

Abstract
Triple negative breast cancer (TNBC) is the most aggressive breast cancer subtype with the worst prognosis and no targeted treatments. TNBC patients are more likely to develop metastases and relapse than patients with other breast cancer subtypes. We aimed to identify TNBC-specific genes and genes associated with lymph node metastasis, one of the first signs of metastatic spread. A total of 33 TNBCs were used; 17 of which had matched normal adjacent tissues available, and 15 with matched lymph node metastases. Gene expression microarray analysis was used to reveal genes that were differentially expressed between these groups. We identified and validated 66 genes that are significantly altered when comparing tumours to normal adjacent samples. Further, we identified 83 genes that are associated with lymph node metastasis and correlated these with miRNA-expression. Pathway analysis revealed their involvement in DNA repair, recombination and cell death, chromosomal instability and other known cancer-related pathways. Finally, four genes were identified that were specific for TNBC, of which one was associated with overall survival. This study has identified novel genes involved in LN metastases in TNBC and genes that are TNBC specific that may be used as treatment targets or prognostic indicators in the future.
Triple negative breast cancer (TNBC) is the most aggressive breast cancer subtype. This subtype is characterised by the absence of the estrogen (ER) and progesterone (PR) receptors, and the human epidermal growth factor receptor 2 (HER2/HER neu). Further characteristics of the TNBC subtype are: a high proportion of germline BRCA1 mutation carriers present with TNBC, high mitotic counts, and a high proportion of somatic p53 mutations1. TNBC accounts for somewhere between 10% and 17% of all breast cancers and is more frequent in women diagnosed at younger ages (under 40 years of age/pre-menopausal2), and those of African-American descent3. Women with TNBC have a poorer prognosis with an increased number and earlier appearance of metastatic disease (on average within the first 2.6 years after initial diagnosis4) compared to other breast cancer subtypes1. Metastatic disease is the most common cause for death from this cancer5,6,7.
The absence of the three receptors significantly reduces targeted treatment options for patients with TNBC and the only treatment remains radiotherapy and chemotherapy3. There are several trials with newer disease specific agents that include, poly (ADP-ribose) polymerase (PARP) inhibitors, angiogenesis inhibitors, EGFR-targeted agents, src kinase inhibitors, an androgen receptor inhibitor (bicalutamide), epigenetic targeting, and PI3K-pathway inhibition1,8. None of these trials have thus far been associated with any significant improvement in TNBC patients. Thus, new treatment targets are urgently needed for women diagnosed with this breast cancer subtype. Taken together the evidence indicates that TNBC is a heterogeneous disease where for example PARP-inhibitors demonstrate good effects in BRCA1 mutation-positive patients diagnosed with TNBC, but not in other TNBC patients9. This effect can be explained by the importance of both genes (BRCA1 and PARP) in DNA repair, such that if both are functionally inactive DNA damage will not be repaired and the affected cell dies9. There is a paucity of studies that have focused on identifying molecular biomarkers that are predictive of metastatic disease in TNBC patients.
microRNAs (miRNAs) are small (18–21 nucleotides) non-coding RNAs, which are able to alter gene expression post-transcriptionally. microRNAs have been found in multiple species, where they are highly conserved, suggesting they play a universal role in the regulation of gene expression. miRNAs regulate multiple biological processes including proliferation, cell death, development, genomic stability, and EMT10,11—key processes in tumour development identified by Weinberg and Hanahan. They not only regulate normal physiological change but are also involved in pathological conditions, such as cancer12.
There are multiple studies that have shown altered miRNA levels in TNBC patients compared to healthy controls and compared to other breast cancer subtypes13,14,15,16. At present there is little known about the influence of miRNAs and metastasis in TNBC, or what genes they control to regulate this process. In this study we have correlated microRNA expression analysis16 with gene expression in a series of TNBC patients where we were able to examine tumour tissue, normal adjacent tissue and lymph node metastases from the same patient and compared that to tumours derived from patients who did not have metastases.
A recent study by Cascione et al. (2013) identified a number of genes as biomarkers that are associated with survival in TNBC using a series of breast tumours and matched lymph node metastases17, however they only examined 230 cancer-related mRNAs, whereas in the current study a total of 11,000 lncRNAs, 24,000 genes and 30,000 coding transcripts were examined. Using this approach we were able to better define gene differences between primary tumours without disease spread and tumours that had lymph node involvement. Especially important are the findings associated with non-coding transcripts (like microRNAs and long non-coding RNAs) that are predicted to play an important role in cancer progression18,19. In this study we have identified a gene expression profile in a series of TNBC patients where we were able to examine tumour tissue, normal adjacent tissue and lymph node metastases from the same patient and compared that to tumours derived from patients who did not have any metastatic disease. We have identified TNBC-specific transcripts for tumour versus normal tissue that has been validated in an independent TNBC cohort as well as the The Cancer Genome Atlas (TCGA) TNBC breast cancer cohort20. Further, we analysed the relationship of these genes with overall survival. Finally, we were able to correlate the gene expression changes from this study with a previous microRNA study using the same breast cancer samples to confirm the relationship between microRNAs and lymph node metastases16.
Results
Differentially expressed genes in all IDC compared to NAT samples
We compared the gene expression of 33 grade 3 primary invasive ductal carcinomas (IDCs) to 17 normal adjacent tissue (NAT) samples (Supplementary Table 1) to reveal gene and lncRNA transcripts significantly associated with TNBC. A total of 185 genes were revealed to have significantly altered expression in tumour samples compared to NAT samples. Unsupervised hierarchical clustering was performed, which showed clear clusters between these two groups (IDC versus NAT). Within the 185 differentially expressed genes 90 were up-regulated in all tumour samples and 95 down-regulated (Supplementary Figure 1A and Supplementary Table 2).
We used the Ingenuity Pathway analysis tool (IPA) to study gene networks and pathways that involve the genes of interest. All 185 significant genes, with their expression values from the tumour versus normal comparison were used to perform a Core Analysis. This analysis was used to interpret the dataset in the context of biological processes that included pathways, upstream regulators and molecular networks. The analysis revealed that the disorder with the highest number of affected genes was Cancer with 119 out of 185 genes. Multiple genes were involved in the process of Cellular Development (60 genes) and Tissue Development (56 genes). The top upstream regulators of our genes of interest are PTGER2, ERBB2, E2F4, and TGFB1 (Supplementary Table 3).
To validate the genes from the tumour versus normal comparison we used an independent cohort containing 16 TNBC samples, 4 NAT and 48 samples of other breast cancer subtypes (Supplementary Table 4). We compared the expression of the 185 differentially expressed genes from our discovery cohort in all TNBC cases (n = 16) to the 4 NAT samples in our second cohort. This validation substantiated the role of 99 of the 185 genes (only 152 of them were on the array that was used for the validation cohort; 99 = 65.13%) that were differentially expressed in the two TNBC cohorts. Additionally, we performed a further validation using the TCGA breast cancer cohort. We compared tumour vs normal samples from 55 TNBC cases and 5 matched normal cases, which validated 95 of the initial 185 significant genes. By comparing all data sets, we identified 66 genes that were differentially expressed in TNBC in each of the cohorts (Table 1). Unsupervised hierarchical clustering of these results is shown in Supplementary Figure 1 A (185 genes in study cohort) B (66 genes in study cohort), C (66 genes in second cohort), D (66 genes in TCGA cohort).
Table 1. 66 common significantly altered genes comparing tumour vs normal samples in the study cohort, our second cohort and the TCGA cohort.
| Transcript ID | Study cohort |
Second cohort |
TCGA |
|||
|---|---|---|---|---|---|---|
| Fold-Change(yes vs. TNBC normal) | p-value(TNBC status) | Fold-Change(IDC vs. NAT) | p-value(TvN) | Fold-Change(IDC vs. NAT) | p-value(TvN) | |
| ABCA10 | −3.07 | 3.59E–21 | −4.69 | 1.76E–09 | −2.71 | 5.51E–10 |
| ABCA6 | −8.96 | 5.42E–23 | −5.01 | 6.37E–06 | −3.83 | 4.95E–10 |
| ABCA8 | −20.18 | 5.12E–29 | −5.41 | 7.23E–05 | −2.58 | 6.91E–10 |
| ABCA9 | −15.16 | 2.85E–35 | −4.55 | 2.43E–06 | −2.54 | 4.50E–08 |
| AGR2 | −13.79 | 5.62E–49 | −20.16 | 3.97E–05 | −2.10 | 3.56E–06 |
| ANKRD30A | −31.96 | 1.48E–25 | −43.09 | 5.71E–12 | −4.91 | 9.08E–12 |
| ANKRD30B | −11.36 | 1.11E–17 | −13.74 | 0.000141 | −2.24 | 0.000458 |
| ANLN | 8.94 | 2.52E–56 | 15.88 | 2.62E–08 | 2.56 | 9.26E–06 |
| ANP32E | 3.06 | 5.47E–40 | 2.58 | 2.37E–06 | 2.05 | 0.000932 |
| APOD | −5.16 | 1.65E–16 | −4.71 | 0.002127 | −7.77 | 9.23E–09 |
| AREG | −16.35 | 1.54E–12 | −7.97 | 0.000308 | −2.06 | 2.80E–06 |
| ASPM | 19.80 | 2.17E–71 | 15.69 | 5.15E–08 | 2.97 | 4.08E–07 |
| ATAD2 | 3.95 | 3.05E–32 | 2.93 | 0.002838 | 2.63 | 2.13E–06 |
| BGN | 3.38 | 1.20E–42 | 8.53 | 1.04E–05 | 2.48 | 1.21E–05 |
| CALU | 3.26 | 1.72E–27 | 2.86 | 3.88E–08 | 2.19 | 3.88E–06 |
| CCT3 | 2.46 | 2.70E–42 | 2.39 | 1.16E–07 | 2.09 | 3.04E–06 |
| CENPF | 18.38 | 9.29E–69 | 14.06 | 1.36E–08 | 2.94 | 6.16E–08 |
| CHML | 2.58 | 1.22E–19 | 3.72 | 1.11E–05 | 2.12 | 2.98E–05 |
| CKS2 | 5.13 | 3.45E–56 | 4.93 | 2.83E–05 | 4.20 | 6.45E–08 |
| COL14A1 | −5.57 | 4.10E–14 | −3.01 | 0.000568 | −3.44 | 5.00E–09 |
| CXCL10 | 3.09 | 8.20E–33 | 24.83 | 6.19E–06 | 4.48 | 1.19E–05 |
| CXCL11 | 9.68 | 9.14E–31 | 11.91 | 0.000101 | 2.03 | 6.14E–05 |
| CXCL9 | 3.20 | 6.32E–17 | 6.31 | 0.005079 | 5.14 | 1.96E–05 |
| DSC2 | 5.31 | 1.63E–29 | 1.89 | 0.006364 | 2.38 | 0.0009 |
| ECT2 | 4.47 | 2.82E–34 | 3.38 | 0.000117 | 2.52 | 4.48E–07 |
| EGR1 | −12.87 | 8.24E–38 | −4.73 | 5.73E–05 | −3.08 | 9.70E–07 |
| FGF10 | −10.16 | 5.14E–16 | −3.04 | 3.45E–05 | −2.34 | 7.78E–11 |
| FN1 | 4.73 | 3.88E–48 | 7.90 | 2.21E–12 | 3.94 | 1.26E–06 |
| FREM1 | −13.48 | 2.05E–36 | −2.37 | 7.86E–06 | −2.18 | 9.51E–09 |
| GPI | 1.87 | 3.91E–09 | 3.66 | 0.000226 | 2.23 | 9.43E–06 |
| HIST1H2BD | 2.60 | 1.71E–32 | 2.30 | 0.004616 | 2.47 | 0.00052 |
| HIST1H3F | 2.65 | 1.06E–38 | 1.61 | 0.002149 | 4.64 | 8.42E–07 |
| IGF1 | −5.83 | 1.42E–18 | −3.65 | 0.000782 | −2.96 | 1.21E–08 |
| IL6ST | −3.20 | 1.92E–23 | −3.39 | 8.60E–08 | −2.36 | 7.53E–06 |
| INHBA | 5.23 | 3.52E–60 | 7.48 | 1.49E–06 | 2.12 | 3.42E–06 |
| JAM2 | −4.20 | 2.88E–29 | −2.47 | 0.001 | −2.08 | 6.94E–08 |
| KIF11 | 6.69 | 6.47E–60 | 8.69 | 1.39E–05 | 2.40 | 1.92E–06 |
| KPNA2 | 3.72 | 6.80E–37 | 5.28 | 4.32E–08 | 2.16 | 9.43E–05 |
| LAMA2 | −4.28 | 4.96E–14 | −2.51 | 0.001598 | −2.02 | 7.43E–07 |
| LIFR | −7.78 | 1.84E–43 | −5.28 | 0.000688 | −2.67 | 3.25E–07 |
| MEG3 | −3.39 | 3.52E–68 | −1.66 | 0.003507 | −2.53 | 6.64E–09 |
| MELK | 14.34 | 1.72E–61 | 14.49 | 1.27E–07 | 2.13 | 2.71E–05 |
| MKI67 | 14.02 | 1.35E–51 | 10.94 | 5.24E–08 | 2.18 | 9.96E–06 |
| MME | −8.64 | 6.89E–40 | −5.17 | 0.001052 | −2.08 | 6.00E–05 |
| MYBL1 | 5.34 | 8.72E–17 | 4.34 | 0.005911 | 2.24 | 0.000728 |
| NUSAP1 | 6.35 | 3.64E–46 | 8.42 | 1.55E–06 | 2.14 | 7.03E–07 |
| OGN | −58.54 | 4.14E–32 | −5.26 | 5.37E–05 | −4.13 | 7.52E–11 |
| PDK4 | −15.78 | 4.50E–36 | −3.74 | 0.006427 | −2.53 | 0.0019 |
| PRDX1 | 1.91 | 8.91E–14 | 1.92 | 0.001102 | 2.00 | 2.67E–05 |
| PRR11 | 2.52 | 2.88E–17 | 9.50 | 1.28E–08 | 2.30 | 1.58E–06 |
| RAD51AP1 | 4.44 | 1.47E–29 | 4.30 | 0.00132 | 2.20 | 7.80E–06 |
| RBMS3 | −5.25 | 2.31E–35 | −2.82 | 0.00085 | −2.17 | 3.32E–07 |
| SLC7A5 | 3.96 | 2.13E–32 | 2.25 | 0.000817 | 2.09 | 0.002088 |
| SMC4 | 3.15 | 1.08E–37 | 2.70 | 7.86E–05 | 2.01 | 1.56E–05 |
| SNRPD1 | 1.91 | 2.42E–26 | 2.39 | 0.000965 | 2.06 | 0.000118 |
| SPARCL1 | −2.16 | 2.33E–13 | −1.93 | 0.001856 | −2.65 | 9.09E–06 |
| SPP1 | 6.68 | 2.97E–12 | 11.48 | 4.91E–06 | 3.40 | 2.30E–06 |
| STIL | 9.28 | 1.33E–38 | 4.23 | 9.29E–05 | 2.14 | 7.21E–06 |
| SULF1 | 2.95 | 9.23E–24 | 5.76 | 2.21E–07 | 3.38 | 7.50E–08 |
| TAT | −3.68 | 3.73E–15 | −60.22 | 2.49E–09 | −2.27 | 6.83E–06 |
| TBC1D9 | −7.35 | 7.69E–50 | −4.12 | 2.20E–07 | −2.25 | 1.37E–07 |
| TOP2A | 8.58 | 4.63E–44 | 15.44 | 4.91E–10 | 4.13 | 6.26E–08 |
| TPX2 | 11.10 | 2.56E–63 | 11.83 | 2.71E–09 | 3.57 | 6.46E–08 |
| TSHZ2 | −3.71 | 3.89E–20 | −4.92 | 3.56E–05 | −2.28 | 2.10E–06 |
| UBE2C | 19.93 | 8.26E–57 | 2.60 | 0.000417 | 2.22 | 1.53E–07 |
| UBE2T | 6.22 | 5.05E–47 | 8.55 | 1.24E–07 | 2.18 | 2.82E–05 |
The 4 TNBC specific genes are written in bold.
Identification of TNBC specific genes
We used our second cohort and the TCGA breast cancer cohort to identify which of the validated genes in each cohort are specific to TNBC. Therefore we performed tumour vs normal comparisons in all non-TNBC cases in both cohorts and compared this to differentially expressed genes in the TNBC cohorts. The results from this analysis are summarised in Table 2.
Table 2. Summary of the gene expression validation in our second cohort and the TCGA TNBC cohort and discovery of TNBC specific genes.
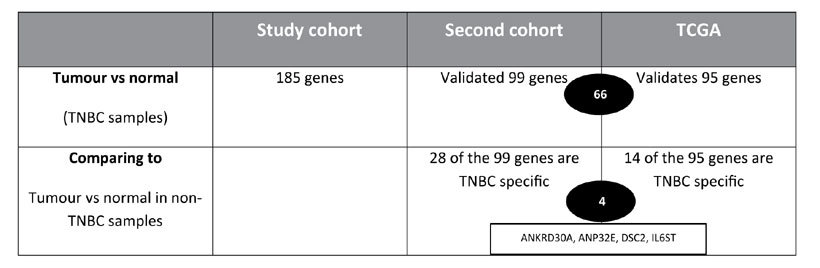
We identified 185 significantly altered genes by comparing all TNBC samples (33) versus all matched normal samples (17) in our study cohort. We then compared all TNBC tumours (16) from our second cohort to all normal samples (4), which validated 99 of the 185 genes. Further, we used the TCGA TNBC cohort to compare 55 TNBC samples to 5 matched normal samples, which validated 95 of our initial 185 genes. By comparing the two sets of validated genes we found 66 common genes. To identify TNBC-specific genes, we compared the 99 validated genes from our second cohort in TNBC samples (16) versus non-TNBC samples (48) and discovered that 28 of these genes are specific to TNBC. In the TCGA cohort we compared the 95 validated genes to the expression of 313 non-TNBC samples, which identified 14 TNBC specific genes. There are 4 TNBC specific genes that are common in both cohorts; these are ANKRD30A, ANP32E, DSC2 and IL6ST.
By performing an IDC versus NAT comparison in our second cohort for all non-TNBC (n = 48) samples and comparing that to the list of genes in the TNBC set (n = 16) we found that, from the 99 validated genes, 28 were found to be differentially expressed in TNBC cases compared to non-TNBC cases, we will refer to these as TNBC specific genes (Fig. 1A; and Supplementary Table 2). Figure 1A shows distinct clustering of the TNBC samples compared to the non-TNBC samples in our second cohort, based on the expression of the 28 TNBC specific genes.
Figure 1. Unsupervised hierarchical clustering of TNBC specific genes in the second cohort and the TCGA cohort.

Through analysis of our second cohort (48 non-TNBC, 16 TNBC samples) we identified 28 TNBC specific genes, which were used for unsupervised hierarchical clustering of TNBC and non-TNBC samples (A). The TCGA breast cancer cohort (313 non-TNBC, 55 TNBC samples) identified 14 TNBC specific genes. The unsupervised hierarchical clustering of the 14 TNBC specific genes successfully clustered the TCGA breast cancer cohort in TNBC patients and non-TNBC patients (B). There are four common TNBC specific genes in both cohorts (our second cohort and the TCGA breast cancer cohort) – these four genes are ANKRD30A, ANP32E, DSC2, IL6ST. (C) shows the unsupervised hierarchical clustering of these four genes in the second cohort and (D) shows the clustering of these genes in the TCGA breast cancer cohort. TNBC samples are shown in light blue and non-TNBC samples are shown in red in the sample tree on the left (y-axis). Genes are clustered along the x-axis. Low gene expression is shown in blue, high expression is shown in red and equivocal expression is shown in grey.
Further to this, the same analysis was done in the TCGA breast cancer cohort (excluding TNBC samples). We compared the gene expression of 313 non-TNBC cases and 43 matched normal samples from the TCGA breast cancer cohort, this was then compared to the 95 validated genes from the TNBC TCGA cohort. In this analysis, we identified that 14 of the 95 validated genes were differentially expressed solely in the TNBC cohort and not in non-TNBC cases and were hence TNBC specific. Figure 1B shows unsupervised hierarchical clustering of the 14 TNBC specific genes in the TCGA cohort.
Finally, by comparing the genes that were found to be TNBC specific genes from our second cohort and the TCGA cohort, we found that four of them (ANKRD30A, ANP32E, DSC2, IL6ST) were common to both TNBC cohorts (Fig. 1). Figure 1C,D show the unsupervised hierarchical clustering of the four validated TNBC specific genes in our study cohort, our second cohort and the TCGA cohort. As can be seen in this analysis, IL6ST and ANKRD30A were expressed at lower levels in TNBC when compared to non-TNBC, while ANPE32E and DSC2 were expressed at higher levels in TNBC when compared to non-TNBC.
TNBC-specific genes associated with overall survival
We used the breast cancer cohort from the Cancer Genome Atlas (TCGA) which contains 55 TNBC samples, of which nine patients died and 46 are still alive. With this information we were able to determine if our 4 TNBC specific genes (ANKRD30A, ANP32E, DSC2, IL6ST) were associated with survival. We performed Kaplan-Meier-Analysis on the four genes that are TNBC specific in our second cohort as well as in the TCGA cohort (ANKRD30A, ANP32E, DSC2, IL6ST). The results from this analysis can be seen in Fig. 2. The survival curves from Fig. 2 show that two of the genes (ANKRD30A, DSC2) show a non-significant trend that low gene expression is associated with shorter survival, ANP32E expression shows no association to survival. High expression of IL6ST shows significant (p = 0.0421) association with better survival outcomes.
Figure 2. Kaplan-Meier analysis of four validated TNBC specific genes.

Kaplan-Meier analysis was performed on the four validated TNBC specific genes (ANKRD30A, ANP32E, DSC2, IL6ST). The analysis of (A) ANKRD30A in TCGA TNBC cohort, Hazard ratio (HR) = 0.5326; p = 0.37. (B) ANP32E in TCGA TNBC cohort; HR = 1.137; p = 0.85. (C) DSC2 in TCGA TNBC cohort; HR = 0.4183; p = 0.21. (D) IL6ST in TCGA TNBC cohort; HR = 0.1921; p = 0.0421*. The Hazard ratio (HR) is smaller than 1 for three genes, which means down-regulated genes decrease overall survival in TNBC, whereas up-regulated genes would increase overall survival. A significant correlation to overall survival can be seen for IL6ST (p < 0.05). The p-value shows the significance of the difference between the survival curves.
Genes associated with lymph node metastasis
Gene expression analysis of the 33 primary TNBC tumours, 15 matched lymph node metastases, and 17 matched normal breast tissues was undertaken to identify which transcripts were associated with various aspects of TNBC. The arrays were analysed using Genomic Suite 6.6 (Partek).
To identify genes responsible for metastasis we performed a categorical comparison. We compared gene expression in three different categories. Category 1: lymph node positive primary tumours (IDC+) versus matched normal adjacent tissue (IDC+ vs NAT+), category 2: lymph node negative primary tumours (IDC-) versus matched normal adjacent tissue (IDC− vs NAT−), and category 3: lymph node metastases versus matched normal adjacent tissue (LNmet vs NAT+) (Fig. 3). The focus of this study is common genes of category 1 and 3, as these genes may be biomarkers for metastatic spread.
Figure 3. Venn Diagram for identification of genes associated with lymph node metastasis.
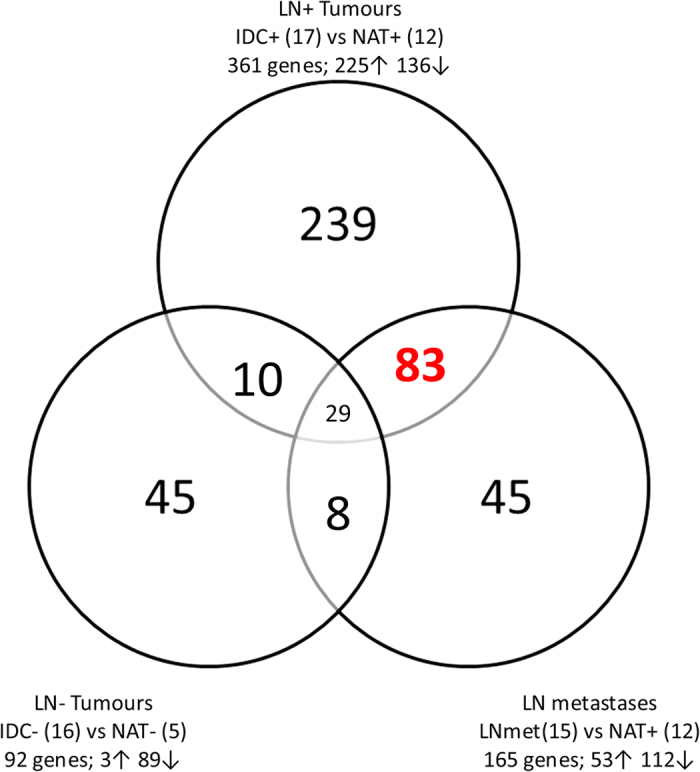
All samples are categorised in lymph node positive primary tumours (IDC+), lymph node negative primary tumours (IDC−), lymph node metastases (LNmet) and matched normal adjacent tissue for lymph node positive cases (NAT+) and lymph node negative cases (NAT−). Gene expression was analysed in all three tumour categories compared to their matched NAT samples. Highlighted in red are the 83 genes that are differentially expressed in IDC+ and LNmet samples but not in IDC− samples.
We identified 361 genes that were differentially expressed in IDC+ tissue compared to matched NAT (category 1), 92 were differentially expressed in IDC- samples compared to their matched NAT (category 2), and 165 had significantly altered expression in LNmet samples compared to matched NAT samples (category 3). There were 83 genes that were specifically associated with LN metastasis, since they were differentially expressed in category 2 and 3. This included 72 protein coding genes and 11 non-coding RNAs (such as MEG3); out of these 36 had increased expression in IDC+ and LNmet samples compared to NAT+ samples, and 47 showed decreased expression in IDC+ and LNmet samples compared to NAT+ samples (Supplementary Table 5).
A second Core analysis was performed in IPA with the 83 significant genes that were potential biomarkers for metastasis. Similarly the top candidate for diseases and disorders was Cancer, with 62 genes involved. The majority of these are involved in cellular processes like cell cycle, DNA replication, recombination, repair, and cell death/survival (Supplementary Table 6). Looking at the upstream regulators of the genes in IPA, we identified a number of activated genes (CSF2, PTGER2, FOXO1 - mostly involved in proliferation, differentiation and invasion) and inhibited genes (TP53, TCF3 - tumour-suppressor, apoptosis regulator). None of these genes were present in the validated TNBC specific gene list but 63% (52 genes) were differentially expressed in tumour versus normal comparisons when all cases were compared in cohort 2 and the TCGA cohort indicating that they may be important biomarkers of metastatic progression in all breast cancers. We also performed survival analysis (using the complete TCGA breast cancer cohort) of the five most up- and five most down-regulated genes from this signature, but were unable to show a significant association to overall breast cancer survival (data not shown).
Our previous study identified miRNAs associated with LN metastasis16. We performed a correlation study (see Material and Methods) to identify microRNAs that could potentially regulate the LN metastasis-associated genes identified in this study. Twenty two microRNAs were found to be significantly altered in lymph node positive primary tumours (IDC+) and lymph node metastases (LNmet) but not in lymph node negative primary tumour (IDC−) samples (Supplementary Table 7). Their expression was correlated with the 83 gene biomarkers using Genomic Suite 6.6 (Partek). The correlation analyses revealed that 17 of these microRNAs can potentially target 50 of the 83 genes. Several genes (31) can be targeted by multiple microRNAs (Supplementary Table 8).
Discussion
The outcome for triple negative breast cancer (TNBC) patients is generally considered to be poor. Nevertheless, TNBC is a heterogeneous disease and the probability of survival and/or relapse is different from patient to patient. Specific gene expression patterns can be used to identify differences in patients with phenotypically similar disease but with very different disease outcomes21,22. The current study aimed to identify expression differences associated with metastatic disease in TNBC. Other reports have provided gene expression profiles for TNBC, but most did not include any comparison to normal adjacent tissue, nor did they separate primary IDCs from lymph node metastases23,24. Other reports identified prognostic markers in TNBC by comparing gene expression profiles of TNBC patients to all other non-TNBC samples25,26, which can be used to assess the general risk of relapse, but not sufficiently specific to identify biomarkers associated uniquely metastatic spread in TNBC. The study herein confirms the more aggressive nature of TNBC and provides an insight into which pathways are most important in differentiating women at risk of relapse. Furthermore, this study used NAT, IDC and LNmet samples that were biopsied at the same time, this allowed the identification of differences in these three tissue types without the influence of further disease progression. Due to the structure of this analysis the criteria meant only a small sample cohort of patients could be identified, but allowed us to reveal significant biological differences between the three different tissue types. This allowed us to examine actual differences between TNBCs that had begun to move away from their primary location compared to those that had not. This is in contrast to studies aimed at identifying differences between distant metastatic lesions that may have been influenced by other tissue specific factors and an increased mutation burden.
In this study we identified and validated TNBC-specific genes, which we were able to associate with overall survival. Further to this, we have revealed genes associated with lymph node metastasis in TNBC and showed that these are potentially targetable by microRNAs with altered expression. This included the lncRNA gene MEG3, which has not previously been reported to be associated with metastatic breast cancer.
We compared IDC to NAT samples to identify tumour-associated genes in TNBC. Although, it has been shown that normal tissue adjacent to TNBC tissue has altered gene expression compared to healthy controls, and that NAT samples already show an expression profile indicating DNA repair deficiency27,28, we reasoned that by comparing NAT to IDC samples we would identify further changes in tumour progression.
The 185 genes that we identified showed clear hierarchical clustering of NAT samples and IDC samples. The pattern included 90 genes that were significantly up-regulated in all tumour samples and 95 that are significantly down-regulated.
We were able to confirm the expression pattern of 66 of the 185 genes in our two validation cohorts (cohort 2 and TCGA), which also allowed us to reveal four genes that are specific for TNBC, by comparing the expression of the validated genes to significantly altered genes in a tumour versus normal comparison in other breast cancer subtypes (Fig. 1 and Supplementary Figure 1).
These four TNBC specific genes are ANKRD30A, ANP32E, DSC2, IL6ST (Fig. 1, Table 1). The expression of these four genes alone clearly clustered TNBC from non-TNBC tumours. Together with the survival data from the TCGA cohort we were able to perform Kaplan-Meier analysis (Fig. 2). Two of the four genes (ANKRD30A, DSC2) show a non-significant trend that higher gene expression is associated with longer survival time. ANP32E does not show any association with overall survival in TNBC patients. Finally, higher expression of IL6ST shows significant association with longer overall survival in TNBC patients. To the best of our knowledge IL6ST has not been associated with overall survival in TNBC before. Nevertheless, it has been shown to be associated to ER expression and with TNBC development/progression29,30,31.
ANKRD30A is well known in breast cancer as NY-BR-1 (Breast Cancer Antigen NY-BR-1)32, nevertheless it has not yet been associated as a TNBC specific marker. Jaeger et al. identified the gene in 2001 as a tissue-specific putative transcription factor in breast tissue by serological screening of a breast cancer library33. It has been found to be highly expressed in the majority of breast cancer cases, but our studies have shown that it has significantly lower expression in TNBC than in the other breast cancer subtypes. Even though our survival data for this gene did not show a significant effect, there is a non-significant trend that lower gene expression appears to be associated with worse overall survival. ANKRD30A has been suggested as a potential target for immunotherapy due to its highly restricted expression pattern in breast cancer it may function as a breast differentiation antigen34. It has also been shown that high ANKRD30A expression is associated with lymph node negativity, as well as with the expression of HER2 and ER35, which supports our findings of lower expression in TNBC and supports the non-significant trend we identified that those with the lowest expression had worst survival.
ANP32E was more highly expressed in TNBC than non-TNBC in our study and has recently been identified as a histone chaperone that removes H2A.Z from chromatin36. H2A.Z is a histone variant which plays a crucial role in various key processes like DNA repair37 and cancer initiation and progression38,39. Together with DSC2, it is part of a six-gene signature predicting breast cancer lung metastasis40. Later it has been shown that these two genes are associated with breast cancer subtype (with ER-negative breast cancers, basal-like breast cancers)41, but neither of them have been stated as TNBC specific markers. DSC2 is a Ca2+-dependent transmembrane cell adhesion protein of desmosomes42. Except for the mentioning in the six-gene signature by Landemaine et al., the function of DSC2 has not been studied in breast cancer.
IL6ST is the signal transducer for interleukin 6 (IL6), ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), and oncostatin M (OSM). We showed that IL6ST was lower in TNBC when compared to non-TNBCs. High expression of IL6ST has been shown to be a good prognostic factor in breast cancer as it increases patients overall survival43, which supports our finding in TNBC where higher expression of IL6ST was shown to be associated with significantly increased survival. Multiple studies identified IL6ST as being positively associated with ER-α expression in breast cancer30,31, which again confirms our findings of decreased levels of this gene in TNBC patients.
The second aim of this study was to identify biomarkers associated with metastasis in TNBC. Lymph node metastases are the first sign of metastatic potential. Even though they are not necessarily associated with prognosis, they provide an ideal tissue to gain knowledge about the mechanisms of tumour progression. In this study, we identified genes that are only differentially expressed in the lymph node positive primary tumours and lymph node metastases (NAT+ v IDC+ and NAT+ v LNmet). By doing so, we identified a unique gene pattern of 83 genes that may be potential biomarkers for metastasis in TNBC. The most significantly down-regulated gene in this comparison was APOD, which is a lipocalin. APOD is used as a biomarker indicating good prognosis in colorectal cancer, if its expression level is increased since APOD induces growth arrest and reduces cell proliferation when its levels are elevated44,45,46. It is known to interact with ERα and can function as a Tamoxifen transporter, which suggests co-expression of ERα and APOD would predict a better outcome47. The fact that its expression is significantly decreased in our cohort (LNmet vs NAT+ fold change = −9.46; IDC+ vs NAT+ fold change = −6.62) confirms the poor prognosis for TNBC patients, especially patients with metastases.
We identified 72 protein coding genes and 11 non-coding RNA (ncRNA) to be associated with lymph node metastasis. The discovery that not just changes in protein coding genes but also in non-coding genes are responsible for cancer development and cancer progression has transformed cancer research in recent years48. The majority of ncRNA species remains to be discovered and many of known ncRNAs still have not been assigned a function. One of the first lncRNAs to be associated with breast cancer survival was HOTAIR49 and as such we have shown that a second lncRNA (MEG3) is also associated with LN metastases further supporting the important role of lncRNAs in disease. MEG3 is known to be down-regulated in multiple cancers and tumour cell proliferation is inhibited by MEG3 expression. It is able to alter the p53 pathway and acts as a tumour suppressor50,51. Recently it has been discovered that MEG3 overexpression leads to G2/M cell cycle arrest and apoptosis, which would explain increased cell growth when MEG3 is down-regulated52. In bladder cancer MEG3 activates autophagy, which leads to increased cell proliferation53. So far the function of MEG3 in triple negative breast cancer is unknown and needs further investigation.
In our previous study we used this TNBC cohort to identify microRNAs (miRNAs) that were associated with lymph node metastasis16. To the best of our knowledge, there has not been a study to correlate altered microRNA expression to gene expression changes in the same cohort, including the comparison of lymph node metastases to primary tumours in TNBC. The inclusion of expression values into the target prediction algorithms within 2 software suites (TargetScan 6.2, microcosm) increases the biological relevance of the prediction. In our cohort, 17 significantly altered microRNAs are likely to contribute to the identified gene expression profile of 83 gene biomarkers for metastasis (Supplementary Table 8). The only other integrated microRNA and mRNA study in a pure TNBC cohort compared expression changes from lymph node metastases to normal adjacent tissue, and primary tumours to normal adjacent tissue, without separation of lymph node positive and lymph node negative tumours and only looking at a set of 230 genes17. Nevertheless we can confirm 6 of their identified genes (SPP1, TOP2A, AREG, EGR1, CD34, and IGF1) as well as one of the identified miRNAs (hsa-miR-101), that they found to be differentially expressed in lymph node metastases compared to primary tumours and to normal tissue. Another miRNA-mRNA integration study in breast cancer by Buffa et al. found three miRNAs (hsa-miR-324, hsa-miR-27b, and hsa-miR-150) to be associated with progression pathways in TNBC54, however this study did not integrate these miRNAs with genes in TNBC alone and did not perform a comparison to normal tissue nor to lymph node metastases. These miRNA were not found to be differentially expressed between tumour versus normal tissue in our analysis and were therefore excluded from further analysis. A number of our miRNAs of interest have been associated with TNBC or tumour progression previously (summarised in Supplementary Table 9). As an example miR-205 is known to be down-regulated in TNBC55, it is a known tumour-suppressor-miR that targets E2F1, LAMC1, suppresses cell proliferation, cell cycle and tumour growth56. Most interesting for this study miR-205 has also recently been associated to lymph node metastasis in a study by Berber et al.57.
We were unable to associate the five most up- and five most down-regulated genes to overall survival in the TCGA cohort. Nevertheless, there are a number of genes within our list of 83 genes, associated to lymph node metastasis, that have been associated with prognosis in other studies. TOP2A is the gene with the highest fold change in our cohort (LNmet vs NAT+ fold change = 4.628; IDC+ vs NAT+ fold change = 5.08). This gene encodes an enzyme that is important for chromosome condensation, chromatin separation during DNA replication. Over-expression of this gene is a known prognostic marker for poor outcome in several malignancies, including TNBC58,59,60. Even though it has been shown that high expression of TOP2A is associated with worse prognosis, it also provides knowledge about the efficacy of treatment since TNBC patients with high TOP2A expression respond better to anthracycline therapy58.
A recent study performed a meta-analysis of global gene expression profiles of TNBC to identify genes of prognostic value61. This study was only looking at over-expressed genes, to identify potential treatment targets. They identified the majority of altered genes in TNBC are involved in chromosomal instability (CIN) and ER signalling. Especially genes causing CIN often lead to aneuploidy in several cancer types62. In general overexpression of these genes and aneuploidy are associated with poor outcome. These studies provided lists of genes correlated with aneuploidy, in which 16 of the genes we identified also appear62,63. These genes are: TPX2, TOP2A, UBE2C, MELK, RAD51AP1, MAD2L1, ATAD2, CKS2, ECT2, GPI, STAT1, CXCL9, CXCL10, CXCL11, SMS, and TNFRSF13B. All of the genes are up-regulated in our cohort, suggesting a poor outcome for the patients and possible aneuploidy. The study from Carter et al. (2006) shows that the gene with the highest level of consistent correlation with total functional aneuploidy is TPX2 which has been supported by Szasz et al. (2013) who used a gene set of 4 CIN genes to measure tumour aneuploidy, two of them are TOP2A and TPX264. Both genes are significantly up-regulated in our cohort, confirming the relationship between aneuploidy and poor outcome for patients with TNBC.
In conclusion, we identified TNBC-specific gene expression profiles and showed novel associations for TNBC-specific genes to prognosis. Additionally, we identified a gene expression profile for genes associated with lymph node metastasis, which we were able to correlate our previous study results of microRNA expression profiles with the profiles of this study. In this way we identified novel microRNA and mRNA interactions, supporting important pathways in TNBC development.
Material and Methods
Study design
A total of 33 grade 3, triple negative, invasive ductal carcinomas (IDCs), 17 matched normal adjacent tissues (NAT), and 15 lymph node metastases (LNmet) were used for gene expression microarray analysis. All samples were formalin-fixed, paraffin-embedded (FFPE) and obtained from the archives of the Hunter Area Pathology Service, John Hunter Hospital, Newcastle, Australia. This cohort has been described previously16. The triple negative phenotype, areas of NAT, invasive cancer and LNmets were confirmed by a pathologist. Further we show significantly lower ER, PR and HER2 expression in the TNBC samples compared to receptor-positive patients in supplementary Figure 2. A 1.5 mm punch biopsy was used to isolate these sections, performed as described previously16. Areas of NAT, IDC and LNmet tissue were biopsied at the same time to examine genetic differences in these three tissue types without the influence of time/further disease progression. The pathologist confirmed that the tumour volume in the core biopsy was >70% of the total core. This study complies with the Helsinki Declaration with ethical approval from the Hunter New England Human Research Ethics Committee (Approval number: 09/05/20/5.02). A waiver of consent was granted for this study in accordance with the National Statement on Ethical Conduct in Research Involving Humans.
RNA extraction
The RNA extraction of all samples was performed as previously described16.
Gene expression microarrays and analysis
The array results have been deposited in Gene Expression Omnibus (GEO) with Accession No. GSE61725.
100 ng total RNA of all FFPE samples was amplified (Ovation FFPE WTA kit) and biotinylated (Encore Biotin module) according to the manufacturers’ instructions (Nugen, San Carlos, California, United States). The samples were hybridised to HuGene 2.0 arrays (Affymetrix, Santa Clara, California, United States) and 17 hours later washed and stained. The Arrays were scanned on a GeneChip Scanner 3000 7G (Affymetrix).
The HumanGene 2.0 arrays (Affymetrix) contain probe features representing 11,000 lncRNAs, 24,000 genes, and 30,000 coding transcripts. The data was imported to Genomic Suite 6.6 (Partek) and a robust multi-array analysis (RMA) was performed, which included log2 transformation, background correction, quantile normalisation and summarisation of the probe features resulting in a set of expression signal intensities.
Unsupervised hierarchical clustering was performed on genes that were found to be significantly different in the comparison of all IDC vs all NAT samples (p-value < 0.5; fold change>1.5 or < − 1.5). Correction for multiple testing was performed using Benjamini–Hochberg procedure.
We compared gene expression in three different categories. Category 1: invasive ductal carcinoma with lymph node metastases versus normal adjacent tissue from patients with lymph node metastases (IDC+ vs NAT+), category 2: IDC- vs NAT-, and category 3: lymph node metastases (LNmet) vs NAT+ (Fig. 3) (“+” = lymph node involvement; “−” = only a primary tumour). The focus of this study was on common genes of category 1 and 3, proposing to be biomarkers for metastasis.
Validation of differentially expressed genes
To validate results of the gene expression arrays, we used an independent cohort with 16 TNBC primary tumours and 4 normal adjacent tissues, and 48 IDCs from patients with breast cancer other than TNBC, all samples were fresh frozen tissue cores. Lymph node metastases were not available from these samples, nevertheless 5 of the TNBC cases had known lymph node involvement. These tumour samples were provided by the Australian Breast Cancer Tissue Bank (Darcy Rd, Westmead, NSW, Australia). This study complies with the Helsinki Declaration with ethical approval from the Hunter New England Human Research Ethics Committee (Approval number: 09/05/20/5.02). A waiver of consent was granted for this study in accordance with the National Statement on Ethical Conduct in Research Involving Humans.
The samples and the method of RNA extraction have been described previously65. The RNA was hybridised to HumanGene 1.0 arrays (Affymetrix) and scanned on a GeneChip Scanner 3000 7G (Affymetrix). The data was imported to Genomic Suite 6.6 (Partek) and analysed as above. Due to the lack of samples from lymph node metastases, only the comparison from tumour versus normal samples could be performed and compared to the previous analyses.
For further validation of the tumour vs normal comparison as well as the identification of TNBC specific genes we used the The Cancer Genome Atlas (TCGA) breast cancer cohort gene expression data20. This data contains raw expression values for 55 TNBC samples with 5 matched NAT samples, and 313 non-TNBC samples with 43 matched NAT samples. The data was imported to Genomic Suite 6.6 (Partek) and analysed as above.
Correlation of genes to overall survival
We performed Kaplan-Meier analysis on the validated TNBC specific genes that we identified. Therefore we used the median gene expression to divide the patients into high and low expression and together with the TCGA survival data we were able to perform the analysis using GraphPad Prism 6 survival analysis tool. P-values were calculated using the log-rank (Mantel-Cox) method and hazard ratios were calculated by the Mantel-Haenszel method.
Pathway analysis
Differentially expressed transcripts were imported with their expression values into Ingenuity Pathway (IPA), where a Core Analysis was performed to identify their involvement in biological processes, pathways and molecular networks.
Correlation to microRNAs
To determine if altered microRNA expression contributed to the identified gene expression profiles, we correlated differentially expressed genes to microRNA microarray expression data from the same sample cohort, a previously published study16.
Partek Genomics Suite 6.6 was used to identify microRNAs that were differentially expressed in lymph node metastases and lymph node positive primary tumours but not in lymph node negative tumours, compared to matched NAT samples. Significance was reached with p-values <0.05 and two-sided fold change >1.5, these parameters identified 22 microRNAs (see Supplementary Table 7). Correction for multiple testing was performed using Benjamini–Hochberg procedure.
These microRNAs were correlated to significantly altered genes identified in this study. The correlation was performed in Partek Genomic Suite 6.6. Pearson Correlation and Spearman’s rank correlation were used to correlate microRNAs with potential target genes. These correlation analyses include the microRNA and gene expression values as well as two target prediction software suites (TargetScan 6.2, and Microcosm).
Statistical analysis
Student’s t-test and ANOVA were used to analyse differences between two or more groups. Significance was regarded for p < 0.05 and two-sided fold change >1.5.
Additional Information
How to cite this article: Mathe, A. et al. Novel genes associated with lymph node metastasis in triple negative breast cancer. Sci. Rep. 5, 15832; doi: 10.1038/srep15832 (2015).
Supplementary Material
Acknowledgments
The authors would like to thank the Pathologist Dr Ricardo Vilain for the review of all tumour and normal tissue specimens used in this study, as well as Ms Tina Hope for her assistance with archival specimens. We would like to acknowledge Dr Carlos Riveros for providing us with the TCGA raw data. Ms Andrea Mathe is supported by a University Postgraduate Award. Ms Brianna Morten is supported by an Australian University Postgraduate Award and an MM Sawyer Scholarship. This study was funded by the National Breast Cancer Foundation and the Hunter Medical Research Institute, through donations from Hunter Breast Cancer Foundation, PULSE and the Bloomfield Foundation.
Footnotes
Author Contributions A.M. study concept and design, carried out experiments, analysis and interpretation of data, drafting of manuscript. M.W.B. carried out gene expression arrays with A.M. and B.M. carried out gene expression arrays of validation cohort, manuscript revision. J.F.F. study design, obtained funding. S.G.B. patient collection, material support. K.A.K. study concept, design and supervision, interpretation of data, obtained funding, manuscript revision. R.J.S. study design, obtained funding, critical revision of the manuscript. All authors have read and approved the final version of this manuscript.
References
- Carey L., Winer E., Viale G., Cameron D. & Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 7, 683–92 (2010). [DOI] [PubMed] [Google Scholar]
- Bauer K. R., Brown M., Cress R. D., Parise C. A. & Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the california cancer registry. Cancer 109, 1721–8 (2007). [DOI] [PubMed] [Google Scholar]
- Reis-Filho J. S. & Tutt A. N. Triple negative tumours: a critical review. Histopathology 52, 108–18 (2008). [DOI] [PubMed] [Google Scholar]
- Dent R. et al. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res Treat 115, 423–8 (2009). [DOI] [PubMed] [Google Scholar]
- Chaffer C. L. & Weinberg R. A. A Perspective on cancer cell metastasis. Science 331, 1559–64 (2011). [DOI] [PubMed] [Google Scholar]
- Cho H. M. et al. microRNA-101 inhibits lung cancer invasion through the regulation of enhancer of zeste homolog 2. Exp Ther Med 2, 963–967 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci P. et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc Natl Acad Sci USA 109, 15312–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddi P. F., Ellis M. J. & Ma C. Molecular basis of triple negative breast cancer and implications for therapy. Int J Breast Cancer 2012, 217185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C. K. et al. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin Cancer Res 16, 4702–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beezhold K. J., Castranova V. & Chen F. Microprocessor of microRNAs: regulation and potential for therapeutic intervention. Mol Cancer 9, 134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. & Ma L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev 31, 653–62 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin G. A. & Croce C. M. MicroRNA signatures in human cancers. Nat Rev Cancer 6, 857–66 (2006). [DOI] [PubMed] [Google Scholar]
- Farazi T. A. et al. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res 71, 4443–53 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen E. A. et al. Biologic profiling of lymph node negative breast cancers by means of microRNA expression. Mod Pathol 23, 1567–76 (2010). [DOI] [PubMed] [Google Scholar]
- Volinia S. et al. Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc Natl Acad Sci USA 109, 3024–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery-Kiejda K. A., Braye S. G., Mathe A., Forbes J. F. & Scott R. J. Decreased expression of key tumour suppressor microRNAs is associated with lymph node metastases in triple negative breast cancer. BMC cancer 14, 51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascione L. et al. Integrated microRNA and mRNA signatures associated with survival in triple negative breast cancer. PLoS One 8, e55910 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner J. R. & Chinnaiyan A. M. The emergence of lncRNAs in cancer biology. Cancer Discov 1, 391–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blenkiron C. et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol 8, R214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacón R. D. & Costanzo M. V. Triple-negative breast cancer. Breast Cancer Res 12 Suppl 2, S3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carotenuto P. et al. Triple negative breast cancer: from molecular portrait to therapeutic intervention. Crit Rev Eukaryot Gene Expr 20, 17–34 (2010). [DOI] [PubMed] [Google Scholar]
- Kuo W. H. et al. Molecular characteristics and metastasis predictor genes of triple-negative breast cancer: a clinical study of triple-negative breast carcinomas. PLoS One 7, e45831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau C. et al. A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res 12, R85 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakha E. A. et al. Prognostic markers in triple-negative breast cancer. Cancer 109, 25–32 (2007). [DOI] [PubMed] [Google Scholar]
- Dent R. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13, 4429–34 (2007). [DOI] [PubMed] [Google Scholar]
- Atkinson R. L. et al. Cancer stem cell markers are enriched in normal tissue adjacent to triple negative breast cancer and inversely correlated with DNA repair deficiency. Breast Cancer Res 15, R77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakhuis B. J., Leemans C. R. & Brakenhoff R. H. Using tissue adjacent to carcinoma as a normal control: an obvious but questionable practice. J Pathol 203, 620–1 (2004). [DOI] [PubMed] [Google Scholar]
- Hartman Z. C. et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res 73, 3470–80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozlu S. et al. Identification of novel genes that co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-PCR approach. Endocr Relat Cancer 13, 1109–20 (2006). [DOI] [PubMed] [Google Scholar]
- Wilson B. J. & Giguere V. Meta-analysis of human cancer microarrays reveals GATA3 is integral to the estrogen receptor alpha pathway. Mol Cancer 7, 49 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissan A. et al. Multimarker RT-PCR assay for the detection of minimal residual disease in sentinel lymph nodes of breast cancer patients. Br J Cancer 94, 681–5 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager D. et al. Identification of a tissue-specific putative transcription factor in breast tissue by serological screening of a breast cancer library. Cancer Res 61, 2055–61 (2001). [PubMed] [Google Scholar]
- Seil I. et al. The differentiation antigen NY-BR-1 is a potential target for antibody-based therapies in breast cancer. Int J Cancer 120, 2635–42 (2007). [DOI] [PubMed] [Google Scholar]
- Varga Z. et al. Preferential nuclear and cytoplasmic NY-BR-1 protein expression in primary breast cancer and lymph node metastases. Clin Cancer Res 12, 2745–51 (2006). [DOI] [PubMed] [Google Scholar]
- Obri A. et al. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 505, 648–53 (2014). [DOI] [PubMed] [Google Scholar]
- Billon P. & Cote J. Precise deposition of histone H2A.Z in chromatin for genome expression and maintenance. Biochim Biophys Acta 1819, 290–302 (2013). [DOI] [PubMed] [Google Scholar]
- Conerly M. L. et al. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res 20, 1383–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S. et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol Syst Biol 4, 188 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landemaine T. et al. A six-gene signature predicting breast cancer lung metastasis. Cancer Res 68, 6092–9 (2008). [DOI] [PubMed] [Google Scholar]
- Culhane A. C. & Quackenbush J. Confounding effects in “A six-gene signature predicting breast cancer lung metastasis”. Cancer Res 69, 7480–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusek R. L., Godsel L. M. & Green K. J. Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J Dermatol Sci 45, 7–21 (2007). [DOI] [PubMed] [Google Scholar]
- Karczewska A., Nawrocki S., Breborowicz D., Filas V. & Mackiewicz A. Expression of interleukin-6, interleukin-6 receptor, and glycoprotein 130 correlates with good prognoses for patients with breast carcinoma. Cancer 88, 2061–71 (2000). [PubMed] [Google Scholar]
- Bajo-Graneras R. et al. Expression and potential role of apolipoprotein D on the death-survival balance of human colorectal cancer cells under oxidative stress conditions. Int J Colorectal Dis 28, 751–66 (2013). [DOI] [PubMed] [Google Scholar]
- Sarjeant J. M. et al. Apolipoprotein D inhibits platelet-derived growth factor-BB-induced vascular smooth muscle cell proliferated by preventing translocation of phosphorylated extracellular signal regulated kinase 1/2 to the nucleus. Arterioscler Thromb Vasc Biol 23, 2172–7 (2003). [DOI] [PubMed] [Google Scholar]
- Do Carmo S., Levros L. C. Jr. & Rassart E. Modulation of apolipoprotein D expression and translocation under specific stress conditions. Biochim Biophys Acta 1773, 954–69 (2007). [DOI] [PubMed] [Google Scholar]
- Soiland H. et al. Co-expression of estrogen receptor alpha and Apolipoprotein D in node positive operable breast cancer—possible relevance for survival and effects of adjuvant tamoxifen in postmenopausal patients. Acta Oncol 48, 514–21 (2009). [DOI] [PubMed] [Google Scholar]
- Mattick J. S. & Makunin I. V. Non-coding RNA. Hum Mol Genet 15 Spec No 1, R17–29 (2006). [DOI] [PubMed] [Google Scholar]
- Gupta R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Ren Z. & Sun P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem 113, 1868–74 (2012). [DOI] [PubMed] [Google Scholar]
- Sun M. et al. Downregulated long noncoding RNA MEG3 is associated with poor prognosis and promotes cell proliferation in gastric cancer. Tumour Biol 35, 1065–73 (2014). [DOI] [PubMed] [Google Scholar]
- Qin R. et al. Long non-coding RNA MEG3 inhibits the proliferation of cervical carcinoma cells through the induction of cell cycle arrest and apoptosis. Neoplasma 60, 486–92 (2013). [DOI] [PubMed] [Google Scholar]
- Ying L. et al. Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol Biosyst 9, 407–11 (2013). [DOI] [PubMed] [Google Scholar]
- Buffa F. M. et al. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res 71, 5635–45 (2011). [DOI] [PubMed] [Google Scholar]
- Sempere L. F. et al. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res 67, 11612–20 (2007). [DOI] [PubMed] [Google Scholar]
- Piovan C. et al. Oncosuppressive role of p53-induced miR-205 in triple negative breast cancer. Mol Oncol 6, 458–72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berber U. et al. miR-205 and miR-200c: predictive micro rnas for lymph node metastasis in triple negative breast cancer. J Breast Cancer 17, 143–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchalova K., Cizkova M., Cwiertka K., Trojanec R. & Hajduch M. Triple negative breast cancer--current status and prospective targeted treatment based on HER1 (EGFR), TOP2A and C-MYC gene assessment. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 153, 13–7 (2009). [DOI] [PubMed] [Google Scholar]
- Lan J. et al. TOP2A overexpression as a poor prognostic factor in patients with nasopharyngeal carcinoma. Tumour Biol 35, 179–87 (2014). [DOI] [PubMed] [Google Scholar]
- Podo F. et al. Triple-negative breast cancer: present challenges and new perspectives. Mol Oncol 4, 209–29 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ejeh F. et al. Meta-analysis of the global gene expression profile of triple-negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis 3, e100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S. L., Eklund A. C., Kohane I. S., Harris L. N. & Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 38, 1043–8 (2006). [DOI] [PubMed] [Google Scholar]
- Huang E. et al. Gene expression predictors of breast cancer outcomes. Lancet 361, 1590–6 (2003). [DOI] [PubMed] [Google Scholar]
- Szasz A. M. et al. The CIN4 chromosomal instability qPCR classifier defines tumor aneuploidy and stratifies outcome in grade 2 breast cancer. PLoS One 8, e56707 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery-Kiejda K. A., Morten B., Wong-Brown M. W., Mathe A. & Scott R. J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 35, 586–96 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.