

Abstract
The growth of tissues and organs is regulated by orchestrated signals from biomolecules such as enzymes and growth factors. The ability to deliver signal molecules in response to particular biological events (e.g., enzyme expression and activation) holds great promise towards tissue healing and regeneration. The current delivery vehicles mainly rely on hydrolysable scaffolds and thin films of protein-containing polymers, which cannot be programmed to respond to biological signals. We report herein an injectable delivery platform based on enantiomeric protein nanocapsules, which can deliver multiple proteins with spatiotemporal control in response to the tissue proteases secreted during wound healing. Exemplified by stroke and diabetic wound healing in mice, sequential delivery of vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) greatly enhances tissue revascularization and vessel maturation, providing effective delivery vehicles for tissue engineering and reparative medicine.
Keywords: protein delivery, sequential release, enzyme responsive, chiral peptides, wound healing
During tissue repair, the orderly presentation of signal proteins in coordination with proteolytic enzymes generally directs the hierarchical remodeling of diseased tissues.[1] For example, angiogenic growth factors in association with tissue-specific protease cascades direct vascular sprouts and subsequent stabilization of blood vessels.[2] In order to deliver multiple signal molecules in a desired sequence, researchers have attempted to develop delivery vehicles with spatiotemporal control by trapping proteins within degradable polymers such as poly(lactide-co-glycolic acid), poly(ε-caprolactone), and hydrogels. Representative examples include the composite films made by the layer-by-layer assembly of proteins and polymers,[3] as well as the composite scaffolds made by electrospinning[4] or by fusing polymer particles that contain desired proteins using organic vapor or high-pressure CO2.[5] For the composite films, the sequential release of the proteins is achieved through a hydrolysable barrier that separates layers of first protein from the subsequent ones; after releasing the first protein, the hydrolysis of the barrier allows for the release of the subsequent proteins. For the composite scaffolds, the sequential release is achieved based on different hydrolysis rates of the polymer moieties within the composites. Although these delivery vehicles enable the sequential elution of multiple proteins, the release process is constitutive which cannot be programmed to respond to any particular biological event. Furthermore, the synthesis of such composites requires harsh chemical processes involving the use of intense mixing and/or organic solvents, which can easily denature growth factors.
Protease-based protein delivery systems were previously studied by others to either directly conjugate proteins to matrices via a protease-sensitive peptide linker or to attach proteins within a bulk hydrogel that is crosslinked by protease-sensitive peptides.[6] However, both approaches expose proteins to the reactive chemical environment, which challenges the stability and bioactivity of proteins.[7] Additionally these approaches are highly dependent on the number and the type of the functional groups on backbone scaffolds for modifications with different peptide linkers, which limits the applicability of multiple proteins with temporal release control and the variety for the selection of buffers or matrices.
We have developed a platform technology for protein delivery based on a mild encapsulation process, in which individual whole proteins are wrapped within an in situ formed thin polymer shell.[8] Since the spatial and temporal patterns of protease expressions are closely related to pathophysiological states,[9] incorporating protease-specific and cleavable peptides within the polymer shells allows for disease-state–specific delivery of signal molecules. Here, we investigated the peptide substrates made from L or D chiral form of amino acids and found that they exhibit similar chemical specificity but distinct proteolytic kinetics (i.e., ~10 fold slower for D enantiomer than the L enantiomer, Supporting Information Table 1). Thus by controlling the L to D ratios of peptide crosslinkers within the shells, the delivery of multiple proteins with spatiotemporal control in response to the proteolytic enzymes in diseased sites could be achieved.
Scheme 1a illustrates our design using plasmin-sensitive peptides as labile crosslinkers. Driven by non-covalent interactions, monomers with neutral 1, positive 2 or negative 3 charges, as well as peptide crosslinkers, are spontaneously enriched around protein molecules. Free-radical polymerization gradually grows a nanogel shell around each protein, leading to the formation of protease-responsive nanocapsules denoted as n(Protein)x%, where Protein denotes the protein core and x% denotes the molar percentage of L peptide in the total L+D peptide crosslinkers used for the nanogel shell (Scheme. 1a). Increasing the ratio of L crosslinker (x%, fast degrading rate) leads to nanocapsules with a faster release kinetic, while decreasing L percentage results in slower releasable nanocapsules (Scheme. 1b). Upon protease degradation the released protein can exert its normal biological function upon nanocapsue degradation and the nanogel polymeric fragments, charged polyacrylamide segments with cleaved peptides, are expected to be biocompatible in the biological milieu as has been previously observed for similar polymers.[10] Furthermore, such nanocapsules can be homogenously dispersed within an injectable hydrogel, providing an injectable delivery platform for enhanced wound healing and tissue repair (Scheme. 1c).
Scheme 1.
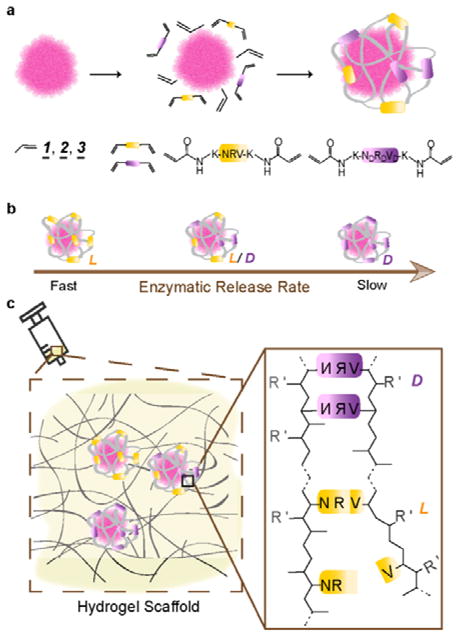
Illustration of chirality-controlled, enzyme-responsive protein nanocapsules with temporal control. (a). The synthesis of the nanocapsules by enriching monomers and crosslinkers around an individual protein molecule and by subsequent in situ polymerization. The monomers can be acrylamide (1, neutral), N-(3-aminopropyl)methacrylamide (2, positively charged), or 2-acrylamino-2-methyl-1-propanesulfonic acid (3, negatively charged). The crosslinkers include the mixtures with designed molar ratios of L (yellow) and D (purple) enantiomers of the peptide Asn-Arg-Val, being the substrate of plasmin. (b) The rate of enzymatic degradations of individual nanocapsule is tuned by varying the ratio of L to D peptide crosslinkers used: faster with more L peptide, slower with more D peptide. (c) Nanocapsules of different proteins and of varying degradation rates can be mixed in matrices (or buffer) of choice for injectable delivery of multiple proteins with precise temporal control in protease-specific disease models.
As an example, nanocapsules containing vascular endothelial growth factor-A 165, designated as n(VEGF)100%, were synthesized using 1 and 2 as the co-monomer and 100% plasmin-labile L-peptide as the crosslinker. These nanocapsules display a spherical morphology and by labeling each of the protein with a gold-nanoparticle, each nanocapsule is found to contain one to two VEGF molecules (Figure 1a). Nanocapsules synthesized with different ratios of enantiomeric peptides, including n(VEGF)100%, n(VEGF)50% and n(VEGF)25%, exhibit similar hydrodynamic diameters, indicating an unbiased polymerization activity between the L and D peptide crosslinkers (Figure 1b). Furthermore, nanocapsules of platelet derived growth factor-BB (PDGF), denoted as n(PDGF)100%, were also synthesized with the nanogel shell being polymerized by 1 and 3 as the co-monomer and 100% L peptide as the crosslinker, which also display a similar nanoscale size under TEM (Supporting Information Figure S1). The plasmin-responsive release capability of the nanocapsule vehicles features a programmable release rate. Figure 1c compares the amount of VEGF released from n(VEGF)100%, n(VEGF)50%, n(VEGF)25% and n(VEGF)0% after the incubation with plasmin for 20 minutes. While a 6-fold increase of VEGF was observed with n(VEGF)100% after plasmin incubation, only 1-fold increase in the quantity of VEGF with n(VEGF)0%, confirming decreasing the L-peptide content within the shells leads to decreasing in vitro release rates. The activity of encapsulated proteins is protected and can be released by isolated enzymes or cell-secreted proteases. First, the degradation of the shell of nanocapsules is triggered by trypsin, followed by a treatment of a protease inhibitor to quench subsequent proteolytic degradation, and the released VEGF was able to induce the phosphorylation of cell-surface receptors to the same extent as free protein did (Figure 1d). This data suggests that 100% of the encapsulated proteins can be retrieved with appropriate amounts of enzymes and that the encapsulated proteins retained 100% of its activity. Second, cells can mediate the release of proteins from nanocapsules with the proteolytic and fibrinolytic proenzymes[11] secreted by cells. We incubated n(PDGF)100%, n(PDGF)0% and free PDGF with human dermal fibroblasts and observed some degree of receptor phosphorylation induced by nanocapsules (Supporting Information Figure S2), which indicated that some degree of encapsulated protein was released from n(PDGF)100% in the timeframe tested.
Figure 1.
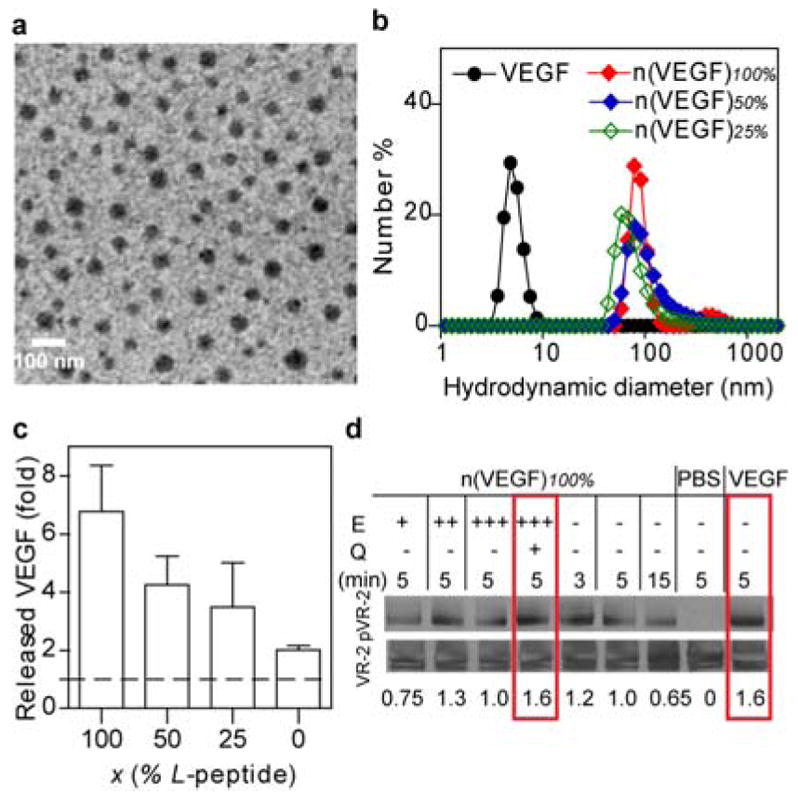
Characterizations of protein nanocapsules. (a) A transmission electron microscopic image of nanocapsules synthesized with gold nanoparticle (AuNP, 4 nm)-labeled VEGF. AuNP is stained with silver enhancer kit to exhibit as dark black dots within the grey spheres of n(VEGF-AuNP)100%. (Scale bar 100 nm) (b) The hydrodynamic sizes of n(VEGF) of different L-to-D ratios via dynamic light scattering. (c) Incubating n(VEGF)100%, n(VEGF)50%, n(VEGF)25% or n(VEGF)0% with plasmin for 20 minutes in ELISA measures the in vitro enzymatic release rates of nanocapsules. (d) Western blotting analysis of the activity of encapsulated VEGF remains identical to free VEGF in inducing receptor phosphorylation. The encapsulated VEGF was first released from n(VEGF)100% at 50 ng/mL via incubation with an enzyme (E), trypsin, at increasing mass ratios of trypsin:n(VEGF)100% = 0.05:1 (labeled as E +), 0.1:1 (labeled as E ++), and 0.2:1 (labeled as E+++). To prevent the released VEGF from being degraded, an inhibitor, aprotinin, was subsequently used to quench (Q) the excessive proteolytic activity of trypsin. Released VEGF, as well as n(VEGF)100% without enzyme pretreatment and free VEGF (50 ng/mL) were incubated with serum-starved human vein endothelial cells for specified amounts of time (min). The activity was indicated by the normalized intensity amounts with the phosphorylation of VEGF receptor-2 normalized by actin.
Therapeutic angiogenesis in vivo, a crucial process towards wound healing, requires continuous presence of VEGF, which is subject to a narrow therapeutic window.[12] However the frequent re-dosing of high concentrations of VEGF (tens of μg) to wound beds has been the standard practice due to inevitable passive leaching of naked proteins from conventional scaffolds and matrices.[13] Here, we tested the ability of n(VEGF)100%/n(VEGF)25% mixture (100 ng each, 200 ng combined) to promote vascularization in a challenging environment: the avascular stroke cavity. VEGF is one of the candidate molecules used in post-stroke neural repair therapies to enhance angiogenesis and functional recovery. However, the delivery of VEGF to ischemic brains is often complicated by the induction of disordered vasculature.[14] Immunohistological analysis of glucose transporter 1 (highly expressed in the brain endothelium) and PDGF receptor-β (highly expressed in pericytes) showed that the bolus delivery of VEGF within a hyaluronic acid (HA) hydrogel did not improve the level of vascularization compared to HA hydrogel alone. In contrast, the n(VEGF)100%/n(VEGF)25% mixture led to statistically significant increases in vascularization and pericyte coverage in both the infarct (inside the stroke cavity) as well as peri-infarct (surrounding the stroke) regions (Figure 2). This result confirms that the nanocapsules are responsive to brain wound environments to allow for active VEGF release at a rate and level sufficient for enhanced vascularization.
Figure 2.
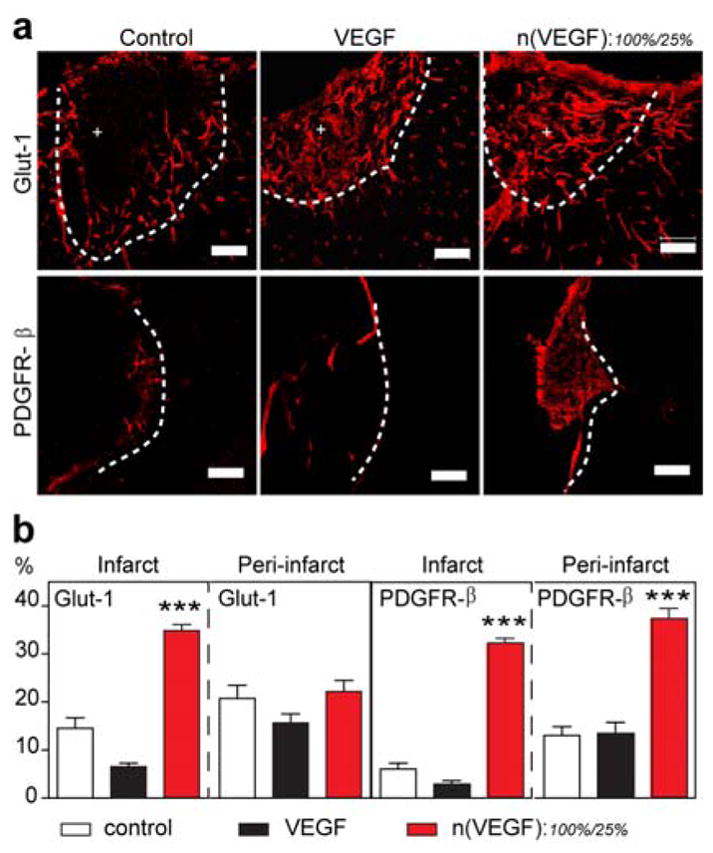
Temporal control of VEGF delivery in mouse stroke model. (a) Representative confocal images of blood vessels (Glut-1+) and their maturity markers (PDGFRβ+) in the infarct (indicated by +) and peri-infarct areas of stroke (separated by the dashed line). The stroke was treated with in situ crosslinked, adhesion peptide-modified hyaluronic acid hydrogels containing no VEGF (control), unencapsulated VEGF (200 ng), or n(VEGF)100% : n(VEGF)25% at 100 ng : 100 ng. (b) Analysis of Glut-1 and PDGFR-β markers for the vascularization in the infarct and peri-infarct areas of stroke. (AVONA with Tukey’s post test, mean ± SEM, N = 3~4, * p<0.05, ** p<0.01, *** p<0.001.) (Scale bar, 100 μm).
In addition to achieving temporal control of one protein release in response to proteases, the nanocapsule technology also enables sequential delivery of multiple proteins by using mixed nanocapsules made with designed protein cores, as well as the types and ratios of L/D crosslinkers. Figure 3a shows that the release profile of n(VEGF)100% and n(PDGF)25% co-delivered to skin wounds from the same scaffold. As expected, VEGF was detected between 0 and 3 days and PDGF was detected between 3 and 6 days demonstrating the sequential release (Figure 3a, Supporting Information Figure S3). Specifically, n(VEGF)100% led to an excess of 200 pg VEGF per mg of tissue than endogenously present VEGF at day 0, but no significant difference on day 3 or day 6, suggesting that n(VEGF)100% was mostly released by day 3 since urokinase plasminogen activator is initially activated in wounds and subsequently deactivated as wound heals and decreases in size.[15] However with the incorporation of D chiral peptide, the excess of PDGF detected with n(PDGF)25% on day 3 and 6 further demonstrated in vivo delayed release enabled by chiral protein nanocapsules. Such time scales are relevant for angiogenesis in mouse skin wounds where the angiogenic peak occurs at 3–5 days.
Figure 3.

In vivo co-delivery of VEGF and PDGF with temporal control in diabetic mouse skin wounds. (a) Tissue-ELISA analysis of wound-mediated release of proteins from n(VEGF)100% and n(PDGF)25% at day 0, 3, and 6 post surgery. (b) Quantification of the gap of granulation tissues in diabetic skin wounds at day 7. Wound was dressed with fibrin matrices that contained (i) no growth factors (control), (ii) un-encapsulated VEGF (200 ng) and PDGF (200 ng), (iii) mixture of n(VEGF)100%/n(VEGF)25%/n(PDGF)100%/n(PDGF)25% at 100 ng/100 ng/100 ng/100 ng (parallel), (iv) mixture of n(VEGF)100%/n(VEGF)25%/n(PDGF)25%/n(PDGF)10% at 100 ng/100 ng/100 ng/100 ng (sequential), and (v) mixture of n(VEGF)25%/n(VEGF)10%/n(PDGF)100%/n(PDGF)25% at 100 ng/100 ng/100 ng/100 ng (reverse sequential). (c) Immunohistochemical analysis of vessel endothelium (CD31+) and pericyte coverage (NG2+) at day 7 and day 10. (d) Representative confocal images of CD31 (upper row) and NG2 (lower row) in the granulation tissue of diabetic skin wounds at day 7. (AVONA with Tukey’s post test, mean ± SEM, N = 4~6, * p<0.05, ** p<0.01.) Scale bar = 50 μm (d).
To further analyze the ability of using enantiomeric nanocapsules to achieve sequential release of growth factors, we sought to demonstrate with the well-documented synergistic effects of VEGF and PDGF[5a] in inducing pericyte coverage of nascent blood vessels in a diabetic skin wound healing model.[16] Because impaired wound closure in diabetic patients is correlated with impaired angiogenesis and angiogenesis is correlated with the ability to sustain granulation tissue, we compared the formation of granulation tissue and related angiogenesis process. The nanocapsule mixture of n(VEGF)100%/n(VEGF)25%/n(PDGF)25%/n(PDGF)10% comprises a sequential delivery strategy of VEGF followed by PDGF, whereas n(VEGF)25%/n(VEGF)10%/n(PDGF)100%/n(PDGF)25% forms the reverse sequence of PDGF release followed by VEGF. Among the two sequential, parallel and bolus delivery strategies, only the sequentially released VEGF then PDGF (condition iv in Figure 3 and Supporting Information Figure S4) led to enhanced formation of granulation tissue and increased vessel density with pericyte coverage. This observation further confirms that these chirally different nanocapsules can be engineered to release proteins in multi-phases in a wound environment. In contrast, nanocapsules that released both VEGF and PDGF in parallel (condition iii in Figure 3) did not show enhanced angiogenesis or mature blood vessels. Interestingly, we observed a non-significant difference in the CD31 expression between wounds treated with the reverse sequential strategy (PDGF first then VEGF, condition v) and the orthogonal sequential delivery (VEGF first then PDGF condition iv), on which a recent report could possibly shed some light where the angiogenesis process in mouse bone healing was promoted by increasing PDGF secretion from preosteoclasts or treatment with exogenous PDGF[17]. Nevertheless the reverse sequential strategy failed to promote pericyte coverage in our study. This last example further exemplifies the ability to control protein release rates with our nanocapsules at disease sites.
In conclusion, we have designed a general platform for multi-protein delivery with spatiotemporal definitions based on enantiomerically engineered protein nanocapsules. Peptide chirality is for the first time utilized to modulate the enzymatic release of proteins and, enabled by the nanocapsule technology, to achieve spatiotemporal control of multiple proteins in cell-instructed microenvironments. These nanocapsules are facilely formed, robust and applicable to proteins in general, providing highly effective protein therapeutic vehicles for disease-specific tissue engineering and regenerative medicine.
Experimental Section
Synthesis of Protein Nanocapsules
The nanocapsules were synthesized using in situ free-radical polymerization. To synthesize n(VEGF), VEGF was diluted in a buffer solution of 10 mM sodium bicarbonate (pH = 8.55) at a final reaction concentration of 100 μg/mL. Acrylamide (AAM) and N-(3-aminopropyl)methacrylamide (APM) and crosslinkers (bisacrylated L/D-KNRVK, or methylene bisacrylamide) were subsequently added to the mixture (molar ratio of VEGF:AAM:APM:crosslinker = 1:3000:3000:600). To synthesize n(PDGF), PDGF was diluted in phosphate buffer saline (PBS) (pH = 7.2–7.4) at a final reaction concentration of 100 μg/mL. Acrylamide, 2-acrylamino-2-methyl-1-propanesulfonic acid (AAMPS) and crosslinkers (bisacrylated L/D-KNVRK) were subsequently added to the mixture (molar ratio of PDGF:AAM:AAMPS:crosslinker = 1:1500:4500:600). Then, freshly prepared ammonium persulfate (APS) (molar ratio of Protein:APS = 1:745) and tetramethylethylenediamine (TEMED) (molar ratio of Protein:TEMED = 1:45000) were added at to initiate in situ polymerization to form the nanocapsules. The reaction was carried out under inert gas for 1.5 hours at 4 °C. The mixture was purified by dialysis against 10 mM phosphate buffer (pH ~7.0). Systemic studies, including size, morphology and structure of the nanocapsules, enzymatic kinetics, cell studies, and animal models (tissue ELISA, focal and permanent stroke model and impaired wound healing in diabetic mice) were carried out to demonstrate the delivery platform, which are detailed in the Supporting Information.
Supplementary Material
Acknowledgments
We thank undergraduate researcher Angela Wong and high school researcher Eric Lin for their hard work. We would also like to thank the Electron Imaging Center for NanoMachines supported by NIH (1S10RR23057 to ZHZ) and the California NanoSystems Institute (CNSI) for the use of instruments. This project was supported by the American Heart Association under grant no. 11GRNT7630021AHA, the National Institutes of Health (NIH) under grant no. R01HL110592 and R01NS079691, and NSF IGERT: Materials Creation Training Program (MCTP) – DGE-0654431.
Footnotes
Author contributions: S.Z. and L.N. performed the experiments, S.Z., L.N., S.T.C., Y.L. and T.S. designed the experiments, S.Z., L.N. and T.S. analyzed the results. S.Z., Y.L. and T.S. wrote the manuscript, with inputs from all authors.
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Leader B, Baca QJ, Golan DE. Nat Rev Drug Discov. 2008;7:21. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Nature. 2008;453:314. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]; Sinclair RD, Ryan TJ. Australas J Dermatol. 1994;35:35. doi: 10.1111/j.1440-0960.1994.tb01799.x. [DOI] [PubMed] [Google Scholar]
- 3.Hsu BB, Jamieson KS, Hagerman SR, Holler E, Ljubimova JY, Hammond PT. Angew Chem Int Ed Engl. 2014;53:8093. doi: 10.1002/anie.201403702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekaputra AK, Prestwich GD, Cool SM, Hutmacher DW. Biomaterials. 2011;32:8108. doi: 10.1016/j.biomaterials.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat Biotechnol. 2001;19:1029. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]; Wang Y, Cooke MJ, Sachewsky N, Morshead CM, Shoichet MS. J Control Release. 2013;172:1. doi: 10.1016/j.jconrel.2013.07.032. [DOI] [PubMed] [Google Scholar]
- 6.Seliktar D, Zisch AH, Lutolf MP, Wrana JL, Hubbell JA. J Biomed Mater Res A. 2004;68:704. doi: 10.1002/jbm.a.20091. [DOI] [PubMed] [Google Scholar]
- 7.Wen J, Anderson SM, Du JJ, Yan M, Wang J, Shen MQ, Lu YF, Segura T. Advanced Materials. 2011;23:4549. doi: 10.1002/adma.201101771. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR. Nat Biotechnol. 2012;30:184. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 8.Yan M, Du J, Gu Z, Liang M, Hu Y, Zhang W, Priceman S, Wu L, Zhou H, Liu Z, Segura T, Tang Y, Lu Y. Nature Nanotechnology. 2009;5:48. doi: 10.1038/nnano.2009.341. [DOI] [PubMed] [Google Scholar]
- 9.Demidova-Rice TN, Hamblin MR, Herman IM. Adv Skin Wound Care. 2012;25:349. doi: 10.1097/01.ASW.0000418541.31366.a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darnell MC, Sun JY, Mehta M, Johnson C, Arany PR, Suo Z, Mooney DJ. Biomaterials. 2013;34:8042. doi: 10.1016/j.biomaterials.2013.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang JC, Wojta J, Binder BR. J Thorac Cardiovasc Surg. 1995;109:1059. doi: 10.1016/S0022-5223(95)70188-5. [DOI] [PubMed] [Google Scholar]
- 12.Ozawa CR, Banfi A, Glazer NL, Thurston G, Springer ML, Kraft PE, McDonald DM, Blau HM. Journal of Clinical Investigation. 2004;113:516. doi: 10.1172/JCI18420. [DOI] [PMC free article] [PubMed] [Google Scholar]; von Degenfeld G, Banfi A, Springer ML, Wagner RA, Jacobi J, Ozawa CR, Merchant MJ, Cooke JP, Blau HM. Faseb Journal. 2006;20:2657. doi: 10.1096/fj.06-6568fje. [DOI] [PubMed] [Google Scholar]
- 13.Galiano RD, Tepper OM, Pelo CR, Bhatt KA, Callaghan M, Bastidas N, Bunting S, Steinmetz HG, Gurtner GC. Am J Pathol. 2004;164:1935. doi: 10.1016/S0002-9440(10)63754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; Greenhalgh DG, Sprugel KH, Murray MJ, Ross R. Am J Pathol. 1990;136:1235. [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. J Clin Invest. 2000;106:829. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ma Y, Zechariah A, Qu Y, Hermann MD. J Neurosci Res. 2012;90:1873. doi: 10.1002/jnr.23088. [DOI] [PubMed] [Google Scholar]
- 15.Wysocki AB, Kusakabe AO, Chang S, Tuan TL. Wound Repair Regen. 1999;7:154. doi: 10.1046/j.1524-475x.1999.00154.x. [DOI] [PubMed] [Google Scholar]
- 16.Frank S, Hubner G, Breier G, Longaker MT, Greenhalgh DG, Werner S. J Biol Chem. 1995;270:12607. doi: 10.1074/jbc.270.21.12607. [DOI] [PubMed] [Google Scholar]
- 17.Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L, Li C, Xie L, Crane J, Wan M, Zhen G, Bian Q, Yu B, Chang W, Qiu T, Pickarski M, Duong le T, Windle JJ, Luo X, Liao E, Cao X. Nat Med. 2014;20:1270. doi: 10.1038/nm.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.