

Abstract
Anaplastic lymphoma kinase-positive (ALK+) anaplastic large cell lymphoma (ALCL) is an aggressive T-cell non-Hodgkin lymphoma characterized by the t(2;5) resulting in overexpression of NPM-ALK, which is known to activate the phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR pathway resulting in cell cycle and apoptosis deregulation. ALK+ ALCL is also characterized by strong AP-1 activity and overexpression of two AP-1 transcription factors, CJUN and JUNB. Here, we hypothesized that a biologic link between AP-1 and AKT kinase may exist, thus contributing to ALCL oncogenesis. We show that JUNB and cJUN bind directly to the AKT1 promoter, inducing AKT1 transcription in ALK+ ALCL. Knockdown of JUNB and CJUN in ALK+ ALCL cell lines downregulated AKT1 mRNA and promoter activity and was associated with lower AKT1 protein expression and activation. We provide evidence that this is a transcriptional control mechanism shared by other cell types even though it may operate in a way that is cell context specific. In addition, STAT3-induced control of AKT1 transcription was functional in ALK+ ALCL and blocking of STAT3 and AP-1 signaling synergistically affected cell proliferation and colony formation. Our findings uncover a novel transcriptional crosstalk mechanism that links AP-1 and AKT kinase, which coordinate uncontrolled cell proliferation and survival in ALK+ ALCL.
Keywords: Anaplastic large cell lymphoma, JUNB, AKT1, ALK+, AP-1, JUNB, cJUN
INTRODUCTION
Anaplastic lymphoma kinase-positive (ALK+) anaplastic large cell lymphoma (ALCL) is a distinct type of aggressive T-cell non-Hodgkin lymphoma, which is characterized by overexpression of ALK due to chromosomal translocations, the most frequent being the t(2;5)(p23;q35)1. This chromosomal aberration leads to the formation of the chimeric protein nucleophosmin (NPM)-ALK, a constitutive oncogenic tyrosine kinase, which is capable of activating multiple oncogenic pathways2 Furthermore, ALCL is characterized by overexpression of CD30, a member of the tumor necrosis factor receptor superfamily3.
JUNB is a member of the activator protein 1 (AP-1) complex, a group of transcription factors that bind to AP-1 DNA recognition elements and induce expression of genes that control a diverse array of cellular functions, spanning from cell growth and apoptosis to cell proliferation and oncogenic transformation4. AP-1 proteins include four subfamilies, Jun, Fos, Maf and ATF, that function as a complex of homodimers and heterodimers, among which cJUN and JUNB are the most well studied4. Previous studies have shown that JUNB suppresses cell cycle progression through either upregulation of the cyclin-dependent kinase inhibitor p16 or direct inhibition of cyclin D15, 6. Furthermore, deregulated JUNB expression has been linked to certain hematological malignancies. JUNB inactivation was found in chronic myeloid leukemia patients7, while transgenic mice specifically lacking JUNB expression in the myeloid lineage developed a chronic myeloid leukemia-like phenotype that eventually progressed to blast crisis8. However, in ALCL and Hodgkin lymphoma, we and others have shown that AP-1 is constitutively active with prominent expression of functional CJUN and JUNB.9–13.
In addition, JUNB was found to interact with the CD30 promoter, inducing CD30 expression in both Hodgkin lymphoma and ALCL14. Another study demonstrated that JUNB is the most important and transcriptionally active of all AP-1 members in ALK+ ALCL and that its activation is highly controlled by NPM-ALK through extracellular signal-regulated kinase 1/2 (ERK1/2) at the transcriptional level and via the mTOR pathway at the translational level15. ETS1 has been identified as the transcription factor that mediates ERK1/2-dependent regulation of JUNB in ALK+ ALCL16, and we have recently shown that JUNB amplification is another mechanism that may lead to the constitutive JUNB expression observed in ALK+ ALCL17. Furthermore, we have demonstrated that CJUN is also highly active in NPM-ALK+ ALCL, since NPM-ALK directly binds to and activates JNK kinase, which, in turn, phosphorylates / activates CJUN.12. Taken together, these findings provide a direct link between AP-1 members and NPM-ALK in ALK+ ALCL. NPM-ALK is also known to activate numerous pathways by recruiting SRC homology 2 or phosphotyrosine binding domain-containing molecules, including the Ras18, the γ-phospholipase19, the JAK-STAT20, the mTOR21 and the PI3K/AKT pathways22.
The AKT family members regulate a diverse array of cellular functions, including apoptosis, cellular proliferation, differentiation, and intermediary metabolism. AKT is also one of most frequently hyperactivated kinases in human cancers23. Aberrant activation of AKT can occur by a variety of mechanisms, including amplification, AKT mutation, and/or alterations in AKT upstream regulators.24. The biologic significance of the AKT in lymphomagenesis has been established in a mouse model25. In ALCL, it has been shown that NPM-ALK mediates its oncogenic function at least in part through phosphorylation and activation of AKT20, 26. In addition, AKT is activated in a substantial subset of ALCL tumors, and AKT1 expression is associated with a significantly lower level of the cyclin-dependent kinase (CDK) inhibitor p27 and a higher rate of tumor cell proliferation.27. Inhibition of AKT in ALCL cells results in cell cycle arrest through increased expression of p27, a negative regulator of the G1-S phase28. Moreover, AKT activation markedly increased mTOR phosphorylation and its downstream effectors, which led to elevated tumor cell survival and apoptosis evasion in ALK+ ALCL28. All these findings imply an important role for AKT1 in the pathogenesis of ALCL.
Transcriptional regulation of AKT1 gene remains largely obscure. Park et al.29 reported that AKT1 is transcriptionally upregulated by the SRC/STAT3 pathway through direct binding of STAT3 on the AKT1 promoter29. In the same study, multiple putative AP-1 binding sites were identified upstream of the AKT1 transcription initiation site. This finding and preliminary data from our laboratory led us to hypothesize that AP-1 transcription factors may be involved in AKT1 gene regulation.
In the present report we provide evidence of AP-1 (JUNB, CJUN)-dependent control of AKT1 transcription and activation in ALK+ ALCL. Notably, AP-1 members induce or suppress AKT1 expression by directly binding on its promoter sequence in a manner that is dictated by cell type specificity. Synergistic action between AP-1 and STAT3 on AKT1 transcription was observed which contributed to increased cell survival and proliferation.
MATERIALS AND METHODS
Cell lines, plasmids and reagents
Three ALK+ ALCL cell lines were used in this study: Karpas 299 (a gift of Dr. M. Kadin, Beth Israel-Deaconess Medical Center, Boston, MA, USA), SR-786, and SUP-M2 (both from ATCC, Rockville, MD, USA). The T-cell acute lymphoblastic leukemia (T-ALL) cell line Jurkat was used as a negative control because of the absence of AP-1 activity. 293T is a cell line derived from human embryonic kidney (HEK) cells (ATCC). All cell lines were grown in Roswell Park Memorial Institute (RPMI)-1640 medium (Life Technologies, Grand Island, NY, USA) as described previously12, supplemented with 10% fetal calf serum, and incubated at 37°C in a humidified atmosphere containing 5% CO2.
The JUNB expression vector pcDNA3.1/V5-His Topo was a gift from Dr. Anupam Agarwal (University of Alabama at Birmingham, Birmingham, AL, USA). The AKT1 luciferase promoter (−4293/+1888/Luc) was a gift from Dr. J. Q. Cheng (University of South Florida College of Medicine, Tampa, FL, USA). STAT 3 inhibitory compound Stattic (Sigma, St. Louis, MO, USA), a small-molecule inhibitor of STAT3 activation and dimerization, and the specific MEK1/2 inhibitor U0126 (Cell Signaling Technology, Beverly, MA, USA) were used.
Transient transfection and luciferase assays
Cells were seeded at a density of 0.5×106 cells/ml 24 hrs before transfection, and 3×106 ALK+ ALCL cells were transfected with the AKT reporter plasmid (−4298/+1888-pGL3 construct), the JUNB expression plasmid, or both along with 10 ng of pRL-CH110 as an internal control, using the Nucleofector solution “V” recommended by Amaxa Biosystems (Lonza, Cologne, Germany). Activities of the AKT1 promoter were analyzed 48 hr after transfection, using the dual luciferase system (Promega, Madison, WI, USA) and the luciferase enzymatic activity was measured with a Monolight 3010 luciferometer (BD Biosciences, San Jose, CA, USA). All the experiments were performed in triplicate and the values were normalized against Renilla luciferase activity. Luciferase activity was expressed as relative to control luciferase activity.
Silencing of JUNB, CJUN, and STAT3 gene expression by siRNA
Transient transfection of Karpas 299, SUP-M2 and SR-786 cells was carried out with Amaxa Nucleofector II, the solution “V”, and the A-030 program as recommended by the manufacturer (Lonza, Basel, Switzerland). Approximately 3×106 cells were transfected with 50 to 300 nM specific siRNA or siControl (scrambled). Whole-cell lysates were prepared 48 hrs after transfection. JUNB siRNA was used as a mixture of four pre-selected siRNAs specifically designed for JUNB (FlexiTube siRNA; Qiagen, Venlo, Netherlands). The specific siRNAs targeting the human CJUN product were pre-designed by Ambion, Inc. (Austin, TX, USA). Small interfering RNA (siRNA) for STAT3, and Control (LUC) were obtained from Dharmacon (Lafayette, CO, USA) (Supplementary Table 1).
Western blot analysis
Western blot analysis was performed using methods described previously30. Antibodies against the following proteins were used: AKT1, pAKT1 (Thr308), pAKT1 (Ser 473), STAT3, p-Tyr705 STAT3, ERK, pERK, pALK and pGSK3α/β (Cell Signaling Technology, Beverly, MA, USA), JUNB and CJUN (Santa Cruz Biotechnology, Santa Cruz, CA, USA), AKT2 and AKT3 (Millipore, Billerica, Massachusetts, MA, USA). β-Actin (Sigma, St Louis, MO) served as a control for protein load and integrity in all immunoblots.
RNA extraction, cDNA synthesis, and real time RT-PCR (RT-qPCR)
For the RT-qPCR analysis, total RNA was extracted using the pure link RNA mini kit from Ambion (Life Technologies, Carlsbad, CA, USA). cDNA was synthesized using the Superscript First Strand Synthesis System (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. mRNA expression levels were quantified by RT-qPCR using the SYBR PCR Master Mix (Applied Biosystems/Life Technologies) in a one-step reaction and the mRNA expression levels were determined by the comparative CT (ΔΔCt) method. GAPDH was used as the endogenous control gene. The primer sequences are listed in Supplementary Table 1. For JUNB, AKT1, AKT2, AKT3 and GAPDH cDNA, the RT-qPCR program included Amplitaq Gold DNA polymerase activation at 95°C (10 min) followed by 40 cycles of DNA denaturation (95°C for 15 sec) and annealing/extension (60°C for 30 sec). All reactions were performed using an ep-realplex Mastercycler (Eppendorf, Hamburg, Germany).
Chromatin immunoprecipitation (ChIP)
ChIP assays and subsequent RT-qPCR analysis were performed as described for the fast ChIP protocol31 The relative occupancy of the immunoprecipitated factor at a locus was estimated using the following equation: 2(Ctmock−Ctspecific), where Ct-mock and Ct-specific are mean threshold cycles of PCR done in triplicate on DNA samples from mock and specific immunoprecipitations. The comparative CT method was used to determine relative expression compared with input antibodies against the proteins JUNB, CJUN, and control IgG (Santa Cruz) and RNA PolII (Millipore Billerica, MA, USA).
Analysis of AKT1 promoter and site-directed mutagenesis: Gene reporter assays
Molecular dissection of the AKT1 promoter was described elsewhere29 and included nine putative AP-1–binding sites upstream of the transcription start site. Mutations were inserted in AP-1 sites using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). More specifically, we changed CA ≥ GT at AP-1 site 7, GT > CA at AP-1 site 8 and GT > CC at AP-1 site 9 (Supplementary Table 1). Some constructs had both mutations (at AP-1 sites 8 and 9, or at AP-1 sites 7 and 9). All mutagenesis was confirmed by DNA sequencing.
Cell proliferation, trypan blue exclusion and colony-formation assays
The (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) assay was used to evaluate proliferation of viable cells, as described previously32. The absorbance was read using a microplate (ELISA) reader mQuant spectrophotometer (BIO-TEK Instruments, Inc., Winooski, VT, USA) at 570 nm. Cell viability was assessed using the trypan blue exclusion assay as described elsewhere32. Cell counting at 48 hrs was performed in triplicate. For colony-formation assay, cells were plated in 24-well plates in 300 µl of MethoCult methylcellulose solution (Sigma, St Louis, MO) at a concentration of 500 cells/well and were incubated for 2 weeks until colony formation was visible. Colonies were counted after staining with p-iodonitrotetrazolium violet (Sigma, St Louis, MO).
Statistical analysis
Data were analyzed using GraphPad Prism software (GraphPad Software, La Jolla, CA). Western blot bands were quantified using ImageJ software (NIH, Bethesda, MD, USA). Comparisons between groups were made using paired or unpaired t-tests as appropriate. P < 0.05 was considered to be statistically significant.
RESULTS
JUNB and CJUN bind to and activate AKT1 promoter in ALK+ ALCL
JUNB gene silencing resulted in decreased levels of AKT1 transcripts in both Karpas 299 and SR-786 cells (Figure 1). Moreover, JUNB depletion attenuated AKT1 protein levels in ALK+ ALCL cell lines, which was accompanied by decreased AKT kinase activity (Figure 1 and Suppl. Figure 1).
Figure 1. JUNB depletion results in suppressed AKT1 mRNA and protein expression in ALK+ ALCL.
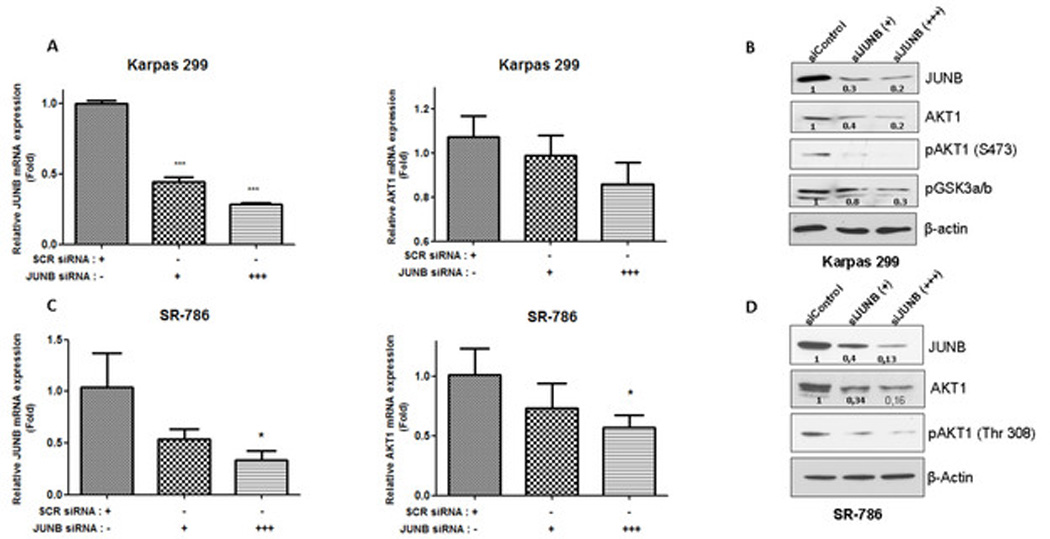
A. Karpas 299 cells were transiently transfected with increasing concentrations of JUNB siRNA, (+,50 nM and +++, 300 nM) or control (scramble, scr) siRNA which resulted in increasingly reduced JUNB (left) and AKT1 (right) mRNA levels as determined by qRT-PCR, which was performed in triplicate. B. AKT1 protein level was decreased following JUNB silencing, which was also associated with reduced levels of Ser473-phosphorylated (activated) AKT1 and reduced phosphorylation of its kinase substrate, GSK3. C. Similar results were obtained with another ALK+ ALCL cell line, SR-786. D. AKT1 protein and Thr308 phosphorylation levels were decreased in SR-786 cells following JUNB gene silencing. The expression level of JUNB and AKT1 mRNA was normalized to the GAPDH mRNA level (control) and it was shown as fold change to control.
*, p<0,05, **, p<0,01, ***, p<0,001 (Student’s t-test).
Previous examination of the genomic sequence spanning 4298 base pairs upstream of the transcription start site of the human AKT1 revealed the presence of nine putative AP-1 binding sites (Figure 2). To investigate the possibility of AP-1 transcription factors binding on the AKT1 promoter, we designed primers spanning those sites and performed ChIP with JUNB and CJUN antibodies using sheared chromatin from the ALK+ ALCL cells. Our results showed that anti-JUNB antibody specifically enriched DNA fragments containing either AP-1 site 8 or AP-1 site 9. The AP-1 sites 1–7 showed minimal or no enrichment with JUNB antibody (not shown). In addition, CJUN co-precipitated with chromatin complexes from the AKT1 promoter. More specifically, a signal was detected in the DNA region spanning AP-1 site 7 (Figure 2). Thus, we provide evidence for direct physical interaction between AKT1 promoter and AP-1 members.
Figure 2. Members of the AP-1 complex bind to and activate AKT1 promoter in ALK+ ALCL.
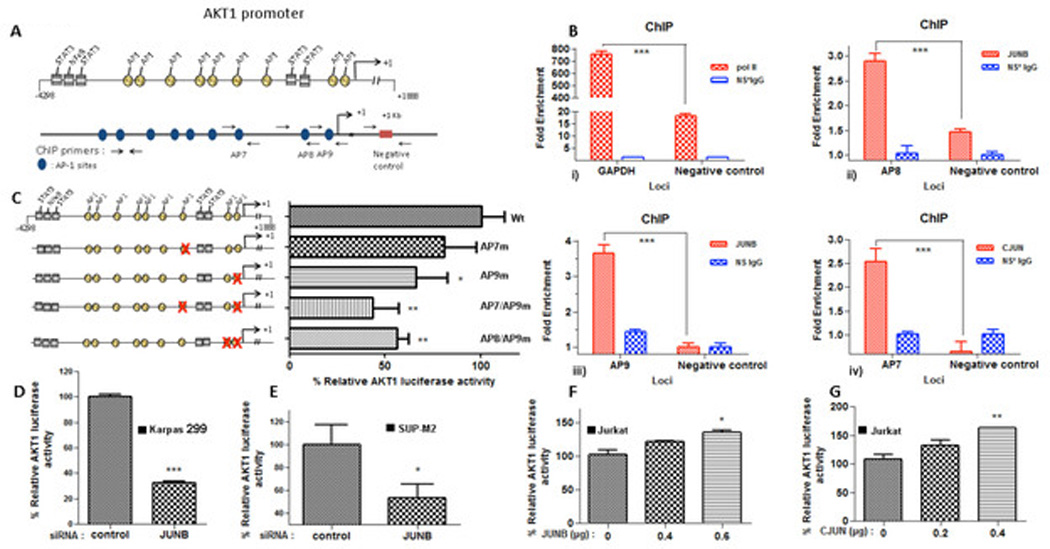
A. Graphical representation of AKT1 promoter sequence. Top: the yellow circles depict putative AP-1 binding sites and the gray boxes depict STAT3/NF-κB binding sites. Bottom, primer design for ChIP. Primers spanning the putative AP-1 binding sites. B. AP-1 (JUNB and CJUN) bind to at least three AP-1 sites on AKT1 promoter. ChIP assays were performed with CJUN and JUNB-specific antibodies as described in “Materials and Methods” using qPCR. i) Pol II enrichment on the GAPDH promoter was used as a positive control for the technique. Studies resulted in the precipitation of AKT1 promoter chromatin containing the AP-1 binding sites 8 (AP8, ii) and 9 (AP9, iii), respectively, in Karpas 299 cells when the anti-JUNB antibody was used. cJUN was found to co-precipitate with AKT1 promoter chromatin containing the putative AP-1 site 7 (AP7, iv) of the AKT1 promoter. The blue bars refer to immunoprecipitation with non-immune rabbit or mouse IgG. The red bars refer to immunoprecipitation using rabbit anti-CJUN or mouse anti-JUNB IgG. Data were analyzed by two-way ANOVA test using a Bonferroni posttest correction. C. Mutation of the specific putative AP-1 sites attenuated the AKT1 promoter activity. A series of mutations were initiated in the full-length luciferase AKT1 promoter. Mutation of the AP1 sites 7 (AP7m), 9 (AP9m) and also double mutants between 7 and 9 (AP7/AP9m) and 8 and 9 (AP8/AP9m) were introduced in the AKT1-Luc promoter. The resulting reporter constructs were then transfected into Karpas 299 cells and subjected to luciferase assay. The promoter activity was normalized against the full length (−4298/+1888) wild type-Wt (unmutated) reporter construct. Mutation of the AP-1 binding sites within the AKT1 promoter abrogated AKT1 reporter activity. D and E. Two ALK+ ALCL cell lines, Karpas 299 (D) and SUP-M2 (E), were co-transfected with JUNB siRNA and the AKT1 reporter and were subjected to luciferase assay. JUNB silencing resulted in the repression of the AKT1 promoter activity in both cell lines. F and G. Overexpression of JUNB (F) and CJUN (G) expression plasmids led to increased AKT1 promoter activity in Jurkat cells in a concentration-dependent manner.
*, p<0,05, **, p<0,01, ***, p<0,001 (Student’s t-test).
To study the functional relevance of those DNA sites to AKT1 regulation, we introduced a series of AP-1 mutations in luciferase-based constructs of the AKT1 promoter and subsequently delivered them into ALK+ ALCL cells. Luciferase activity was measured and values were normalized against the control (unmutated, wild-type AKT1-Luc promoter). Our data confirmed that AP-1 mutation sites 7 and 9 reduced the overall luciferase activity of the AKT promoter, while double mutants (AP7/AP9m and AP8/AP9m) had an additive effect, suggesting that those DNA regions represent important regulatory sites for various AP-1 transcription factors.
Next, we tested the ability of the AKT1 promoter to remain active following JUNB depletion. Briefly, ALK+ ALCL cell lines were co-transfected with the wild-type AKT1-Luc promoter construct and JUNB siRNA or control siRNA. Data from the ALK+ ALCL cell lines Karpas 299 (Figure 2D) and SUP-M2 (Figure 2E) suggested that JUNB loss is associated with a profound decrease in AKT1 promoter activity. Moreover, we examined whether JUNB overexpression is sufficient to augment AKT1 transactivation. Because JUNB protein is highly expressed in ALK+ ALCL cells and because these cells exhibit strong AP-1 activity10, we utilized the T-cell leukemia cell line Jurkat, which is characterized by low JUNB protein expression and almost undetectable AP-1 binding activity17 JUNB (Figure 2F) and CJUN (Figure 2G) expression plasmids were transfected into Jurkat cells together with the AKT1-Luc promoter construct. Both JUNB and CJUN overexpression led to a significant increase in luciferase activity in a concentration-dependent manner.
JUNB-induced control of AKT1 transcription is cell type-specific
To examine whether JUNB-mediated transcriptional regulation of AKT1 represents a universal mechanism, we examined the effects of JUNB overexpression and depletion on AKT1 expression in a different, non cancerous cellular context using the HEK293T cells, which expresses low levels of JUNB. Surprisingly, forced expression of JUNB (Figure 3A) resulted in a dose-dependent decrease of total AKT1 protein level. Furthermore, JUNB transfection was associated with significantly weaker AKT1 promoter activity (Figure 3B). By contrast, JUNB silencing completely abolished the JUNB-induced repression of the AKT1 promoter and led to a remarkable increase in its transcriptional activity (Figure 3C).
Figure 3. JUNB-mediated transactivation of AKT1 is cell context-dependent.

A. Forced expression of JUNB resulted in a concentration-dependent decrease of total AKT1 protein levels in HEK293T cells. B. Forced expression of JUNB repressed AKT1 promoter activity in HEK293T cells. C. AKT1 promoter activity was restored following JUNB knockdown. D. HEK293T cells were co-transfected with JUNB expression plasmid and the AKT1-Luc constructs harboring either wild-type (Wt) or mutated AP-1 sites 7 (AP7m), 8 (AP8m), or 9 (AP9m). Mutation at site 8 or 9 led to abrogation of the JUNB-dependent AKT1 promoter repression, while AP7 mutation did not result in de-repression of promoter activity. E. cJUN transactivates AKT1 promoter in HEK293T cells. F. Compared with wild-type AP-1, mutation of AP-1 sites 7 or 8 results in decreased AKT1 promoter activity in co-transfection assays with cJUN expression plasmid, whereas mutation at AP-1 site 9 didn’t change the cJUN-induced activation status of the AKT1 promoter. G. NPM-ALK can partly induce JUNB-mediated AKT1 trans-repression. Ectopic expression of NPM-ALK plasmid but not the kinase-dead (K210R) NPM-ALK vector resulted in significant repression of AKT1 promoter. H. Western blot analysis following transfection of NPM-ALK and the kinase-dead (K210R) expression plasmids showed that the JUNB protein levels were increased following NPM-ALK ectopic expression in HEK293T cells. Also, there was concomitant AKT1 protein upregulation. pALK was used as a positive control for the kinase activity of NPM-ALK.
*, p<0,05, **, p<0,01, ***, p<0,001 (Student’s t-test).
To determine whether JUNB suppresses AKT1 activity through direct binding on the gene promoter, HEK293T cells were co-transfected with JUNB plasmid and either wild-type (containing all the AP-1 sites) or a mutated (harboring mutations at AP-1 site 7, 8, or 9) AKT1-Luc promoter plasmid. Luciferase assay revealed that when AP-1 site 8 and 9 were mutated from the promoter either individually or together (Supplementary Figure 1B), the total AKT1 promoter activity increased significantly as compared to wild-type AKT1 promoter suggesting a direct JUNB-mediated transcriptional regulation of AKT1 (Figure 3D). However, transfection of CJUN resulted in higher AKT1 promoter activity in 293T cells (Figure 3E), which is consistent with the tumor-promoting properties of CJUN. Moreover, mutation of AP-1 site 7 or 8 on the AKT1 promoter, resulted in a marked decrease in luciferase activity, compared with the wild-type AKT1 promoter in the presence of CJUN (Figure 3F).
Since JUNB is positively regulated by NPM-ALK at the transcriptional and translational level in ALK+ ALCL15, we studied the contribution of NPM-ALK to the regulation of AKT1 expression in 293T cells. We observed that transient transfection of NPM-ALK but not the kinase-dead (K210R) expression vector led to a substantial decrease in AKT1 promoter activity (Figure 3G). In addition, NPM-ALK induced JUNB mRNA (Supplementary Figure 1D) and protein levels but it was also associated with elevated total AKT1 protein levels (Figure 3H), suggesting that NPM-ALK–induced AKT1 overexpression is mediated through a post-transcriptional mechanism. Furthermore, JUNB overexpression in 293T cells (Supplementary Figure 2A) was found to increase the cyclin-dependent kinase inhibitor (CDKI) p16INK4a (Supplementary Figure 2B) and decrease the protein levels of cyclin D1 and pAKT1 (ser473) (Supplementary Figure 2C), while it was associated with reduction in overall cell numbers. Of note, viability didn’t change significantly (Supplementary Figure 2D). Taken together, our data are indicative of a similar JUNB-AKT1 interaction that may be attained through direct protein-DNA binding, even though the effect of JUNB on AKT1 transcription is the opposite in a non cancerous cell system (HEK293T) from what was observed in ALK+ ALCL.
JUNB and CJUN synergize to transcriptionally activate AKT1, AKT2 and AKT3
It is known that JUNB and CJUN knockdown has been linked to attenuation of cell viability (Trypan blue), growth (MTS) and colony formation (colony assay) in ALCL cell lines (Karpas 299 and SR-786)17 and cell cycle arrest12 To investigate the possible cooperation between JUNB and CJUN in driving AKT1 transcription, we silenced both genes in the ALK+ ALCL cells, which showed further suppression of AKT1 transactivation compared with silencing of each one alone (Figure 4B). Next, we examined whether JUNB or CJUN deficiency could also modulate the transcription of the other two AKT isoforms, namely AKT2 and AKT3, whose genes are mapped on different chromosomes. Using JASPAR database33, we identified several potential AP-1 binding sites in the gene promoters of AKT2 and AKT3 (Supplementary Table 2). Western blotting data, revealed that simultaneous JUNB and CJUN gene silencing in two ALK (+) ALCL cell lines, Karpas 299 (Figure 4C) and SR-786 (Figure 4D) resulted in lower protein levels for all the AKT isoforms. Moreover, RT-qPCR analysis showed a significant decrease in the AKT3 mRNA after JUNB silencing, which was further potentiated by simultaneous JUNB/CJUN knockdown in ALK+ ALCL cells (Figure 4E). Similar results were seen for the AKT2 gene, although JUNB silencing alone showed a less prominent effect on AKT2 mRNA (Figure 4F). Our results suggest that in ALK+ ALCL, JUNB and CJUN can synergistically activate the transcription of AKT1 and the other members of the gene family, AKT2 and AKT3.
Figure 4. Regulation of AKT genes through cooperation of AP-1 members in ALK+ ALCL.

A. Simultaneous cJUN and JUNB silencing was performed using siRNA for both genes and was confirmed by Western blot analysis. B. Combined JUNB and cJUN knockdown had an additive effect on the reduction of AKT1 promoter activity resulting in decreased AKT1 levels (not shown). C and D. In two NPM-ALK(+) ALCL cell lines, Karpas 299 (C) and SR-786 (D), CJUN/JUNB silencing attenuated AKT1, AKT2 and AKT3 protein levels. Moreover, phosphorylation of GSK3 and AKT1 kinase activation (as shown here by T308 and S473 phosphorylation of AKT1) were substantially lowered. Of note, transient JUNB gene inhibition, increased AKT2 protein levels in both cell lines. E and F. Finally, silencing both JUNB and CJUN led to a substantial decrease in AKT3 and (E) AKT2 (F) mRNA expression levels. qRT-PCR was performed in triplicates, and the values were normalized against the expression levels of GAPDH.
p<0,05, **, p<0,01, ***, p<0,001 (Student’s t-test).
JUNB enhances STAT3-induced activity of the AKT1 promoter
STAT3 binding on the exon 1 region of the AKT1 promoter has been shown to be of crucial importance to the transcriptional activity of AKT1 in other cell types29. Here, we verified that STAT3 ablation resulted in a significant decrease in the promoter activity of AKT1 in ALK+ ALCL (Figure 5A). Simultaneous STAT3 and JUNB silencing showed that STAT3-induced transcriptional activity of AKT1 was further potentiated by JUNB (Figure 5A) and was associated with decreased cell viability in two ALK+ ALCL cell lines tested, Karpas 299 (Figure 5C) and SR-786 (Figure 5D). Moreover, treatment with Stattic, a potent STAT3 inhibitor34profoundly decreased STAT3 phosphorylation at Tyr705 in a dose-dependent manner, which correlated well with AKT1 inhibition and was also accompanied by JUNB downregulation (Figure 5E). Previous studies have shown that the JUNB promoter harbors STAT3 binding sites and that STAT3 transcriptionally modulates JUNB expression35, 36, thus, inhibition of STAT3 activation should attenuate JUNB levels. In addition, treatment with the ERK inhibitor, U0126, diminished ERK phosphorylation and coincided with reduced AKT1 expression (Figure 5E). Of interest, following treatment with Stattic we noticed a significant induction in phosphorylation levels of ERK; a possible explanation for this finding is uncertain. Furthermore, we used a gene reporter assay to measure the AKT1-Luc promoter activity in ALK+ ALCL cells, following treatment with Stattic or U0126, or both inhibitors (Figure 5F). AKT1 activity was significantly inhibited in a concentration-dependent manner. Combined drug treatment further repressed AKT1 promoter activity, as an additive effect (Figure 5F).
Figure 5. AP-1 and STAT3 induced AKT1 promoter activity in ALK+ ALCL cells.

A. JUNB enhanced STAT3-induced promoter activity on the AKT1 promoter. B. Western blotting showed that simultaneous depletion of JUNB and STAT3 results in additional reduction of the AKT1 protein level. C and D. in two ALCL cell lines, Karpas 299 (C) and SR-786 (D), loss of JUNB or STAT3 results in a notable reduction in cell viability, as determined by Trypan blue staining. Moreover, simultaneous depletion of JUNB/STAT3 had an additive effect in cell viability reduction, compared to the loss of each transcription factor alone. Results are shown in triplicates E. Western blotting data from cells treated with both inhibitors. Stattic leads to a concentration-dependent reduction in the protein level of phosphorylated STAT3 and does not affect the total STAT3 protein level. Interestingly, pERK is increased 20-fold in respect to the control (DMSO) when the maximum dose (20 µM) of Stattic inhibitor is used, while JUNB protein levels are also significantly decreased and AKT1 protein is almost undetectable. UO126 treatment (10 µM) diminished the pERK protein level, while the total ERK level did not change substantially. JUNB was severely affected by UO126 exposure and AKT1 protein expression was also reduced. F. Pharmacologic inhibition of JUNB and STAT3 results in reduced AKT1 promoter activity. UO126 (MEK1/2 inhibitor), and Stattic (STAT3 inhibitor) were used to treat cells transfected with the AKT1-Luc promoter construct. Luciferase assay was performed 48 hours later. The combination treatment of UO126 and Stattic had a profound effect on the activity of the AKT1 promoter. Stattic: +. 5 µM, ++, 10µM, +++, 20µM. UO126 : +, 50µM, ++, 100 µM.
p<0,05, **, p<0,01, ***, p<0,001 (Student’s t-test).
JUNB and STAT3 depletion attenuates cell proliferation and survival of ALK+ ALCL cells
Next, we examined whether STAT3- or JUNB-induced AKT1 repression would biologically affect our ALK+ ALCL model. Treatment with both inhibitors, Stattic and U0126 in doses described by others16, 37–39, attenuated proliferation of ALK+ ALCL cells in a concentration-dependent manner and showed synergistic effects (Figure 6A and 6B). Pharmacologic inhibition of STAT3 and JUNB also resulted in a substantial decrease in cell viability, although no synergy between the drugs was observed (Figure 6C and 6D). In addition, treatment of ALK+ ALCL cells with Stattic and U0126 severely impaired colony formation (Figure 6E and 6F).
Figure 6. Biologic effect of JUNB/STAT3 pharmacologic inhibition on ALK+ ALCL in Karpas 299 and SR-786.
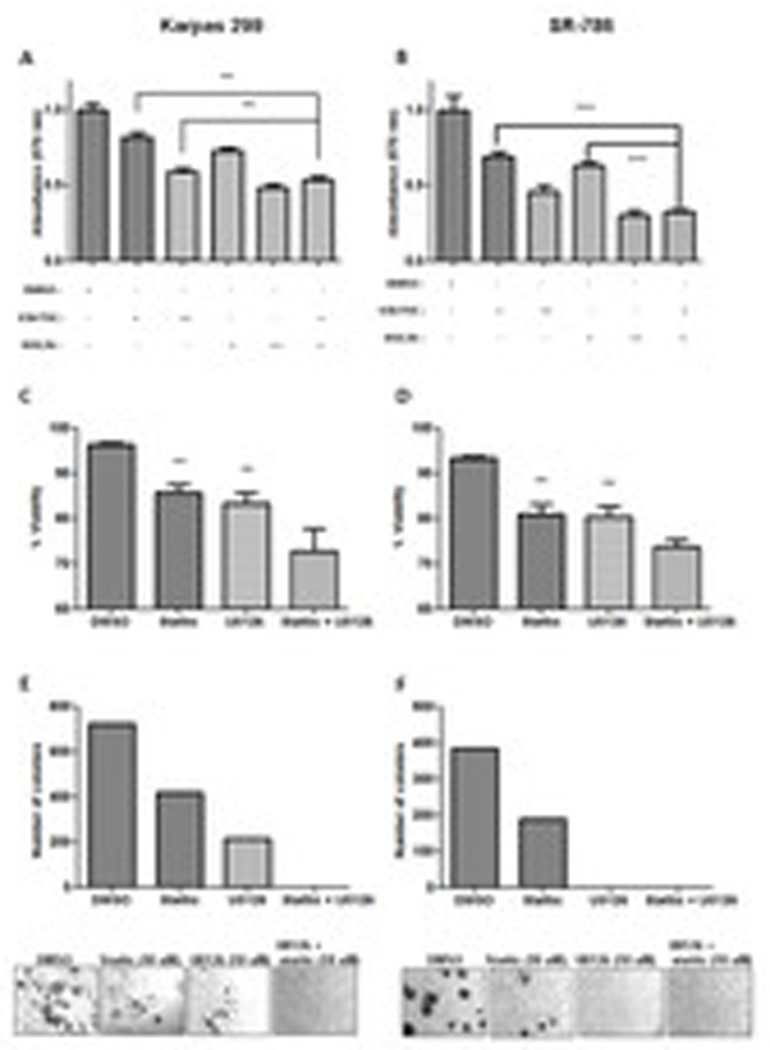
A, and B. ALK+ ALCL cell lines Karpas 299 (A) and SR-786 (B) were treated with Stattic (10 µM), U0126 (10 µM), or both pharmacologic inhibitors. The proliferation of viable cells (MTT assay) was then measured 48 hours after treatment. Stattic and U0126 treatment in ALK+ ALCL resulted in dose-dependent cell proliferation arrest. Moreover, the combined treatment had an additive effect on the proliferation of both cell lines. C and D. Cell viability assessed by the trypan blue exclusion assay in Karpas 299 (C) and SR-786 cells (D) following combined treatment with Stattic and U0126 shows increased cell death compared to each drug alone. E. Colony formation of Karpas 299 cells was obliterated after treatment with both Stattic and U0126. F. Treatment with Stattic resulted in decreased number of colonies formed, while U0126 completely diminished visible colonies in SR-786. When both drugs were added in the medium (10 µM each) the results were similar with U0126 treatment alone (no colonies were visible).
p<0,05, **, p<0,01, ***, p<0,001 (Student’s t-test).
DISCUSSION
Cumulative data suggest that AP-1 plays an important role in the pathogenesis of ALK+ ALCL10, 12, 15. We recently reported that JUNB amplification is frequent among ALK+ ALCL patients and that this mechanism, among others, contributes to overexpression of JUNB that is observed in all ALK+ ALCL tumors17. Moreover, we uncovered the tumor-promoting properties of JUNB/CD30 axis in ALK+ ALCL, which are mediated mainly through deregulation of cell cycle progression.17 In the present study, we hypothesized that AP-1 transcription factors might have a role in the regulation of AKT1 in ALK+ ALCL.
In a seminal study,29 investigators first identified that AKT1 promoter contain putative AP-1 binding sites. We show here that AKT1 is a direct transcriptional target of JUNB and CJUN, which are the AP-1 transcription factors highly expressed and activated in ALK+ ALCL. We demonstrated that both proteins bind to AP-1 binding sites on the AKT1 promoter and thus coordinate its transactivation in ALK+ ALCL cells. Gene silencing of JUNB resulted in reduced AKT1 mRNA and protein levels and is also associated with reduced AKT1 promoter activity and AKT activation. Using side-directed mutagenesis and gene reporter assays, we identified the specific AP-1 sites on the AKT1 promoter, which are crucial for JUNB (sites 8 and 9) or CJUN (site 7) binding leading to AKT1 gene expression. Moreover, simultaneous JUNB and CJUN gene silencing by siRNA delivery resulted in a marked decrease in AKT1 reporter activity as well AKT protein expression and activation.
Although several published studies have significantly enriched our knowledge for the post-translational regulation of AKT140, 41 and its kinase activity, our understanding for the regulation of AKT1 at the transcriptional level remains poor. Evidence supports the idea of E2F-mediated modulation of AKT1 activation through direct binding of the promoter of the adaptor protein Grb2-associated binder 2 (Gab2)42. Moreover, cAMP response element-binding protein (CREB) was shown to bind to CRE binding sites on the AKT1 promoter, activating its transcription in forskolin-treated cells43. In other cell system such as colorectal cancer, the Wnt/β-catenin pathway has been shown to play a pivotal role in AKT1 transcriptional control44. However, in that study the AKT1 promoter was unresponsive to Wnt/β-catenin in non cancerous HEK293T cells. Those findings are partially in agreement with our data showing opposite effects of JUNB on AKT1 promoter in HEK293T cells as compared to ALK+ ALCL cells. More specifically, we found that in non-cancerous HEK293T cells, forced expression of JUNB repressed AKT1 transactivation associated with AKT1 protein downregulation. The JUNB inhibitory effect on AKT1 promoter activity was JUNB-specific because it was reversed when JUNB siRNA was introduced into the cells. By contrast, CJUN overexpression continued to activate the AKT1 promoter in the same non-cancerous cell system. The differential effects in JUNB-mediated AKT1 transcriptional control between ALK+ ALCL and HEK293T cells could be attributed to the fact that in the context of ALK+ ALCL, the expression levels and combination of AP-1 proteins may differ dramatically as compared to non cancerous cells. Their relative abundance is crucial in modulating specific gene transcription, which is responsible for engaging cells in a precise destiny. Notably, JUNB-selective regulation of transcription, in a positive or a negative manner, has been shown to depend on the interacting partner and the promoter context45.
AKT2 and AKT3, the other two homologs of the AKT family of highly conserved proteins46–48, are characterized by broad expression, sometimes with a distinct pattern of tissue specificity for each isoform. In a previous survey on lymphomas, all three AKT isoforms were ubiquitously expressed in ALCL, both ALK+ and ALK−, and in other CD30+ lymphomas49. However, our knowledge of the transcriptional regulation of AKT2 and AKT3 is fairly limited. In this study we show data of transcriptional control of AKT2 and AKT3 by the AP-1 members CJUN and JUNB in ALK+ ALCL. Specifically, we demonstrated that JUNB or CJUN gene silencing led to reduced AKT2 and AKT3 mRNA levels and was accompanied by similar changes in the protein level. Moreover, we showed that combined JUNB and CJUN loss had a more profound impact on AKT1, AKT2, AKT3 mRNA and protein levels. A previous study has shown that the AKT2 promoter is under transcriptional control of MyoD, although at least one AP-1 site was identified. Using JASPAR database analysis we identified the presence of multiple CJUN/JUNB binding sites on AKT2 and AKT3 gene promoters. Although further studies are required to confirm these data with functional studies, the present study provides first evidence that both CJUN and JUNB may regulate AKT2 and AKT3 transcription, possibly in a manner similar to that of AKT1.
It was previously reported that SRC-mediated activation of STAT3 facilitates binding of STAT3 dimmers on the AKT1 promoter, thereby inducing its transcription29. JAK/STAT pathway is known to be highly activated by NPM-ALK in ALK+ ALCL leading to increased cell survival, and uncontrolled cell cycle progression30, 50, 51. In this study, we provide evidence that STAT3 induces transcription of AKT1 for the first time in the context of ALK+ ALCL. Notably, STAT3 and JUNB could synergize for the AKT1 transcriptional control in ALK+ ALCL. This synergistic pattern had also biologic significance, since simultaneous depletion of JUNB and STAT3 by siRNA in both ALK+ ALCL cell lines severely impaired cell viability. Interestingly, synergy between STAT3 and members of the JUN family has been reported in transcriptional regulation of target genes in other cell systems52–56.
In summary, we have shown that, in addition to the PI3K-associated activation of AKT, a crosstalk between AP1 transcription factors, JAK/STAT and AKT exists and further contributes to tumor cell proliferation and survival in ALK+ ALCL (Figure 7). Uncovering novel crosstalks between oncogenic pathways crucial for ALK+ ALCL oncogenesis will enrich our understanding of the molecular mechanisms underlying resistance to therapy.
Figure 7. Proposed model of AP-1/STAT3/AKT crosstalk mechanism in ALCL.

On the basis of our findings shown here and the current knowledge from published data, we propose the following model in ALK+ ALCL. The PI3K/AKT pathway is constitutively active in ALK+ ALCL due largely to the direct physical interaction of NPM-ALK with PI3K. This PI3K-dependent pathway is highly conducive to determining the phenotype of ALCL, which is characterized by apoptosis and cell cycle deregulation. In addition, AKT is regulated at the transcriptional level by AP-1 and STAT3 through protein-DNA interactions in the upstream or downstream region of the AKT1 promoter, respectively. AP-1 members of the Jun family, JUNB and CJUN, play important roles in the pathogenesis of ALK+ ALCL. CJUN is activated through phosphorylation by JNKs in a NPM-ALK-dependent manner, whereas JUNB levels are regulated by NPM-ALK at the transcriptional level through MEK/ERK and Ets-1 transcription factor, and at the translational level through mTOR15, 16. Moreover, the JAK/STAT pathway is directly activated by NPM-ALK, and STAT3 is overexpressed and highly activated in ALK+ALCL22, 50. JUNB/CJUN and STAT3, in turn transcriptionally regulate AKT1 and possibly AKT2 and AKT3, thus contributing to AKT overexpression in ALK+ ALCL. Taken together, in addition to the ALK kinase-associated activation of downstream kinases, a functional crosstalk between AP1 transcription factors, JAK/STAT and AKT exists and further contributes to cell cycle and apoptosis deregulation and ultimately to oncogenesis of ALK+ ALCL.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Jin Q. Cheng (Departments of Pathology and Interdisciplinary Oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL) for providing the AKT1-Luc promoter-constructs. We also thank Dr. Anupam Agarwal (University of Alabama at Birmingham, Birmingham, AL) for providing the JUNB expression plasmid pcDNA3.1/V5-His Topo.
This study was supported by grants from the National Cancer Institute (R01-CA90853 to FXC), The University of Texas MD Anderson Functional Proteomics Core Facility (NCI Cancer Center Support Grant CA16672), and an institutional research grant (IRG) of The University of Texas MD Anderson Cancer Center, Houston, Texas. VA is supported by a Hellenic Association for Molecular Cancer Research scholarship. GZR is a recipient of ‘ARISTEIA-II’ grant (#4757) from Research Funding Program: Investing in knowledge society through the European Social Fund).
The abbreviations used are
- ALCL
anaplastic large cell lymphoma
- ALK
anaplastic lymphoma kinase
- NPM-ALK
nucleophosmin ALK
- AP-1
activator protein-1
- AKT
protein kinase B
- STAT3
signal transducer and activator of transcription 3
- CML
chronic myeloid leukemia
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Delsol GRE, Stein H, Wright D, Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology and Genetics of Tumors of Haematopoietic and Lymphoid Tissues. World Health Organization Classification of Tumours. IARC Press. 2001:230–235. [Google Scholar]
- 2.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008 Jan;8(1):11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 3.Stein H, Foss HD, Durkop H, Marafioti T, Delsol G, Pulford K, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000 Dec 1;96(12):3681–3695. [PubMed] [Google Scholar]
- 4.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002 May;4(5):E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 5.Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000 May 2;19(9):2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Passegue E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000 Jun 15;19(12):2969–2979. doi: 10.1093/emboj/19.12.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang MY, Liu TC, Chang JG, Lin PM, Lin SF. JunB gene expression is inactivated by methylation in chronic myeloid leukemia. Blood. 2003 Apr 15;101(8):3205–3211. doi: 10.1182/blood-2002-05-1598. [DOI] [PubMed] [Google Scholar]
- 8.Passegue E, Jochum W, Schorpp-Kistner M, Mohle-Steinlein U, Wagner EF. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell. 2001 Jan 12;104(1):21–32. doi: 10.1016/s0092-8674(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 9.Rassidakis GZ, Thomaides A, Atwell C, Ford R, Jones D, Claret FX, et al. JunB expression is a common feature of CD30+ lymphomas and lymphomatoid papulosis. Mod Pathol. 2005 Oct;18(10):1365–1370. doi: 10.1038/modpathol.3800419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002 Aug 1;21(15):4104–4113. doi: 10.1093/emboj/cdf389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drakos E, Leventaki V, Schlette EJ, Jones D, Lin P, Medeiros LJ, et al. c-Jun expression and activation are restricted to CD30+ lymphoproliferative disorders. Am J Surg Pathol. 2007 Mar;31(3):447–453. doi: 10.1097/01.pas.0000213412.25935.e4. [DOI] [PubMed] [Google Scholar]
- 12.Leventaki V, Drakos E, Medeiros LJ, Lim MS, Elenitoba-Johnson KS, Claret FX, et al. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood. 2007 Sep 1;110(5):1621–1630. doi: 10.1182/blood-2006-11-059451. [DOI] [PubMed] [Google Scholar]
- 13.Leventaki V, Drakos E, Karanikou M, Psatha K, Lin P, Schlette E, et al. c-JUN N-terminal kinase (JNK) is activated and contributes to tumor cell proliferation in classical Hodgkin lymphoma. Hum Pathol. 2014 Mar;45(3):565–572. doi: 10.1016/j.humpath.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe M, Ogawa Y, Itoh K, Koiwa T, Kadin ME, Watanabe T, et al. Hypomethylation of CD30 CpG islands with aberrant JunB expression drives CD30 induction in Hodgkin lymphoma and anaplastic large cell lymphoma. Lab Invest. 2008 Jan;88(1):48–57. doi: 10.1038/labinvest.3700696. [DOI] [PubMed] [Google Scholar]
- 15.Staber PB, Vesely P, Haq N, Ott RG, Funato K, Bambach I, et al. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood. 2007 Nov 1;110(9):3374–3383. doi: 10.1182/blood-2007-02-071258. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe M, Itoh K, Togano T, Kadin ME, Watanabe T, Higashihara M, et al. Ets-1 Activates Overexpression of JunB and CD30 in Hodgkin's Lymphoma and Anaplastic Large-Cell Lymphoma. Am J Pathol. 2011 Nov 19; doi: 10.1016/j.ajpath.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atsaves V, Lekakis L, Drakos E, Leventaki V, Ghaderi M, Baltatzis GE, et al. The oncogenic JUNB/CD30 axis contributes to cell cycle deregulation in ALK+ anaplastic large cell lymphoma. Br J Haematol. 2014 Aug 22; doi: 10.1111/bjh.13079. [DOI] [PubMed] [Google Scholar]
- 18.Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin's lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol. 1997 Apr;17(4):2312–2325. doi: 10.1128/mcb.17.4.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai RY, Dieter P, Peschel C, Morris SW, Duyster J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol. 1998 Dec;18(12):6951–6961. doi: 10.1128/mcb.18.12.6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nieborowska-Skorska M, Slupianek A, Xue L, Zhang Q, Raghunath PN, Hoser G, et al. Role of signal transducer and activator of transcription 5 in nucleophosmin/anaplastic lymphoma kinase-mediated malignant transformation of lymphoid cells. Cancer Res. 2001 Sep 1;61(17):6517–6523. [PubMed] [Google Scholar]
- 21.Cho-Vega JH, Vega F, Medeiros LJ. An attractive therapeutic target, mTOR pathway, in ALK+ anaplastic large cell lymphoma. Adv Anat Pathol. 2008 Mar;15(2):105–112. doi: 10.1097/PAP.0b013e318166139f. [DOI] [PubMed] [Google Scholar]
- 22.Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001 Mar 1;61(5):2194–2199. [PubMed] [Google Scholar]
- 23.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005 Nov 14;24(50):7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 24.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 25.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004 Mar 18;428(6980):332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 26.Duyster J, Bai RY, Morris SW. Translocations involving anaplastic lymphoma kinase (ALK) Oncogene. 2001 Sep 10;20(40):5623–5637. doi: 10.1038/sj.onc.1204594. [DOI] [PubMed] [Google Scholar]
- 27.Rassidakis GZ, Feretzaki M, Atwell C, Grammatikakis I, Lin Q, Lai R, et al. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005 Jan 15;105(2):827–829. doi: 10.1182/blood-2004-06-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vega F, Medeiros LJ, Leventaki V, Atwell C, Cho-Vega JH, Tian L, et al. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res. 2006 Jul 1;66(13):6589–6597. doi: 10.1158/0008-5472.CAN-05-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park S, Kim D, Kaneko S, Szewczyk KM, Nicosia SV, Yu H, et al. Molecular cloning and characterization of the human AKT1 promoter uncovers its up-regulation by the Src/Stat3 pathway. J Biol Chem. 2005 Nov 25;280(47):38932–38941. doi: 10.1074/jbc.M504011200. [DOI] [PubMed] [Google Scholar]
- 30.Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, et al. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003 Aug 21;22(35):5399–5407. doi: 10.1038/sj.onc.1206849. [DOI] [PubMed] [Google Scholar]
- 31.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1(1):179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 32.Tian L, Peng G, Parant JM, Leventaki V, Drakos E, Zhang Q, et al. Essential roles of Jab1 in cell survival, spontaneous DNA damage and DNA repair. Oncogene. 2010 Nov 18;29(46):6125–6137. doi: 10.1038/onc.2010.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014 Jan;42(Database issue):D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006 Nov;13(11):1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Kojima H, Nakajima K, Hirano T. IL-6-inducible complexes on an IL-6 response element of the junB promoter contain Stat3 and 36 kDa CRE-like site binding protein(s) Oncogene. 1996 Feb 1;12(3):547–554. [PubMed] [Google Scholar]
- 36.Coffer P, Lutticken C, van Puijenbroek A, Klop-de Jonge M, Horn F, Kruijer W. Transcriptional regulation of the junB promoter: analysis of STAT-mediated signal transduction. Oncogene. 1995 Mar 2;10(5):985–994. [PubMed] [Google Scholar]
- 37.Ito M, Zhao N, Zeng Z, Chang CC, Zu Y. Synergistic growth inhibition of anaplastic large cell lymphoma cells by combining cellular ALK gene silencing and a low dose of the kinase inhibitor U0126. Cancer Gene Ther. 2010 Sep;17(9):633–644. doi: 10.1038/cgt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto K, Mizumoto A, Nishimura K, Uda A, Mukai A, Yamashita K, et al. Association of toxicity of sorafenib and sunitinib for human keratinocytes with inhibition of signal transduction and activator of transcription 3 (STAT3) PloS one. 2014;9(7):e102110. doi: 10.1371/journal.pone.0102110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin L, Hutzen B, Lee HF, Peng Z, Wang W, Zhao C, et al. Evaluation of STAT3 signaling in ALDH+ and ALDH+/CD44+/CD24− subpopulations of breast cancer cells. PloS one. 2013;8(12):e82821. doi: 10.1371/journal.pone.0082821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002 Jul;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 41.Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005 Nov 14;24(50):7401–7409. doi: 10.1038/sj.onc.1209099. [DOI] [PubMed] [Google Scholar]
- 42.Chaussepied M, Ginsberg D. Transcriptional regulation of AKT activation by E2F. Mol Cell. 2004 Dec 3;16(5):831–837. doi: 10.1016/j.molcel.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Misra UK, Pizzo SV. Upregulation of AKT1 protein expression in forskolin-stimulated macrophage: evidence from ChIP analysis that CREB binds to and activates the AKT1 promoter. J Cell Biochem. 2007 Mar 1;100(4):1022–1033. doi: 10.1002/jcb.21086. [DOI] [PubMed] [Google Scholar]
- 44.Dihlmann S, Kloor M, Fallsehr C, von Knebel Doeberitz M. Regulation of AKT1 expression by beta-catenin/Tcf/Lef signaling in colorectal cancer cells. Carcinogenesis. 2005 Sep;26(9):1503–1512. doi: 10.1093/carcin/bgi120. [DOI] [PubMed] [Google Scholar]
- 45.Hsu JC, Cressman DE, Taub R. Promoter-specific trans-activation and inhibition mediated by JunB. Cancer Res. 1993 Aug 15;53(16):3789–3794. [PubMed] [Google Scholar]
- 46.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, et al. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci U S A. 1992 Oct 1;89(19):9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brodbeck D, Cron P, Hemmings BA. A human protein kinase Bgamma with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J Biol Chem. 1999 Apr 2;274(14):9133–9136. doi: 10.1074/jbc.274.14.9133. [DOI] [PubMed] [Google Scholar]
- 48.Bellacosa A, Franke TF, Gonzalez-Portal ME, Datta K, Taguchi T, Gardner J, et al. Structure, expression and chromosomal mapping of c-akt: relationship to v-akt and its implications. Oncogene. 1993 Mar;8(3):745–754. [PubMed] [Google Scholar]
- 49.Fillmore GC, Wang Q, Carey MJ, Kim CH, Elenitoba-Johnson KS, Lim MS. Expression of Akt (protein kinase B) and its isoforms in malignant lymphomas. Leuk Lymphoma. 2005 Dec;46(12):1765–1773. doi: 10.1080/10428190500159944. [DOI] [PubMed] [Google Scholar]
- 50.Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002 Feb 7;21(7):1038–1047. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- 51.Ruchatz H, Coluccia AM, Stano P, Marchesi E, Gambacorti-Passerini C. Constitutive activation of Jak2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Exp Hematol. 2003 Apr;31(4):309–315. doi: 10.1016/s0301-472x(03)00007-9. [DOI] [PubMed] [Google Scholar]
- 52.Nishikawa T, Hagihara K, Serada S, Isobe T, Matsumura A, Song J, et al. Transcriptional complex formation of c-Fos, STAT3, and hepatocyte NF-1 alpha is essential for cytokine-driven C-reactive protein gene expression. J Immunol. 2008 Mar 1;180(5):3492–3501. doi: 10.4049/jimmunol.180.5.3492. [DOI] [PubMed] [Google Scholar]
- 53.Schuringa JJ, Timmer H, Luttickhuizen D, Vellenga E, Kruijer W. c-Jun and c-Fos cooperate with STAT3 in IL-6-induced transactivation of the IL-6 respone element (IRE) Cytokine. 2001 Apr 21;14(2):78–87. doi: 10.1006/cyto.2001.0856. [DOI] [PubMed] [Google Scholar]
- 54.Zhang X, Wrzeszczynska MH, Horvath CM, Darnell JE. Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Molecular and cellular biology. 1999;19(10):7138–7146. doi: 10.1128/mcb.19.10.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ginsberg M, Czeko E, Muller P, Ren Z, Chen X, Darnell JE., Jr Amino acid residues required for physical and cooperative transcriptional interaction of STAT3 and AP-1 proteins c-Jun and c-Fos. Mol Cell Biol. 2007 Sep;27(18):6300–6308. doi: 10.1128/MCB.00613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci U S A. 1995 Sep 26;92(20):9097–9101. doi: 10.1073/pnas.92.20.9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.