

Abstract
We have shown that dietary fish oil is protective against experimentally-induced colon cancer and the protective effect is enhanced by co-administration of pectin. However, the underlying mechanism(s) have not been fully elucidated. We hypothesized that fish oil with butyrate, a pectin fermentation product, protects against colon cancer initiation by decreasing cell proliferation and increasing differentiation and apoptosis through a p27Kip1 mediated mechanism. Rats were provided diets of corn or fish oil, with/without butyrate, and terminated 12, 24 or 48 h post azoxymethane (AOM) injection. Proliferation (Ki-67), differentiation (Dolichos Biflorus Agglutinin), apoptosis (TUNEL) and p27Kip1 (cell cycle mediator) were measured in the same cell within crypts in order to examine the coordination of cell cycle as a function of diet. DNA damage (N7-methylguanine) was determined by quantitative immunohistochemical analysis. Dietary fish oil decreased DNA damage by 19% (P=0.001) and proliferation by 50% (P=0.003) and increased differentiation by 56% (P=0.039) compared to corn oil. When combined with butyrate, fish oil enhanced apoptosis 24 h post AOM injection compared to a corn oil/butyrate diet (P=0.039). There was an inverse relationship between crypt height and apoptosis in fish oil/butyrate group (r= −0.53, P=0.040). Corn oil/butyrate group showed a positive correlation between p27Kip1 expression and proliferation (r= 0.61, P=0.035). These results indicate the in vivo effect of butyrate on apoptosis and proliferation is dependent on dietary lipid source. These results demonstrate the presence of an early coordinated colonocyte response by which fish oil and butyrate protects against colon tumorigenesis.
Keywords: fish oil, butyrate, colon cancer, cell
Introduction
There is accumulating epidemiological, clinical and experimental evidence showing that dietary modification is an important factor in the prevention of colon cancer (1). We have previously shown that, using an AOM-induced colon carcinogenesis model, a fish oil/pectin diet protects against colon cancer by lowering tumor incidence (2), and DNA damage at the initiation stage of colon carcinogenesis (3) compared to a corn oil/cellulose or corn oil/pectin diet.
Dietary fish oil, rich in n−3 polyunsaturated fatty acids (PUFA), i.e., eicosapentaenoic acid (EPA, 20:5n−3) and docosahexaenoic acid (DHA, 22:6n−3), protects against colon cancer by altering cell kinetics (i.e. proliferation, differentiation and apoptosis) (3–8). Pectin is a soluble fiber and it is fermented by colonic microflora, which produces short chain fatty acids (e.g., acetate, propionate and butyrate), CO2, methane and other gases (9). Among the short chain fatty acids, butyrate is used as a major energy source by colonocytes (10, 11). Even though some controversies remain concerning the in vivo responses of colonocytes to butyrate exposure (9), butyrate has been shown to impact cell kinetics, e.g. cell proliferation, differentiation and apoptosis, which regulate development of colon carcinogenesis (12–16). Therefore, we hypothesized that the combination of fish oil with butyrate would protect against colon cancer initiation through an integrated pattern of changes in cell proliferation, differentiation, apoptosis and p27kip1 protein levels. This hypothesis was further supported by a very recent study (17) showing that pescovegetarians (vegetarians who consume fish and seafood products) have a much lower risk of developing colorectal cancer, indicating potential benefit from the interaction of n−3 PUFA and dietary fiber.
To date, a number of investigators have measured proliferation and apoptosis in the same preclinical model. Even though data generated at the animal target tissue level are informative, they show only an averaged overall trend. Measuring these variables in the same cell offers the potential to determine the coordination of physiological phenomena in a cell. Furthermore, even though p27Kip1 is a well-established inhibitor of cell proliferation, p27Kip1 may also regulate differentiation and apoptosis (18, 19). However, the connection of p27Kip1 with proliferation, differentiation and apoptosis is not clear. By determining these four variables in the same cell, we were able to provide an opportunity to gain greater understanding of the regulation of cell kinetics in individual cells. To our knowledge, this study is the first to determine all four of these variables in the same cell.
Materials and Methods
Animals and study design
The animal use protocol was approved by the University Animal Care Committee of Texas A&M University. Forty-eight male weanling Sprague Dawley rats (100–120g) (Harlan, Houston, TX) were individually housed and maintained in a temperature and humidity-controlled animal facility. This study was a 2×2×4 factorial design with two types of fat (corn oil or fish oil), with or without butyrate, and at four time points (0, 12, 24 or 48 h) post carcinogen injection. Rats were provided with the defined diets for 3 wk before carcinogen azoxymethane (AOM, 15 mg/kg of body weight, s.c.). Rats had free access to food and water at all times and 48h food intake was measured after 2 wk of receiving diets. Body weights were recorded weekly throughout the study.
Diets
The four defined diets (Table 1) differed in lipid source (corn oil or fish oil) and the administration of butyrate pellets. The major differences between the fatty acid compositions of the two lipid sources were significantly higher amounts of EPA (20:5, n−3) and DHA (22:6, n−3) in the fish oil compared to corn oil diet, and higher amounts of 18:2 (n−6) in the corn oil diet. Diets were prepared based on the standard AIN-76A formulation with modification of 15% fat amount, corn oil or fish oil (Vacuum-deodorized Menhaden fish oil, OmegaPure, Houston, TX) (20). Diets contained equivalent amounts of antioxidants; 26 mg α-tocopherol, 14 mg γ-tocopherol and 2 mg tertiary butylhydroquinone (TBHQ)/100g diet. Gastro-resistant slow-release butyrate pellets (S.A. Valpharma, Serravalle, Italy) designed to be primarily released in the colon (20) were supplemented (1.5 g/100 g diet) into the diets. A higher butyrate concentration in the feces was reported in rats fed with these pellets (20).
Table 1.
Composition of experimental diets*
| Ingredient | CO (g) | COB (g) | FO (g) | FOB (g) |
|---|---|---|---|---|
| Dextrose | 51.06 | 51.06 | 51.06 | 51.06 |
| Casein | 22.35 | 22.35 | 22.35 | 22.35 |
| Cellulose | 6.00 | 6.00 | 6.00 | 6.00 |
| Corn Oil | 15.00 | 15.00 | 3.50 | 3.50 |
| Fish Oil | 0.00 | 0.00 | 11.50 | 11.50 |
| Salt mix, AIN-76A | 3.91 | 3.91 | 3.91 | 3.91 |
| Vitamin mix, AIN-76A | 1.12 | 1.12 | 1.12 | 1.12 |
| Butyrate pellet | 0.00 | 1.50 | 0.00 | 1.50 |
| Total | 100.00 | 101.50 | 100.00 | 101.50 |
Diets contained equivalent amounts of antioxidants; 26 mg α-tocopherol, 14 mg γ-tocopherol and 2 mg tertiary butylhydroquinone (TBHQ)/100g diet. The butyrate diets were supplemented with Gasto-resistant slow-release butyrate pellets (1.5 g/100g of diet, Valpharma, Serravalle, Italy).
Tissue acquisition
Rats were euthanized by CO2 asphyxiation, and the entire colon was immediately resected. After removal of the rectum, 1 cm of the distal colon was fixed in 4 % paraformaldehyde and another 1 cm was fixed in 70% ethanol. From the paraformaldehyde-fixed tissue section and serial sections were cut; one was used for colocalization of proliferation and p27Kip1, and the other for colocalization of differentiation and apoptosis. Since ethanol fixed tissue was used for quantitative localization of N7-methylguanine, N7-methylguanine was not part of the colocalization protocol but was measured in a tissue section adjacent to that used for the paraformaldehyde fixed sections.
Colocalization of cell proliferation and p27Kip1
Antigen was retrieved by microwave treatment in 0.1 M sodium citrate buffer (pH 6.0). To block non-specific background staining, tissue sections were incubated with normal rabbit serum (Jackson, West Grove, PA) in normal sheep serum (Jackson). A mixed solution of monoclonal anti-Ki-67 antibody (BD Biosciences, San Diego, CA) and rabbit anti-p27Kip1 antibody was used as the primary antibodies. Slides were incubated with biotinylated sheep anti-mouse IgG (Jackson) and Texas-Red Streptavidin (Vector laboratory Inc., Burlingame, CA). Subsequently, slides were incubated with fluorescein-labeled goat anti-rabbit IgG (Jackson). Finally, slides were counterstained and mounted with DAPI/antifade (Vector) and were stored at 4 °C before imaging. Omission of each or both primary antibodies was used as a negative control. At least 20 crypt columns per animal were chosen for quantitative analysis.
Images of colonic crypts were visualized using a Nikon Eclipse TE300 microscope (Nikon Inc., Melville, NY, USA) equipped with an FITC filter (excitation 490–505 nm/ emission 515–545 nm) and a Texas-Red filter (excitation 560–585 nm/ emission 600–652 nm). Images were captured using a Micromax 5 MHz cooled digital CCD camera (Princeton Instruments, Trenton, NJ, USA) with a constant exposure time and 2 × 2 binning. The position of Ki-67 positive stained cell and the staining intensity of p27Kip1 were assessed by cell position within the crypt using a MetaMorph Imaging System (Version 4.6r3, Universal Imaging Corp., Downingtown, PA, USA). For cell proliferation, the proliferation index was calculated as 100 times the number of labeled cells per crypt column divided by the total number of cells per crypt column (21). Proliferative zone was calculated as 100 times the position of the highest labeled cell divided by the number of cells per crypt column (21). For p27Kip1 quantitation, nuclei on one side of a crypt column were outlined and the staining intensity was measured. Background staining intensity was subtracted from the staining intensity of the nuclei. Optimum offset and gain were determined by preanalysis of multiple darkly- and lightly-stained tissues to maximize the distribution of stain intensity so that small differences in staining were quantifiable. For accuracy and consistency purposes, once established, the settings remained constant for all images.
Colocalization of differentiation and apoptosis
Apoptosis was detected using ApopTag fluorescein in situ apoptosis kit (Intergen, Purchase, NY) and differentiation was determined using Dolichos Biflorus Agglutinin (DBA) glycoprotein staining. DBA has a carbohydrate specificity to α-N-actylgalactosamine (22). This carbohydrate residue is thought to increase with normal differentiation of colonic epithelial cells (23, 24).
Antigen sites in these tissue sections were retrieved by treatment in Proteinase K (5 µg/ml). Tissues were incubated with non-fat milk and then incubated with biotinylated DBA (10 µg/ml, Vector). Slides were incubated with Texas-Red conjugated streptavidin (Vector). Preincubation of 0.2 M N-acetyl-D-galactosamine (Sigma, St. Louis, MO) with DBA or omission of DBA was used as a negative control for DBA staining. TdT enzyme was applied to tissue sections and then sections were incubated in anti-digoxigenin-fluorescein in blocking solution. Omission of TdT enzyme was used as a negative control and DNAse I treated tissue section was used as a positive control for apoptosis. At least 20 crypt columns per animal were randomly chosen for quantitative analysis. The position of DAB-stained cells and apoptotic cells were recorded using a MetaMorph Image system. Images were captured using the same inverted fluorescent Nikon microscope as described above. The differentiation index and apoptosis index were calculated as previously described (24).
In vivo measurement of N7-methylguanine DNA adducts
DNA damage was measured by quantitative immunohistochemistry using a rabbit polyclonal antibody to N7-methylguanine (gift from Dr. Geoff Margison, Paterson Institute for Cancer Research, Manchester, UK). Tissue sections were placed in prewarmed 50 mM NaOH/40% ethanol at 55 °C to denature DNA and neutralized with 5 % acetic acid/40 % ethanol. Sections were incubated with the primary antibody followed by biotinylated goat anti-rabbit IgG. The antibody-antigen complex was visualized using the DAKO Liquid DAB (diamino-benzidine tetrahydrochloride) Substrate-Chromagen System (DAKO, Carpinteria, CA). Liver N7-methylguanine DNA adducts in AOM-injected animals were used as a positive control. Omission of primary antibody was used as a negative control. Images of colonic crypts were visualized on a MICROSTAR IV, Reichert light microscope, captured by a digital camera (Sony DXC-970 MD, color 3CCD) and staining intensity (assessed by cell position) was analyzed using NIH Image software (NIH Image, version 1.61). Each nucleus on one side of a crypt column was outlined and the staining intensity was measured. Background staining intensity was subtracted from the staining intensity of the nuclei.
Statistical analyses
Proliferation, p27Kip1, differentiation, apoptosis and DNA damage data were analyzed using three-way ANOVA to determine the effect of fat, butyrate and time. When p < 0.05 for the interactions, means of all diet groups were separated using Fisher’s Protected Least Significant Difference (LSD) test. When p < 0.05 for the effects of fat, fiber or time but not for the interaction, overall means for fat, fiber or time were separated using the Fisher’s LSD test. The correlations between variables were tested using Pearson’s correlations, and statistical significance was assessed using Fisher’s distribution, with calculations performed using PROC CORR in SAS (SAS Institute Inc. Cary, NC).
Results
There were no significant differences in food intake or body weight gain among any of the four treatment groups (data not shown).
Images of colocalization of proliferation, p27Kip1, differentiation and apoptosis
Figure 1A shows a representative image of colocalization of proliferation (shown in orange) and p27 (shown in green). Proliferation was predominantly localized in the lower region of the crypt. Green fluorescence (p27Kip1 expression levels) in epithelial cells was quantitatively assessed in each epithelial cell.
Fig. 1.
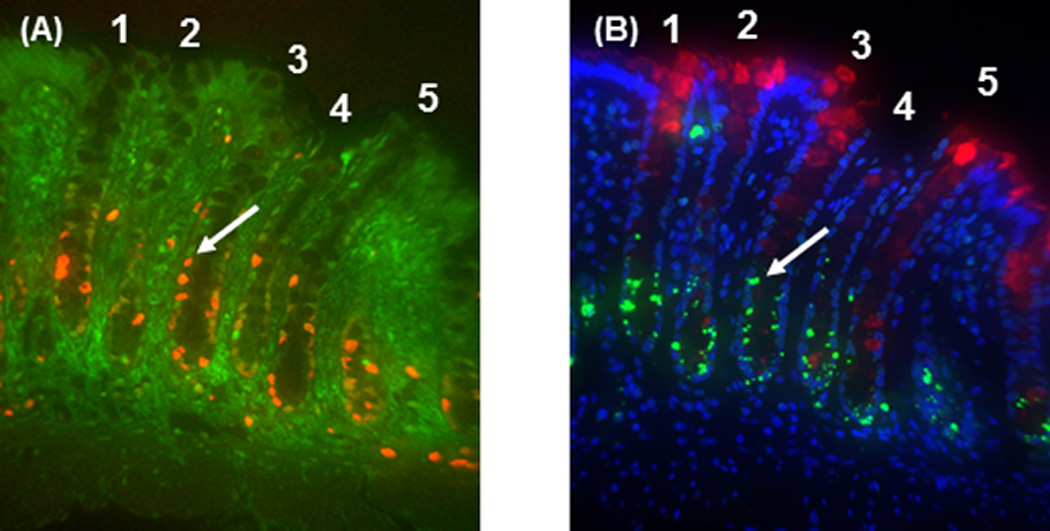
Colocalization photomicrograph (400×) of proliferation, p27Kip1, differentiation and apoptosis in serial sections. A: Proliferation (Ki-67, orange) was predominantly localized to the lower part of the crypts and p27Kip1 (green) staining was localized in the nuclei of colonic cells within crypts. B: lectin binding (red) was primarily located in the upper part of the crypt and apoptotic cells (green) were found in the lower region of the crypt 9 h after AOM injection. Blue staining represents DAPI counterstaining. Panels A and B are derived from serial sections and crypts 1–5 in panel A are the same crypts shown in panel B. Arrows show the same cell labeled for cell proliferation and p27 Kip1 in panel A, and apoptosis in panel B.
In each serial section (Fig. 1B), differentiation (shown in red) and apoptosis (shown in green) were colocalized. Lectin DBA was primarily localized in the upper part of the crypt where the greatest numbers of differentiated cells are expected. The steady state level of apoptosis was low and frequently found in the upper portion of the crypt. In contrast, following carcinogen injection, apoptosis increased particularly in the bottom part of the colonic crypt (Fig. 1B).
Carcinogen effects on DNA damage, proliferation, p27Kip1, differentiation and apoptosis
N7-methylguanine DNA adducts, as an indicator of DNA damage, increased by12 h following AOM injection and then started to decrease by 48 h post AOM injection (P < 0.001, Fig. 2A). Cell proliferation decreased at 12 h after carcinogen injection and returned to basal levels at 48 h after AOM injection (P < 0.001, Fig. 2B). In contrast, the level of p27Kip1 was not changed by AOM injection (Fig. 2C). Differentiation was increased at 24 h post AOM injection (P = 0.014, Fig. 2D) and apoptosis was maximized at the same time point (P < 0.001, Fig. 2E).
Fig. 2.
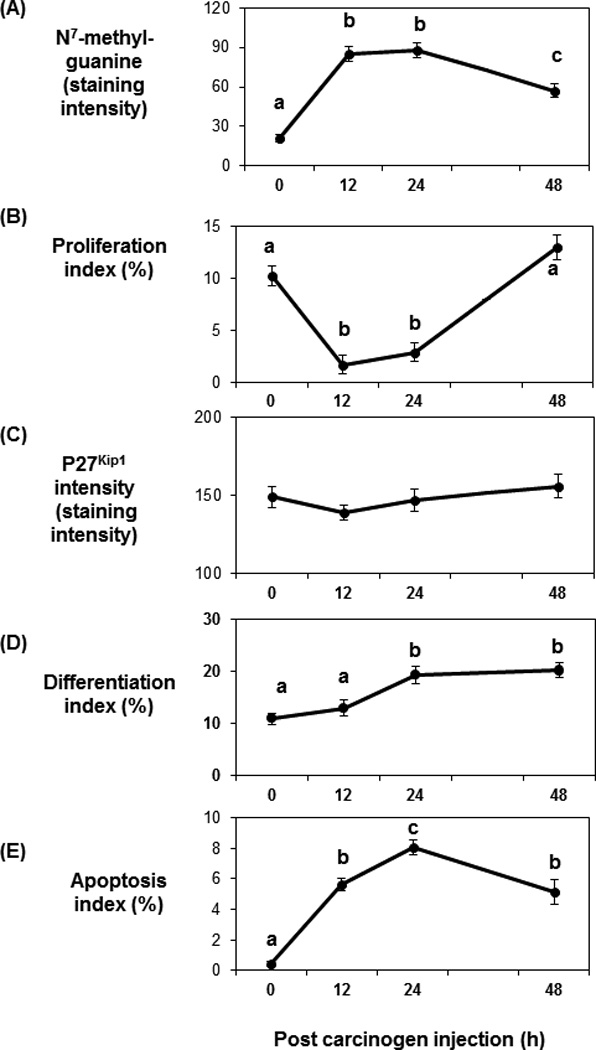
Carcinogen effects on DNA adduct level, cell proliferation, p27Kip1, differentiation and apoptosis over time. A: DNA adduct levels were increased 12 h post-AOM injection and decreased by 48 h after AOM injection (P < 0.001). B: Cell proliferation decreased 12 h after AOM injection and increased at 48 h post-AOM injection (P < 0.001). C: Carcinogen injection did not affect p27Kip1 level. D: Differentiation increased 24 h after AOM injection (P = 0.014). E: Maximum apoptosis was achieved at 24 h post AOM injection (P < 0.001). Data are presented as means ± SE. Means without a common letter are significantly different.
Dietary fat and butyrate effects on DNA damage, proliferation, p27Kip1, differentiation and apoptosis
Dietary fish oil resulted in lower DNA adduct levels compared to corn oil throughout all time points (P = 0.001, Fig. 3A). Even though there was no butyrate effect on cell proliferation, there was an obvious main effect of dietary lipid after carcinogen injection. Dietary fish oil decreased cell proliferation index compared to the corn oil diet (P = 0.003, Fig. 3B main graph). At 48 h post carcinogen injection, there was a compensatory increase in cell proliferation above that observed at 0 h in corn oil-fed rats (10.2% to 18.5%) but not in fish oil-fed rats (9% to 8.41%) (P = 0.010) (Fig. 3B, inset). The pattern of changes in proliferative zone over time, as a function of dietary fat type, was similar to proliferation index over time (data not shown). There was no significant effect of dietary fat or butyrate on p27Kip1. Dietary fish oil increased differentiation compared to the corn oil diet (P = 0.039, Fig. 3C), and butyrate treatment also increased differentiation (P = 0.041) (Fig 3C, inset). There was no main effect of dietary fat or butyrate on apoptosis. When the data were analysized using a subplot analysis for the different time points, the fish oil/butyrate diet increased the apoptotic index compared to the other groups at 24 h post carcinogen injection (P = 0.039, Fig. 3D).
Fig. 3.
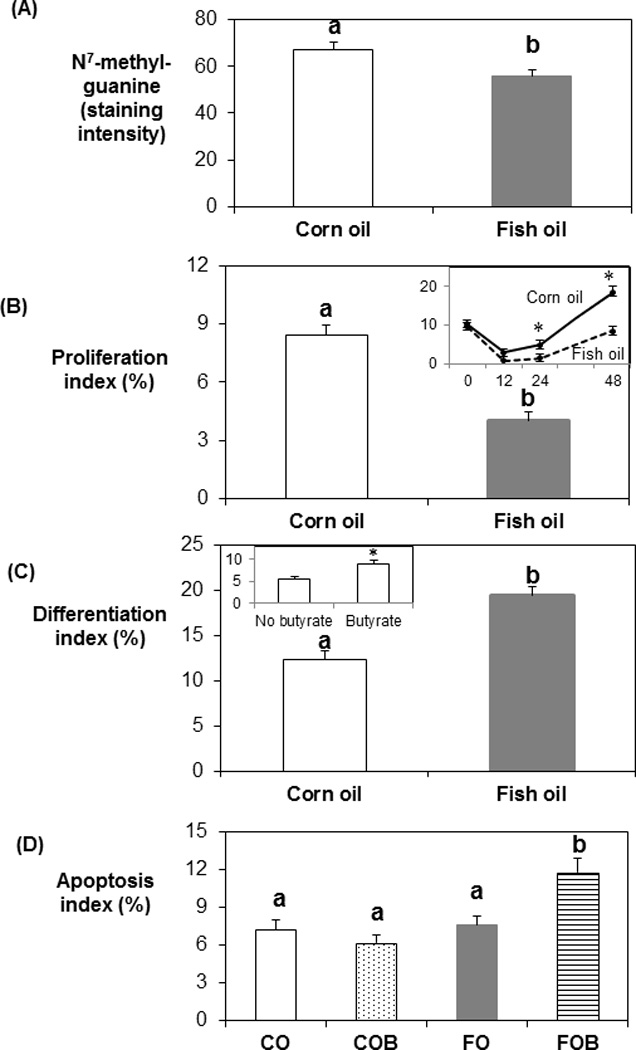
Dietary fat effects on DNA adduct level, cell proliferation, p27Kip1, differentiation and apoptosis. Dietary fish oil resulted in lower DNA damage (A, P = 0.001), cell proliferation (B, P = 0.003) and elevated differentiation (C, P = 0.039) compared to the corn oil diet. At 48 h post carcinogen injection, there was a compensatory increase of cell proliferation beyond that observed at 0 h in corn oil-fed rats but not in fish oil-fed rats (P = 0.010, B inset). Butyrate treatment increased differentiation (P = 0.041, C inset). Fish oil/butyrate diet increased apoptosis, relative to the other three groups at 24 h post carcinogen injection (D, P = 0.039). CO: corn oil; CO/B: corn oil/butyrate, FO: fish oil; FO/B: fish oil/butyrate. Data are presented as means ± SE. Bars without a common letter are significantly different.
Correlation among DNA adducts, proliferation, p27Kip1, differentiation and apoptosis
After carcinogen injection, there was a decrease of proliferation and increase of apoptosis (Fig. 2B & 2E), which was associated with a decrease of crypt height (Fig. 4A). There was no significant correlation between proliferation and crypt height (data not shown). There was an inverse relationship between crypt height and apoptosis in fish oil/butyrate treated animals (coeffiecient = −0.52, P = 0.040) (Fig. 4B). However, a significant inverse relationship was not detected in corn oil/butyrate group (Fig. 4B).
Fig. 4.
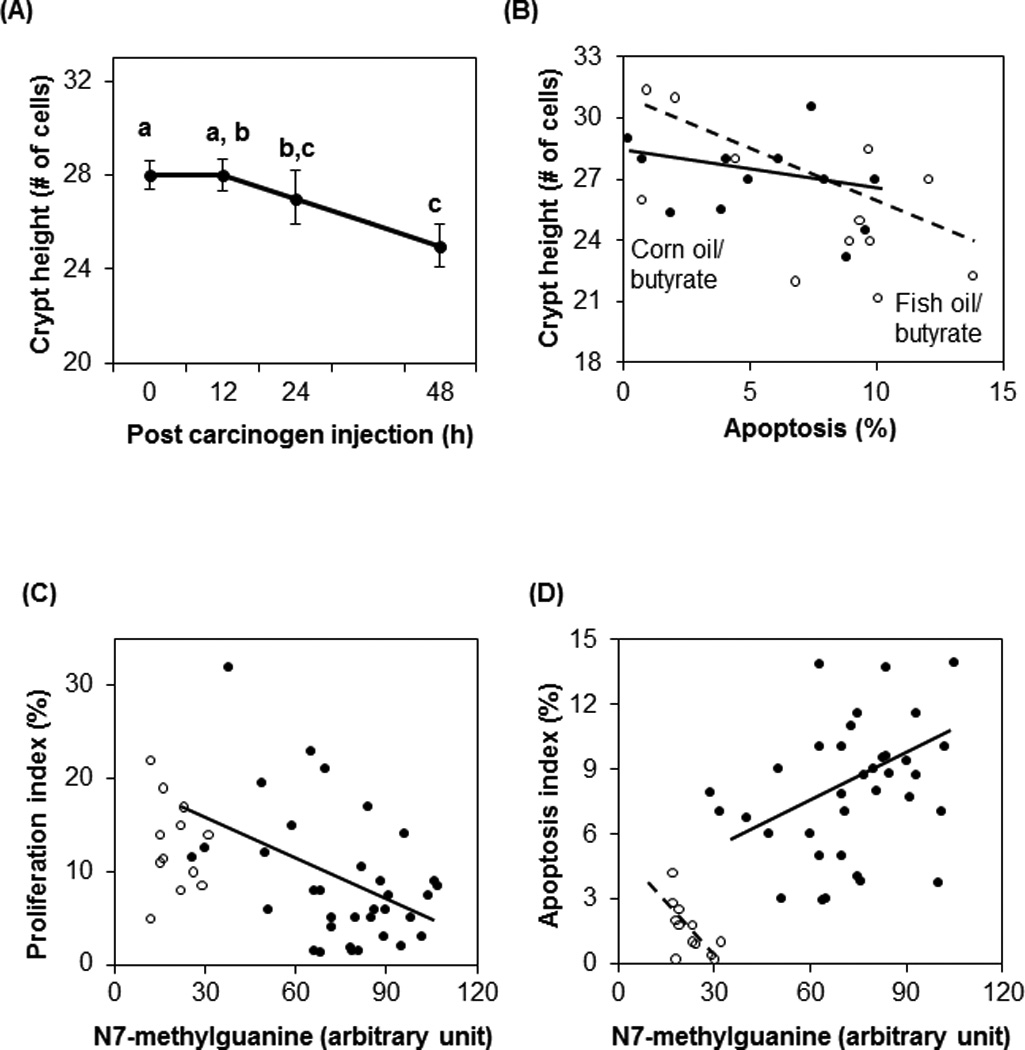
Correlation between crypt height and hours post carcinogen injection (A), between crypt height and apoptosis (B), between DNA damage and proliferation (C), and between DNA damage and apoptosis (D). (A) After carcinogen injection, crypt height decreased (P = 0.011). Data are presented as means ± SE. Means without a common letter are significantly different. (B) There was a negative relationship between crypt height and apoptosis in the fish oil/butyrate group (correlation coefficient = −0.53, P = 0.040) (○,broken line). The correlation between crypt height and apoptosis in corn oil/butyrate-fed rats was not significant (●, solid line). (C) There was no correlation between DNA adduct level and cell proliferation in saline animals (○). After carcinogen injection, there was an inverse relationship between DNA adduct level and cell proliferation (correlation coefficient = −0.42, P = 0.010) (●, solid line). (D) In saline rats, there was an inverse relationship between DNA damage and apoptosis (correlation coefficient = −0.70, P = 0.012) (○, broken line). In contrast, there was a positive relationship between DNA damage and apoptosis after carcinogen injection (correlation coefficient = 0.36, P = 0.033) (*, solid line).
As expected, DNA adduct levels were low in the saline injected rats, and there was no correlation between adduct levels and proliferation (Fig. 4C). However, in carcinogen injected animals, there was an inverse relationship between DNA adduct levels and cell proliferation (coefficient = −0.42, P = 0.010). In saline groups, there was an inverse relationship between DNA damage and apoptosis (coefficient = −0.70, P = 0.012) (Fig. 4D). In contrast, there was a positive relationship between DNA damage and apoptosis after carcinogen injection (coefficient = 0.36, P = 0.033).
To further explore the interactive effects of dietary fat and butyrate treatment on p27Kip1, a positive relationship between p27Kip1 level and proliferation in the corn oil/butyrate group (coefficient = 0.61, P = 0.035) was observed (Fig. 5). In contrast, there was no significant correlation between p27Kip1 level and proliferation in fish oil/butyrate diet (Fig. 5). Interestingly, at the same expression level of p27Kip1, the corn/butyrate diet was associated with a higher level of proliferation compared to the fish oil/butyrate diet.
Fig 5.
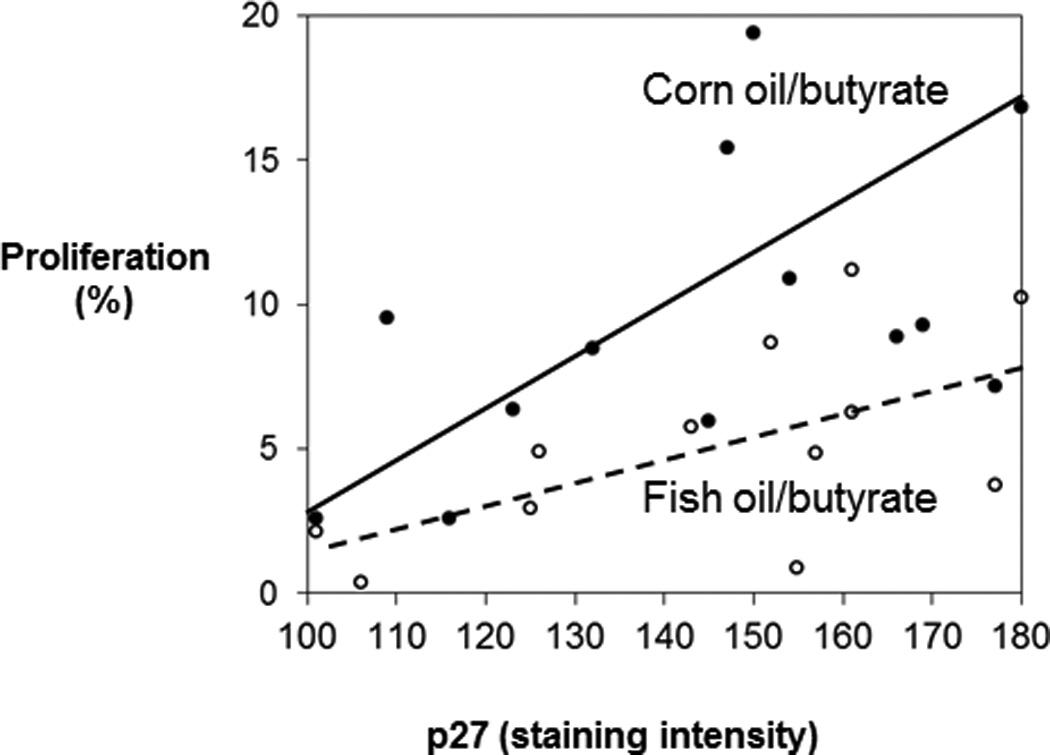
Correlation between p27Kip1 and proliferation as a function of diet. There was a positive relationship between p27Kip1 and proliferation in corn oil/butyrate fed animals (correlation coefficient = 0.61, P = 0.035) (●, solid line). The correlation between p27Kip1 and cell proliferation in fish oil/butyrate-fed rats was not significant (○, broken line).
Discussion
The normal colon is a dynamic tissue dependent upon an equilibrium among cell proliferation, differentiation and death (25). Colon tumors are initiated by nuclear damage leading to a disturbance of this steady-state. In this study, the relationships among cell proliferation, differentiation, apoptosis and a cell cycle mediator p27Kip1 were determined using a colocalization technique that allowed for the determination of all four variables in the same cell. To the best of our knowledge, this study is the first work using this approach to examine regulation of cell kinetics during cancer initiation and the response to diet. The associations between DNA damage and the four variables were also assessed.
Accumulating evidence indicates that dietary fish oil protects against colon cancer (2–8). The current data also strongly indicate that dietary fish oil decreased DNA damage and cell proliferation, and increased differentiation. At 48 h post AOM injection, corn oil-fed animals had a colonocyte proliferation index that was elevated above the levels observed at 0 h (control). In comparison, fish oil-fed rats exhibited restoration of colonocyte proliferation to a level similar to those at 0 h, which may result in lower propagation of DNA damaged cells in the fish oil-fed animals. The apoptotic index was increased, but only when rats received the combination of fish oil and butyrate.
Even though it is known that butyrate modulates cell cycle kinetics, the effect of butyrate on colon cancer development is still debated (9). Results from some studies suggest that butyrate is chemopreventive by decreasing tumor growth via a reduction in cell proliferation and an increase in differentiation and apoptosis (13, 20, 26). The administration of 1% butyrate in drinking water has been reported to increase the percentage of rats with colonic tumors (27). Others report no benefit of butyrate administration with respect to aberrant crypt foci (ACF) formation when slow-release butyrate pellets were provided to rats consuming a corn oil diet (28). However, our laboratory has shown that butyrate in combination with fish oil decreased ACF formation compared to corn oil/butyrate diet (20). These studies (20, 28) in combination with data from our study suggest that whether or not butyrate is protective against colon carcinogenesis depends on the type of dietary fat consumed. Consistent with this finding, dietary fish oil decreased cell proliferation and increased differentiation compared to the corn oil diet. However, when butyrate was combined with fish oil, this diet also increased apoptosis, whereas the pro-apoptotic effect of butyrate was not present when it was combined with corn oil.
Apoptosis plays an important role in tissue homeostasis by eliminating damaged cells, suggesting its importance in cancer therapy and the prevention of carcinogenesis (29, 30). With respect to molecular mechanisms of action, mounting data confirm that the inhibition of this process may be a critical event in the development of colonic tumors (3, 24, 29, 30). Cyclooxygenase-2 (COX-2) is an enzyme that converts arachidonic acid (20:4, n−6) to prostaglandin E2, and has been reported to be involved in colonic tumor development (31, 32). Studies have shown that prostanglandins produced by COX-2 promote colon cell proliferation and inhibit apoptosis (32). The increase of cell proliferation and resistance to apoptosis that occurs in colon cancer cell lines and in animal models of the disease was reversed by Sulindac, a COX-2 specific inhibitor (33, 34). When COX-2 is over expressed, butyrate does not induce apoptosis in rat intestinal epithelial cells (35). We and others have previously shown that n−3 enriched fish oil compared to n−6 rich corn oil reduces the level of mucosal arachidonic acid (36), COX-2 expression (37, 38) and alters the relative levels of prostaglandin E2 and prostaglandin E3 (39) in rat colonocytes. Thus, fish oil compared to corn oil decreases COX-2 expression and its pro-tumorigenic metabolites, thereby generating an environment in which butyrate can induce apoptosis. Further studies are needed to understand the mechanisms whereby fish oil and butyrate are able to effect changes in proliferation and apoptosis through COX and prostaglandin pathways.
The enhancement of apoptosis with fish oil feeding in combination with butyrate was also verified in experiments using ex vivo isolated epithelial cells. Cells from fish oil-fed rats incubated with butyrate induced apoptosis via alteration of mitochondrial function (decrease of mitochondrial membrane potential, cytochrome C release from mitochondria and increase of caspase 3 activity) compared to corn oil and butyrate incubated cells (40). In addition, it was reported that fish oil/pectin (butyrate generating fiber) diets decrease anti-apoptotic bcl-2 expression followed by increased apoptosis (41). The enhanced apoptosis was attributed to a reduction of bcl-2 via methylation of bcl-2 promoter region in fish oil/pectin group (42). In contrast, corn oil with butyrate treatment increased bcl-2 levels in mouse colonic cells (43). The proapoptotic effects of fish oil/pectin diet were also associated with the suppression of peroxisome proliferator-activated receptorδ (PPARδ) and PGE2 and increase of PGE3 (39) and modulation of microRNA and mRNA expression profiles (44).
In the present study, when butyrate was combined with a corn oil diet, there was a positive relationship between p27Kip1 and cell proliferation. The increase in p27Kip1 levels might compensate for the increase of cell proliferation induced by carcinogen administration since p27Kip1 is an inhibitor of cyclin dependent kinase, which blocks progression of the cell cycle (18, 19). However, the increase of p27Kip1 was not sufficient to suppress the elevated cell proliferation associated with the corn oil/butyrate group since the cell proliferation was higher compared to the fish oil/butyrate group.
Butyrate may produce conflicting effects on the growth of normal versus cancerous colon cells through its impact on acetyl CoA/histone acetyltransferases (HATs) or as a histone deacetylase (HDAC) inhibitor (45). In normal colon, butyrate acts as a primary energy source and does not inhibit cell proliferation by epigenetic changes via activation of acetyl CoA/HATs. In contrast, in cancer cells butyrate slows cell proliferation but induces differentiation and apoptosis by functioning as a HDAC inhibitor. Further studies are needed to examine the interaction of butyrate with lipid sources with respect to histone acetylation and cell kinetics.
Collectively, data from our study indicate that an enhanced early coordinated response to carcinogen may be one mechanism by which fish oil and butyrate protect against colon tumorigenesis. Our data also suggest that the effects of butyrate may depend in part on the type of fat in the diet. This may partly explain the controversy and inconsistency of butyrate effects on cell cycle kinetics and colon cancer development across in vivo and in vitro studies. Although our data do not address the cellular modifications in the colon that contribute to tumorigenesis, such as epigenetic modifications or stem cell-specific mutations, it does describe outcomes of those modifications and global changes in one form of DNA mutations. The ability of this combination of nutrients to alter global and gene-specific epigenetic states (20, 41, 46) at various stages of tumorigenesis and to modulate downstream events such as proliferation and apoptosis indicate the involvement of multiple mechanisms that contribute to risk reduction. However, further studies are needed to investigate the impacts of fish oil and butyrate on colon adult stem cell damage and epigenetic state.
Acknowledgments
We gratefully acknowledge Dr. Robert Burghardt for the helpful discussions. We also thank Ms. Stella S. Taddeo for excellent laboratory assistance and Ms. Kimberly Paulhill for assistance with data analysis. We also acknowledge the generous donation of dietary corn oil by Degussa BioActives (Champaign, IL), gastro-resistant slow-release butyrate pellets by Valpharma (Serravalle, Italy) and American Institute for Cancer Research. This work was supported by NIH CA61750, CA82907, RO1 CA129444 and CA57030, NSBRI NASA NCC9-58 and NIEHS 1P30ES023512.
Footnotes
Disclosure of Potential Conflicts of Interest: Authors declare no conflicts of interest.
References
- 1.Barone M, Lofano K, De Tullio N, Licinio R, Albano F, Di Leo A. Dietary, endocrine, and metabolic factors in the development of colorectal cancer. J Gastrointest Cancer. 2012;43:13–19. doi: 10.1007/s12029-011-9332-7. [DOI] [PubMed] [Google Scholar]
- 2.Chang WCL, Chapkin RS, Lupton JR. Fish oil blocks azoxymethane-induced rat colon tumorigenesis by increasing cell differentiation and apoptosis rather than decreasing cell proliferation. J Nutr. 1998;128:491–497. doi: 10.1093/jn/128.3.491. [DOI] [PubMed] [Google Scholar]
- 3.Hong MY, Lupton JR, Morris JS, Wang N, Carroll RJ, Davidson LA, et al. Dietary fish oil reduces O6-methylguanine DNA adduct levels in rat colon in part by increasing apoptosis during tumor initiation. Cancer Epidemiol Biomarkers Prev. 2000;9:819–826. [PubMed] [Google Scholar]
- 4.Toit-Kohn JL, Louw L, Engelbrecht AM. Docosahexaenoic acid induces apoptosis in colorectal carcinoma cells by modulating the PI3 kinase and p38 MAPK pathways. J Nutr Biochem. 2009;20:106–114. doi: 10.1016/j.jnutbio.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Neilson AP, Djuric Z, Ren J, Hong YH, Sen A, Lager C, et al. Effect of cyclooxygenase genotype and dietary fish oil on colonic eicosanoids in mice. J Nutr Biochem. 2012;23:966–976. doi: 10.1016/j.jnutbio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S, Sandler DP, Galanko J, Martin C, Sandler RS. Intake of polyunsaturated fatty acids and distal large bowel cancer risk in whites and African Americans. Am J Epidemiol. 2010;171:969–979. doi: 10.1093/aje/kwq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West NJ, Clark SK, Phillips RK, Hutchinson JM, Leicester RJ, Belluzzi A, et al. Eicosapentaenoic acid reduces rectal polyp number and size in familial adenomatous polyposis. Gut. 2010;59:918–925. doi: 10.1136/gut.2009.200642. [DOI] [PubMed] [Google Scholar]
- 8.Cockbain AJ, Toogood GJ, Hull MA. Omega-3 polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut. 2012;61:135–149. doi: 10.1136/gut.2010.233718. [DOI] [PubMed] [Google Scholar]
- 9.Lupton JR. Microbial degradation products influence colon cancer risk: the butyrate controversy. J Nutr. 2004;134:479–484. doi: 10.1093/jn/134.2.479. [DOI] [PubMed] [Google Scholar]
- 10.Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13:517–526. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Astbury SM, Corfe BM. Uptake and metabolism of the short-chain fatty acid butyrate, a critical review of the literature. Curr Drug Metab. 2012;13:815–821. doi: 10.2174/138920012800840428. [DOI] [PubMed] [Google Scholar]
- 12.Xiao M, Liu YG, Zou MC, Zou F. Sodium butyrate induces apoptosis of human colon cancer cells by modulating ERK and sphingosine kinase 2. Biomed Environ Sci. 2014;227:197–203. doi: 10.3967/bes2014.040. [DOI] [PubMed] [Google Scholar]
- 13.Zuo L, Lu M, Zhou Q, Wei W, Wang Y. Butyrate suppresses proliferation and migration of RKO colon cancer cells though regulating endocan expression by MAPK signaling pathway. Food Chem Toxicol. 2013;62:892–900. [PubMed] [Google Scholar]
- 14.Cho Y, Turner ND, Davidson LA, Chapkin RS, Carroll RJ, Lupton JR. Colon cancer cell apoptosis is induced by combined exposure to the n-3 fatty acid docosahexaenoic acid and butyrate through promoter methylation. Exp Biol Med. 2014;239:302–310. doi: 10.1177/1535370213514927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonçalves P, Martel F. Butyrate and colorectal cancer: the role of butyrate transport. Curr Drug Metab. 2013;14:994–1008. doi: 10.2174/1389200211314090006. [DOI] [PubMed] [Google Scholar]
- 16.Leonel AJ, Alvarez-Leite JI. Butyrate: implications for intestinal function. Curr Opin Clin Nutr Metab Care. 2012;15:474–479. doi: 10.1097/MCO.0b013e32835665fa. [DOI] [PubMed] [Google Scholar]
- 17.Orlich MJ, Singh PN, Sabaté J, Fan J, Sveen L, Bennett H, et al. Vegetarian Dietary Patterns and the Risk of Colorectal Cancers. JAMA Intern Med. 2015 doi: 10.1001/jamainternmed.2015.59. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 19.Mayo C, Lloreta J, Real FX, Mayol X. In vitro differentiation of HT-29 M6 mucus-secreting colon cancer cells involves a trychostatin A and p27(KIP1)-inducible transcriptional program of gene expression. J Cell Physiol. 2007;212:42–50. doi: 10.1002/jcp.20999. [DOI] [PubMed] [Google Scholar]
- 20.Crim KC, Sanders LM, Hong MY, Taddeo SS, Turner ND, Chapkin RS, et al. Upregulation of p21Waf1/Cip1 expression in vivo by butyrate administration can be chemoprotective or chemopromotive depending on the lipid component of the diet. Carcinogenesis. 2008;29:1415–1420. doi: 10.1093/carcin/bgn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong MY, Nulton E, Shelechi M, Hernández LM, Nemoseck T. Effects of dark chocolate on azoxymethane-induced colonic aberrant crypt foci. Nutr Cancer. 2013;65:677–685. doi: 10.1080/01635581.2013.789542. [DOI] [PubMed] [Google Scholar]
- 22.Nash R, Neves L, Faast R, Pierce M, Dalton S. The lectin Dolichos biflorus agglutinin recognizes glycan epitopes on the surface of murine embryonic stem cells: a new tool for characterizing pluripotent cells and early differentiation. Stem Cells. 2007;25:974–982. doi: 10.1634/stemcells.2006-0224. [DOI] [PubMed] [Google Scholar]
- 23.Boland C, Montgomery C, Kim Y. Alterations in human colonic mucin occurring with cellular differentiation and malignant transformation. Proc Natl Acad Sci USA. 1982;79:2051–2055. doi: 10.1073/pnas.79.6.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong MY, Chang WCL, Chapkin RS, Lupton JR. Relationship among colonocyte proliferation, differentiation and apoptosis as a function of diet and carcinogen. Nutr Cancer. 1997;28:20–29. doi: 10.1080/01635589709514548. [DOI] [PubMed] [Google Scholar]
- 25.Leiszter K, Galamb O, Sipos F, Krenács T, Veres G, Wichmann B, et al. Sporadic colorectal cancer development shows rejuvenescence regarding epithelial proliferation and apoptosis. PLoS One. 2013;8:e74140. doi: 10.1371/journal.pone.0074140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen HM, Lin YW, Wang JL, Kong X, Hong J, Fang JY. Identification of potential target genes of butyrate in dimethylhydrazine-induced colorectal cancer in mice. Nutr Cancer. 2013;65:1171–1183. doi: 10.1080/01635581.2013.828087. [DOI] [PubMed] [Google Scholar]
- 27.Freeman H. Effects of differing concentrations of sodium butyrate on 1,2-dimethylhydrazine-induced rat intestinal neoplasia. Gastroenterology. 1986;91:596–602. doi: 10.1016/0016-5085(86)90628-1. [DOI] [PubMed] [Google Scholar]
- 28.Caderni G, Luceri C, De Filippo C, Salvadori M, Giannini A, Tessitore L, Dolara P. Slow-release pellets of sodium butyrate do not modify azoxymethane (AOM)-induced intestinal carcinogenesis in F344 rats. Carcinogenesis. 2001;22:525–527. doi: 10.1093/carcin/22.3.525. [DOI] [PubMed] [Google Scholar]
- 29.West NJ, Courtney ED, Poullis AP, Leicester RJ. Apoptosis in the colonic crypt, colorectal adenomata, and manipulation by chemoprevention. Cancer Epidemiol Biomarkers Prev. 2009;18:1680–1687. doi: 10.1158/1055-9965.EPI-09-0006. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Yu J. Role of apoptosis in colon cancer biology, therapy, and prevention. Curr Colorectal Cancer Rep. 2013;9 doi: 10.1007/s11888-013-0188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahmoud AS, Umair A, Azzeghaiby SN, Alqahtani FH, Hanouneh S, Tarakji B. Expression of Cyclooxygenase-2 (COX-2) in Colorectal Adenocarcinoma: an Immunohistochemical and Histopathological Study. Asian Pac J Cancer Prev. 2014;15:6787–6790. doi: 10.7314/apjcp.2014.15.16.6787. [DOI] [PubMed] [Google Scholar]
- 32.Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol. 2010;2010:215158. doi: 10.1155/2010/215158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tai WP, Hu PJ, Wu J, Lin XC. The inhibition of Wnt/β-catenin signaling pathway in human colon cancer cells by sulindac. Tumori. 2014;100:97–101. doi: 10.1700/1430.15823. [DOI] [PubMed] [Google Scholar]
- 34.Martin JE, Young GP, LE Leu RK, Hu Y. Comparing the effects of COX and non-COX-inhibiting NSAIDs on enhancement of apoptosis and inhibition of aberrant crypt foci formation in a rat colorectal cancer model. Anticancer Res. 2013;33:3581–3588. [PubMed] [Google Scholar]
- 35.Tsujii M, Dubois RN. Alteration in cellular adhesions and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 36.Lee DY, Lupton JR, Aukema HM, Chapkin RS. Dietary fat and fiber alter rat colonic mucosal lipid mediators and cell proliferation. J Nutr. 1993;123:1808–1817. doi: 10.1093/jn/123.11.1808. [DOI] [PubMed] [Google Scholar]
- 37.Lupton JR, Chang WCL, Hong MY, Chapkin RS. Fat/fiber interactions on colonic cytokinetics: Relationship to colon cancer. Asia Pacific J Clin Nutr. 1999;8:S37–S40. [Google Scholar]
- 38.Bagga D, Wang LW, Farias-Eisner R, Galaspy JA, Reddy ST. Differential effects of prostaglandin derived from w-6 and w-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc Natl Acad Sci USA. 2003;100:1751–1756. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanamala J, Glagolenko A, Yang P, Carroll RJ, Murphy ME, Newman RA, et al. Dietary fish oil and pectin enhance colonocyte apoptosis in part through suppression of PPARdelta/PGE2 and elevation of PGE3. 2008;29:790–796. doi: 10.1093/carcin/bgm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong MY, Chapkin RS, Barhoumi R, Burghardt RC, Turner ND, Henderson CE, et al. Fish oil increases mitochondrial phospholipid unsaturation upregulation reactive oxygen species and apoptosis in rat colonocytes. Carcinogenesis. 2002;23:1919–1925. doi: 10.1093/carcin/23.11.1919. [DOI] [PubMed] [Google Scholar]
- 41.Hong MY, Chapkin RS, Davidson LA, Turner ND, Morris JS, Carroll RJ, et al. Fish oil enhances targeted apoptosis during colon tumor initiation in part by downregulating bc l-2. Nutr Cancer. 2003;46:44–51. doi: 10.1207/S15327914NC4601_06. [DOI] [PubMed] [Google Scholar]
- 42.Cho Y, Turner ND, Davidson LA, Chapkin RS, Carroll RJ, Lupton JR. A chemoprotective fish oil/pectin diet enhances apoptosis via Bcl-2 promoter methylation in rat azoxymethane-induced carcinomas. Exp Biol Med (Maywood) 2012;237:1387–1393. doi: 10.1258/ebm.2012.012244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turk HF, Kolar SS, Fan YY, Cozby CA, Lupton JR, Chapkin RS. Linoleic acid and butyrate synergize to increase Bcl-2 levels in colonocytes. Int J Cancer. 2011;128:63–71. doi: 10.1002/ijc.25323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah MS, Schwartz SL, Zhao C, Davidson LA, Zhou B, Lupton JR, et al. Integrated microRNA and mRNA expression profiling in a rat colon carcinogenesis model: effect of a chemo-protective diet. 2011;43:640–654. doi: 10.1152/physiolgenomics.00213.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho Y, Turner ND, Davidson LA, Chapkin RS, Carroll RJ, Lupton JR. Colon cancer cell apoptosis is induced by combined exposure to the n-3 fatty acid docosahexaenoic acid and butyreate through protmoter methylation. Exp Biol Med. 2014;239:302–310. doi: 10.1177/1535370213514927. [DOI] [PMC free article] [PubMed] [Google Scholar]