based on the assumption that the strong diurnal variation in drinking water consumption in mice (Dole et al., 1983) would result in roughly equal periods of high and low nicotine exposure. The resulting nAChR current means were significantly different among the experimental groups (‡p = 0.0007): the vehicle control wildtype group was significantly different from the other three groups (each comparison p < 0.05), and the other three groups were not significantly different from each other (each comparison p > 0.05).
Percent desensitization of nAChR currents by acute nicotine was calculated for each neuron: [ACh current before acute nicotine − ACh current after acute nicotine]/[ACh current before acute nicotine] × 100. Analysis of acute nicotine-induced receptor desensitization data in young mice found significant effects of genotype (Fig. 5B, two way ANOVA, main effect of genotype, F(1,46) = 35.51, p < 0.0001), where wildtype layer VI neurons appear less sensitive to desensitization by this paradigm than α5−/− neurons. This general pattern is similar to our previous findings in adult mice (Bailey et al., 2010) and to what we see in adulthood in this study (data not shown, main effect of genotype, F(1,27) = 55.34, p < 0.0001). Interestingly, developmental in vivo nicotine does not appear to alter the degree of acute nicotine desensitization in brain slice at either age (young: main effect of developmental in vivo nicotine, F(1,46) = 0.05, p = 0.8, adult: main effect of developmental in vivo nicotine, F(1,27) = 1.57, p = 0.2). In wildtype mice, however, the ability of acute nicotine exposure to desensitize receptors to nAChR currents was significantly greater in young mice compared to adult mice (data not shown, main effect of age, F(1,36) = 10.25, p = 0.003). This greater sensitivity to desensitization in young wildtype mice underscores the vulnerability of the developing brain to nicotine.
We further explored the postulate that nAChR desensitization from developmental in vivo nicotine results in wildtype nAChR currents that resemble those from α5−/− mice. This desensitization would occur predominantly at night since mice show strong diurnal variation in water consumption, with the majority of drinking in the dark cycle and minimal consumption in the light cycle (Dole et al., 1983). As such, layer VI neurons in mice exposed to developmental nicotine would likely experience high nicotine and have acutely desensitized nAChR currents at night, and would likely experience low nicotine and have recovered nAChR currents during the day. Accordingly for the developmental nicotine groups only, we have calculated hypothetical “in vivo averages” for the nAChR currents in young mice, which combine the nAChR currents for each neuron before and after desensitization with acute nicotine in brain slice in an attempt to model the daily average nAChR current over 24 h. These averages were then compared with the normal, non-desensitized nAChR currents measured in the vehicle control groups at this age. Results of this analysis for young mice are shown in Fig. 5C and indeed suggest that the contribution of acute nicotine desensitization in developing wildtype mice causes their α4β2* nAChRs to function in a manner similar to those of α5−/− mice in vivo. One-way ANOVA identified a significant difference amongst the experimental groups (p = 0.0007). Moreover, the Tukey’s post-hoc test for multiple comparisons found that nAChR currents from vehicle control wildtype mice were significantly different from each of the other three experimental groups (each comparison p < 0.05), whereas these other groups were not significantly different from each other (each comparison p > 0.05).
3.8. Summary of results
The changes in apical dendrite morphology and nAChR currents that result from developmental in vivo nicotine in wildtype and α5−/− mice are summarized in Table 2. This synopsis suggests that our model of “in vivo average” nAChR currents described above is consistent with the striking ability of developmental in vivo nicotine to make wildtype layer VI neurons resemble those of α5−/− mice morphologically. Since layer VI nAChR currents would only experience periods of strong desensitization until the end of in vivo nicotine treatment at P21, our findings raise the possibility that mechanisms underlying postnatal apical dendrite maturation can be manipulated prior to this age. By contrast, a different pattern of results arise from α5−/− mice. In these mice, developmental in vivo nicotine appears capable of inducing a substantial upregulation of nAChR currents which is unmasked by washout of acute nicotine. Like young wildtype mice exposed to vehicle control, young α5−/− mice have a predominantly immature apical dendritic phenotype. Despite strong desensitization of nAChRs in young α5−/− mice by acute nicotine, these mice would likely have periods of normalized, high nAChR currents during the day, which may be capable of triggering the apical dendrite maturation that results in their normalized pattern of apical dendrite morphology by adulthood.
Table 2.
Summary of apical dendrite phenotype and nAChR current amplitudes in young and adult wildtype and α5−/− mice treated developmentally with in vivo nicotine or vehicle control.
| Young mice | Adult mice | |
|---|---|---|
| Wildtype (Vehicle control) |
|
|
| Wildtype (In vivo nicotine) |
|
|
| α5−/− (Vehicle control) |
|
|
| α5−/− (In vivo nicotine) |
|
|
Bold, italic text is used to indicate specific nAChR current amplitude hypothesized to influence adult apical dendrite morphology. High nAChR currents in young mice may trigger the apical dendrite retraction necessary for the mature pattern of adult apical dendrite morphology.
4. Discussion
We have found that developmental in vivo nicotine in mice leads to persistent alterations in morphology and nAChR currents in mPFC layer VI neurons, a population of neurons that have been implicated in the control of attention behavior. While changes were observed in young mice near the completion of the in vivo nicotine treatment, differences remained prominent in fully mature mice that had not been exposed to nicotine for more than two months. The direction of the alterations in neuronal morphology and nAChR currents occurred in a chrna5 genotype-dependent manner, highlighting an important role for α5 subunit-containing nAChRs in normal mPFC development and their potential as a target for the teratogenic effects of nicotine.
4.1. Consequences of developmental in vivo nicotine on nAChR currents
Further research is required to identify mechanisms underlying the observed changes in nAChR currents following developmental in vivo nicotine treatment, as nicotine exhibits a complex pharmacology at nAChRs including agonist activation and desensitization, and can also lead to changes in expression, trafficking and post-translational modification of receptor subunits (Changeux, 2010; Lester et al., 2009; Marks et al., 2011; Quick and Lester, 2002). Of note, the inclusion of the α5 subunit within the α4β2* nAChR boosts its activation by nicotine and also protects to some degree against desensitization by this drug (Bailey et al., 2010). In the adult frontal cortex, chronic in vivo nicotine treatment leads to an upregulation of α4β2* nAChRs containing α4 and β2 subunits (Besson et al., 2007; Marks et al., 2011; Sparks and Pauly, 1999) but not those containing the α5 subunit (Mao et al., 2008). Similar results are seen in the developing brain, where chronic in vivo nicotine treatment increases expression of α4 and β2 subunits without altering α5 subunit content (Counotte et al., 2012; Lv et al., 2008). Very little is known about the effects of the inclusion of an α5 subunit on the ability of in vivo nicotine to alter nAChR post-translational modification or its ability to pharmacologically chaperone nAChRs through the endoplasmic reticulum (Srinivasan et al., 2011). However, several of these mechanisms of nAChR modulation may contribute to the ability of developmental in vivo nicotine to create a lasting increase in nAChR currents in corticothalamic neurons of α5−/− mice expressing α4 and β2 subunits.
4.2. Potential links between nAChR signaling and apical dendrite morphology
We find that developmental in vivo nicotine also exerts significant changes on the morphology of layer VI neurons in a manner strikingly dependent on chrna5 genotype. In wildtype mice, developmental in vivo nicotine maintains an immature pattern of apical dendrite morphology into adulthood, and also results in decreased nAChR currents. Morphologically, it could be argued that mPFC layer VI neurons from wildtype mice exposed to developmental in vivo nicotine come to resemble neurons from vehicle control α5−/− mice in adulthood. Electrophysiologically, our recordings in the presence of acute nicotine suggest that the average nAChR currents in layer VI neurons of the developmentally-treated young wildtype mice are similar to vehicle control young α5−/− mice in vivo. Of relevance, the developmental period between the age of the young and adult mice is normally accompanied by significant cerebral cortical circuit refinement (Berardi et al., 2000; Hensch, 2004). The elevated nAChR signaling in layer VI neurons of normal, young wildtype mice may be a necessary trigger for apical dendrite retraction. In particular, nAChR signaling can mediate retraction (Pugh and Berg, 1994) of neuronal processes in culture and, more specifically, is associated with a shortening of mPFC layer VI neuron apical dendrites during this time (Bailey et al., 2012). The electrophysiological results from this current study show that this normally-elevated nAChR signaling is decreased following developmental in vivo nicotine, suggesting a potential mechanism for our morphological findings in wildtype mice. It is also important to note that developmental in vivo nicotine exposure alters the morphology of apical but not basal dendrite trees, suggesting that nicotine interferes with signaling pathways that influence the maturation of apical dendrites only. Such pathways may underlie differential alterations in apical versus basal dendrite trees observed during normal postnatal development (Romand et al., 2011) and accelerated aging (Shimada et al., 2006) of rodent cerebral cortical pyramidal neurons.
In the α5−/− mice, the relationship between nAChR currents and apical dendrite morphology appears somewhat more complicated. Our initial results suggested the possibility that developmental in vivo nicotine restores normal morphological patterns through its seeming normalization of nAChR currents in these neurons during development. However, with exposure of the brain slice to acute nicotine, it appears that mPFC layer VI neurons α5−/− mice are even more sensitive to suppression of their nAChR currents. On average, it would seem that the nAChR currents in young α5−/− mice exposed to nicotine in vivo would remain similar to those in α5−/− mice treated with vehicle. Consistent with this interpretation, apical dendritic morphology of young α5−/− mice exposed to developmental in vivo nicotine did not differ significantly from young α5−/− vehicle control mice. However, after developmental nicotine exposure is complete, adult α5−/− mice show normalized apical dendritic morphology. This normalization suggests that having high nAChR currents at least part of the day during development may be sufficient to trigger dendritic retraction. Alternatively, it is possible that normalization of apical dendrite morphology in the adult α5−/− mice may reflect an entirely different set of neurobiological processes. For example, selective apical dendrite retraction of other populations of prefrontal cortical neurons has been demonstrated with the experience of stress (Brown et al., 2005; Cook and Wellman, 2004; Goldwater et al., 2009) or accelerated aging (Shimada et al., 2006). It is possible that wildtype and α5−/− offspring in this current study may have experienced distinct environments during development because they were generated from separate homozygous lines, however, these lines were less than five generations removed from shared heterozygous parents and there was no noticeable effect of genotype on maternal physiology or care for offspring.
4.3. Implications for prefrontal attention circuits
By altering the apical dendrite complexity of mPFC layer VI neurons, developmental in vivo nicotine has the potential to disrupt the circuitry that supports attention. Cortical neurons typically receive laminar-specific presynaptic local cortical and long-range inputs (Llano and Sherman, 2009). Furthermore, cortical layer VI neurons appear capable of detecting and responding to excitatory input received by their distal apical dendrites (Ledergerber and Larkum, 2010; Zarrinpar and Callaway, 2006). Following chronic developmental in vivo nicotine in wildtype mice, the unusual preponderance of neurons with apical dendrites that stretch across the width of the mPFC to the pial surface suggests changes in the overall profile of synaptic inputs to these neurons, likely significantly altering the function of prefrontal cortical circuits to which these neurons contribute.
Pyramidal neurons within layer VI of the mPFC are implicated in modulating attention behavior through their prominent projections both to more superficial mPFC layers and to the mediodorsal thalamus (Briggs and Usrey, 2008; Gabbott et al., 2005; Zikopoulos and Barbas, 2006). It should also be noted that some neurons in this population project to additional subcortical regions including the dorsal striatum and lateral hypothalamus (Gabbott et al., 2005). While nAChR signaling in mPFC layer VI neurons is important for normal attention performance in adult mice (Guillem et al., 2011), the implications of nAChR signaling during development for attentional performance are not well understood. Our results suggest that perturbation of developmental nAChR signaling in mPFC layer VI neurons will have repercussions both for the excitation and underlying morphological structure of a neuronal population that contributes to normal attention behavior. These findings suggest a potential neurobiological mechanism of the attention deficits observed in humans and in animal models following developmental exposure to nicotine in vivo (Counotte et al., 2011; Heath et al., 2010; Pauly and Slotkin, 2008; Schneider et al, 2011; Thakur et al., 2013).
Supplementary Material
Acknowledgments
This work was supported by grants to EKL from the Canadian Institutes of Health Research (MOP 89825), the Canada Research Chairs Program, and the Canadian Foundation for Innovation. We thank Dr. Mariella De Biasi for the gift of the nAChR α5 subunit knockout mice and Dr. Raad Nashmi for the gift of the nAChR α4-YFP knock-in mice. We thank Dr. Nathalie Goodfellow for reviewing the manuscript and contributing useful suggestions.
Abbreviations
- nAChR
nicotinic acetylcholine receptor
- α5−/−
genetic deletion for the nicotinic acetylcholine receptor α5 subunit (chrna5 null)
- chrna5
gene name for the nicotinic acetylcholine receptor α5 subunit
- mPFC
medial prefrontal cortex
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.neuropharm.2013.09.003.
References
- Alves NC, Bailey CD, Nashmi R, Lambe EK. Developmental sex differences in nicotinic currents of prefrontal layer VI neurons in mice and rats. PLoS One. 2010;5:e9261. doi: 10.1371/journal.pone.0009261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, Alves NC, Nashmi R, De Biasi M, Lambe EK. Nicotinic α5 subunits drive developmental changes in the activation and morphology of prefrontal cortex layer VI neurons. Biol Psychiatry. 2012;71:120–128. doi: 10.1016/j.biopsych.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, De Biasi M, Fletcher PJ, Lambe EK. The nicotinic acetylcholine receptor α5 subunit plays a key role in attention circuitry and accuracy. J Neurosci. 2010;30:9241–9252. doi: 10.1523/JNEUROSCI.2258-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi N, Pizzorusso T, Maffei L. Critical periods during sensory development. Curr Opin Neurobiol. 2000;10:138–145. doi: 10.1016/s0959-4388(99)00047-1. [DOI] [PubMed] [Google Scholar]
- Besson M, Granon S, Mameli-Engvall M, Cloez-Tayarani I, Maubourguet N, Cormier A, Cazala P, David V, Changeux JP, Faure P. Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A. 2007;104:8155–8160. doi: 10.1073/pnas.0702698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs F, Usrey WM. Emerging views of corticothalamic function. Curr Opin Neurobiol. 2008;18:403–407. doi: 10.1016/j.conb.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SM, Henning S, Wellman CL. Mild, short-term stress alters dendritic morphology in rat medial prefrontal cortex. Cereb Cortex. 2005;15:1714–1722. doi: 10.1093/cercor/bhi048. [DOI] [PubMed] [Google Scholar]
- Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- Cohen G, Roux JC, Grailhe R, Malcolm G, Changeux JP, Lagercrantz H. Perinatal exposure to nicotine causes deficits associated with a loss of nicotinic receptor function. Proc Natl Acad Sci U S A. 2005;102:3817–3821. doi: 10.1073/pnas.0409782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60:236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Goriounova NA, Li KW, Loos M, van der Schors RC, Schetters D, Schoffelmeer AN, Smit AB, Mansvelder HD, Pattij T, Spijker S. Lasting synaptic changes underlie attention deficits caused by nicotine exposure during adolescence. Nat Neurosci. 2011;14:417–419. doi: 10.1038/nn.2770. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Goriounova NA, Moretti M, Smoluch MT, Irth H, Clementi F, Schoffelmeer AN, Mansvelder HD, Smit AB, Gotti C, Spijker S. Adolescent nicotine exposure transiently increases high-affinity nicotinic receptors and modulates inhibitory synaptic transmission in rat medial prefrontal cortex. FASEB J. 2012;26:1810–1820. doi: 10.1096/fj.11-198994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dole VP, Ho A, Gentry RT. An improved technique for monitoring the drinking behavior of mice. Physiol Behav. 1983;30:971–974. doi: 10.1016/0031-9384(83)90264-0. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Rogers SW. Nicotinic receptor subunit α5 modifies assembly, up-regulation, and response to pro-inflammatory cytokines. J Biol Chem. 2010;285:26049–26057. doi: 10.1074/jbc.M110.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, Morrison JH. Structural and functional alterations to rat medial pre-frontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164:798–808. doi: 10.1016/j.neuroscience.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillem K, Bloem B, Poorthuis RB, Loos M, Smit AB, Maskos U, Spijker S, Mansvelder HD. Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science. 2011;333:888–891. doi: 10.1126/science.1207079. [DOI] [PubMed] [Google Scholar]
- Heath CJ, King SL, Gotti C, Marks MJ, Picciotto MR. Corticothalamic connectivity is vulnerable to nicotine exposure during early postnatal development through α4/β2/α5 nicotinic acetylcholine receptors. Neuropsychopharmacology. 2010;35:2324–2338. doi: 10.1038/npp.2010.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- Kassam SM, Herman PM, Goodfellow NM, Alves NC, Lambe EK. Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J Neurosci. 2008;28:8756–8764. doi: 10.1523/JNEUROSCI.2645-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff MA, Levine RJ, Clemens JD, DerSimonian R, Wilkins DG. Serum cotinine concentration and self-reported smoking during pregnancy. Am J Epidemiol. 1998;148:259–262. doi: 10.1093/oxfordjournals.aje.a009633. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Onksen J, Lindstrom J. Roles of accessory subunits in α4β2(*) nicotinic receptors. Mol Pharmacol. 2008;74:132–143. doi: 10.1124/mol.108.046789. [DOI] [PubMed] [Google Scholar]
- Kvalvik LG, Nilsen RM, Skjaerven R, Vollset SE, Midttun O, Ueland PM, Haug K. Self-reported smoking status and plasma cotinine concentrations among pregnant women in the Norwegian Mother and Child Cohort Study. Pediatr Res. 2012;72:101–107. doi: 10.1038/pr.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledergerber D, Larkum ME. Properties of layer 6 pyramidal neuron apical dendrites. J Neurosci. 2010;30:13031–13044. doi: 10.1523/JNEUROSCI.2254-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Banghart MR, Dougherty DA, Goate AM, Wang JC. Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J. 2009;11:167–177. doi: 10.1208/s12248-009-9090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Frosch MP, Phillips MD, Tauck DL, Aizenman E. Nicotinic antagonists enhance process outgrowth by rat retinal ganglion cells in culture. Science. 1988;239:1293–1296. doi: 10.1126/science.3344435. [DOI] [PubMed] [Google Scholar]
- Llano DA, Sherman SM. Differences in intrinsic properties and local network connectivity of identified layer 5 and layer 6 adult mouse auditory corticothalamic neurons support a dual corticothalamic projection hypothesis. Cereb Cortex. 2009;19:2810–2826. doi: 10.1093/cercor/bhp050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J, Mao C, Zhu L, Zhang H, Pengpeng H, Xu F, Liu Y, Zhang L, Xu Z. The effect of prenatal nicotine on expression of nicotine receptor subunits in the fetal brain. Neurotoxicology. 2008;29:722–726. doi: 10.1016/j.neuro.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ. The α4β2α5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem. 2008;104:446–456. doi: 10.1111/j.1471-4159.2007.05011.x. [DOI] [PubMed] [Google Scholar]
- Marks MJ, McClure-Begley TD, Whiteaker P, Salminen O, Brown RW, Cooper J, Collins AC, Lindstrom JM. Increased nicotinic acetylcholine receptor protein underlies chronic nicotine-induced up-regulation of nicotinic agonist binding sites in mouse brain. J Pharmacol Exp Ther. 2011;337:187–200. doi: 10.1124/jpet.110.178236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercelina-Roumans PE, Schouten H, Ubachs JM, van Wersch JW. Cotinine concentrations in plasma of smoking pregnant women and their infants. Eur J Clin Chem Clin Biochem. 1996;34:525–528. doi: 10.1515/cclm.1996.34.7.525. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley TD, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA. Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osadchy A, Kazmin A, Koren G. Nicotine replacement therapy during pregnancy: recommended or not recommended? J Obstet Gynaecol Can. 2009;31:744–747. doi: 10.1016/S1701-2163(16)34281-5. [DOI] [PubMed] [Google Scholar]
- Pauly JR, Slotkin TA. Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr. 2008;97:1331–1337. doi: 10.1111/j.1651-2227.2008.00852.x. [DOI] [PubMed] [Google Scholar]
- Pauly JR, Sparks JA, Hauser KF, Pauly TH. In utero nicotine exposure causes persistent, gender-dependant changes in locomotor activity and sensitivity to nicotine in C57Bl/6 mice. Int J Dev Neurosci. 2004;22:329–337. doi: 10.1016/j.ijdevneu.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; 2001. [Google Scholar]
- Pidoplichko VI, De Biasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Pugh PC, Berg DK. Neuronal acetylcholine receptors that bind alpha-bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci. 1994;14:889–896. doi: 10.1523/JNEUROSCI.14-02-00889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Romand S, Wang Y, Toledo-Rodriguez M, Markram H. Morphological development of thick-tufted layer v pyramidal cells in the rat somatosensory cortex. Front Neuroanat. 2011;5:5. doi: 10.3389/fnana.2011.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, Garg S, Garg PK. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci U S A. 2010;107:5190–5195. doi: 10.1073/pnas.0909184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, De Biasi M. The nicotinic acetylcholine receptor subunit α5 mediates short-term effects of nicotine in vivo. Mol Pharmacol. 2003;63:1059–1066. doi: 10.1124/mol.63.5.1059. [DOI] [PubMed] [Google Scholar]
- Schneider T, Ilott N, Brolese G, Bizarro L, Asherson PJ, Stolerman IP. Prenatal exposure to nicotine impairs performance of the 5-choice serial reaction time task in adult rats. Neuropsychopharmacology. 2011;36:1114–1125. doi: 10.1038/npp.2010.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Tsuzuki M, Keino H, Satoh M, Chiba Y, Saitoh Y, Hosokawa M. Apical vulnerability to dendritic retraction in prefrontal neurones of ageing SAMP10 mouse: a model of cerebral degeneration. Neuropathol Appl Neurobiol. 2006;32:1–14. doi: 10.1111/j.1365-2990.2006.00632.x. [DOI] [PubMed] [Google Scholar]
- Sparks JA, Pauly JR. Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology (Berl) 1999;141:145–153. doi: 10.1007/s002130050818. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Pantoja R, Moss FJ, Mackey ED, Son CD, Miwa J, Lester HA. Nicotine up-regulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J Gen Physiol. 2011;137:59–79. doi: 10.1085/jgp.201010532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J. Ca2+ permeability of the (α4)3(β2)2 stoichiometry greatly exceeds that of (α4)2(β2)3 human acetylcholine receptors. Mol Pharmacol. 2007;71:769–776. doi: 10.1124/mol.106.030445. [DOI] [PubMed] [Google Scholar]
- Thakur GA, Sengupta SM, Grizenko N, Schmitz N, Page V, Joober R. Maternal smoking during pregnancy and ADHD: a comprehensive clinical and neurocognitive characterization. Nicotine Tob Res. 2013;15:149–157. doi: 10.1093/ntr/nts102. [DOI] [PubMed] [Google Scholar]
- Tong VT, Jones JR, Dietz PM, D’Angelo D, Bombard JM. Trends in smoking before, during, and after pregnancy – Pregnancy Risk Assessment Monitoring System (PRAMS), United States, 31 sites, 2000–2005. MMWR Surveill Summ. 2009;58:1–29. [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Expression of α5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J Comp Neurol. 2005;481:19–30. doi: 10.1002/cne.20357. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Callaway EM. Local connections to specific types of layer 6 neurons in the rat visual cortex. J Neurophysiol. 2006;95:1751–1761. doi: 10.1152/jn.00974.2005. [DOI] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Prefrontal projections to the thalamic reticular nucleus form a unique circuit for attentional mechanisms. J Neurosci. 2006;26:7348–7361. doi: 10.1523/JNEUROSCI.5511-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.