

Abstract
The recent identification of IDH mutations in gliomas and several other cancers suggests that this pathway is involved in oncogenesis; however effector functions are complex and yet incompletely understood. To study the regulatory effects of IDH on hypoxia-inducible-factor 1-alpha (HIF1A), a driving force in hypoxia-initiated angiogenesis, we analyzed mRNA expression profiles of 288 glioma patients and show decreased expression of HIF1A targets on a single-gene and pathway level, strong inhibition of upstream regulators such as HIF1A and downstream biological functions such as angio- and vasculogenesis in IDH mutant tumors. Genotype/imaging phenotype correlation analysis with relative cerebral blood volume (rCBV) MRI – a robust and non-invasive estimate of tumor angiogenesis – in 73 treatment-naive patients with low-grade and anaplastic gliomas showed that a one-unit increase in rCBV corresponded to a two-third decrease in the odds for an IDH mutation and correctly predicted IDH mutation status in 88% of patients. Together, these findings (1) show that IDH mutation status is associated with a distinct angiogenesis transcriptome signature which is non-invasively predictable with rCBV imaging and (2) highlight the potential future of radiogenomics (i.e. the correlation between cancer imaging and genomic features) towards a more accurate diagnostic workup of brain tumors.
Somatic mutations of the isocitrate dehydrogenase (IDH) 1 and 2 genes are highly frequent in lower-grade (WHO grades II-III) gliomas1 and secondary glioblastomas2 and were found to be an early event in gliomagenesis3 with profound effects on the molecular and genetic route of oncogenic progression4,5 and on clinical outcome6,7,8. IDH mutations were also observed in several other cancers, including acute myeloid leukemia9, T-cell lymphoma10, chondrosarcoma, enchondroma11, and cholangiocarcinoma12. The identification of IDH mutations in multiple cancers suggests that this pathway is involved in oncogenesis.
Mechanistically, IDH mutations induce a neomorphic enzyme activity converting α-ketoglutarate (α-KG) to (R)-2-hydroxyglutarate ((R)-2HG)13. One model for how mutant IDH may contribute to oncogenesis and the one that has perhaps gained the most traction is transformation by α-KG antagonism48. (R)-2HG is structurally and chemically very similar to α-KG and it has been proposed that (R)-2HG competitively inhibits the function of α-KG-dependent dioxygenases, such as the JmjC domain-containing histone demethylases (JmjC-KDMs), which cause histone demethylation5,14,15, and the ten-eleven translocation (TET) family of DNA hydroxylases, which cause DNA demethylation5,16 and are considered a major mechanism in the tumorigenesis of IDH mutant gliomas4,17.
Beside these epigenetic modications, (R)-2HG has also been shown to regulate the activity of α-KG-dependent dioxygenases, specifically EGLN prolyl 4-hydroxylases, that are responsible for targeting hypoxia-inducible factor 1-alpha (HIF1A) – a driving force in tumorigenesis in general and angiogenesis in particular18 – for ubiquitination by the von Hippel-Lindau (VHL) tumor suppressor and proteasomal degradation19. However, unlike the general assumption that (R)-2HG would competitively inhibit the activity of EGLN prolyl 4-hydroxylases and thus would lead to increased levels of HIF1A, it has on the contrary been shown that (R)-2HG acts as an activator rather than an inhibitor of EGLN prolyl 4-hydroxylases, ultimately leading to decreased levels of HIF1A20,21,22. Although these results were initially surprising, it suggests that low levels of HIF1A suppress specific hallmarks of cancer required for aggressive behaviour (such as angiogenesis) in IDH mutant gliomas20,23 which fits with the indolent clinical course of these tumors6,7,8.
The aim of the present study was to study the regulatory effects of IDH-1/2 on HIF1A and related downstream signaling with mRNA expression and functional analysis and subsequent genotype / imaging phenotype correlation analysis to assess whether the observed molecular findings translate into distinct phenotypes, which can be detected non-invasively on MRI and thus would allow prediction of the IDH mutation status.
Results
IPA of upstream regulators revealed significant inhibition of HIF1A in IDH-1/2 mutant tumors (Table 1a and Supplementary Table 3) and correspondingly increased expression of EGLN prolyl 4-hydroxylases – specifically EGLN 1 and EGLN3 which are the principal prolyl hydroxylases for degradation of HIF1A24 – on a single-gene level (Supplementary Fig. 1). Moreover both IPA of upstream regulators and single-gene analysis (Table 1a and Supplementary Fig. 1) concordantly demonstrated significantly decreased expression of HIF1A-regulated pro-angiogenic key targets such as vascular endothelial growth factor A (VEGFA), angiopoietin-2 (ANGPT2) and platelet-derived growth factor A (PDGFA). We then studied the differential activity of HIF1A dependent pathways between IDH-1/2 mutant and wild-type tumors using a novel method for assessing the enrichment of gene sets in individual samples called gene set variation analysis (GSVA)25. This approach evaluates the enrichment of specific gene sets within the individual samples rather than on a group-level, which is a distinct advantage over gene set enrichment analysis. GSVA unveiled highly differential expression of HIF1A dependent gene sets, with inhibition of hypoxia-mediated signaling in general, and specifically vasculo- and angiogenesis in IDH-1/2 mutant tumors (Fig. 1; empirical Bayes moderated-t p-values for individual gene sets are provided in Supplementary Table 2). These findings were further supported by the IPA downstream effects analysis, which demonstrated a significant inhibition of HIF1A mediated pro-angiogenic biological functions such as development, migration and movement of endothelial cells, development of blood vessel, vasculogenesis, angiogenesis, and (neo)vascularization in IDH-1/2 mutant tumors (Table 1b and Supplementary Table 3).
Table 1. Ingenuity pathway analysis of upstream regulators (A) and downstream biological functions (B).
| (A) Upstream regulators in IDH-1/2 mutant tumors | ||||
|---|---|---|---|---|
| Upstream regulator | Molecule type | p value of overlap | Activation z score | Predicted activation state |
| Vegf | Group | 4.11E-30 | −7.287 | Inhibited |
| PDGF BB | Complex | 3.94E-36 | −5.17 | Inhibited |
| HIF1A | Transcription regulator | 6.54E-10 | −4.65 | Inhibited |
| VEGFA | Growth factor | 1.27E-10 | −4.306 | Inhibited |
| Pdgf | Complex | 2.94E-10 | −3.646 | Inhibited |
| ANGPT2 | Growth factor | 6.79E-09 | −3.361 | Inhibited |
| (B) Downstream biological functions in IDH-1/2 mutant tumors | ||||
| Biological function | p value of overlap | Activation z score | Predicted activation state | |
| Development of blood vessel | 7.87E-30 | −4.185 | Decreased | |
| Migration of endothelial cells | 3.26E-15 | −3.799 | Decreased | |
| Vasculogenesis | 8.89E-28 | −3.717 | Decreased | |
| Movement of endothelial cells | 2.66E-16 | −3.540 | Decreased | |
| Angiogenesis | 9.39E-28 | −3.556 | Decreased | |
| Neovascularization | 1.53E-10 | −2.451 | Decreased | |
| Vascularization | 6.85E-12 | −2.319 | Decreased | |
| Development of endothelial cells | 6.58E-12 | −2.020 | Decreased | |
Both concordantly demonstrate a significant decrease in key angiogenic regulators such as HIF1A, VEGFA, PDGF or ANGPT2 (A) and consequently angiogenic biological processes (B) in IDH-1/2 mutant tumors. Full results are available in the data supplement (Supplementary Table 3).
Figure 1. Gene set variation analysis (GSVA) of differentially activated gene sets identified inhibition of hypoxia, vasculo- and angiogenesis signaling pathways in IDH-1/2 mutant tumors and activation in IDH-1/2 wild-type tumors.

Rows represent samples of 288 patients with low-grade diffuse or anaplastic gliomas from The Cancer Genome Atlas (TCGA). Columns represent GSVA enrichment scores for the individual gene set.
To assess whether these differential genetic signatures between IDH-1/2 mutant and wild-type tumors translate into distinct phenotypes, which can be detected non-invasively, rCBV imaging – a robust and clinically meaningful estimate of tumor angiogenesis26,27,28 – was analyzed from a local dataset in 73 treatment-naive patients with low-grade diffuse and anaplastic gliomas. Using a semi-automated radiogenomic imaging approach (see Methods) we demonstrated that IDH-1/2 mutant and wild-type tumors are both associated with distinct imaging phenotypes, with IDH-1/2 mutant tumors clustering at a significantly lower rCBV (median of the 50st percentile, 1.09; interquartile range, 0.80–1.47) as compared to their wild-type counterpart (median of the 50st percentile, 2.08; interquartile range, 1.49–2.57), suggesting indeed decreased angiogenesis in IDH-1/2 mutant tumors as was predicted from the IPA downstream effects analysis of gene expression data (Fig. 2; representative cases are shown in Fig. 3). In detail, rCBV was significantly different between IDH-1/2 mutant and wild-type tumors for all evaluated percentiles, with the 90st percentile yielding the highest z-score (4.89; p < 0.01) (Table 2). To quantify the predictive value of rCBV for determining IDH-1/2 mutation status we analyzed the cumulative rCBV histogram distribution with cross-validated logistic-regression models. Thereby, the highest AUC was again obtained for the 90st percentile of the cumulative rCBV histogram distribution (92% (95% CI, 84–100%)) where a one-unit increase in rCBV corresponded to a decrease in the odds for presence of an IDH-1/2 mutation by 69% and correctly predicted IDH-1/2 mutation status in 88% of patients (positive predictive value (PPV): 89%, negative predictive value (NPV): 78% – detailed results are available in the Table 2).
Figure 2. Relative cerebral blood volume (rCBV) imaging heat-map of 73 treatment-naive patients with low-grade diffuse and anaplastic gliomas.
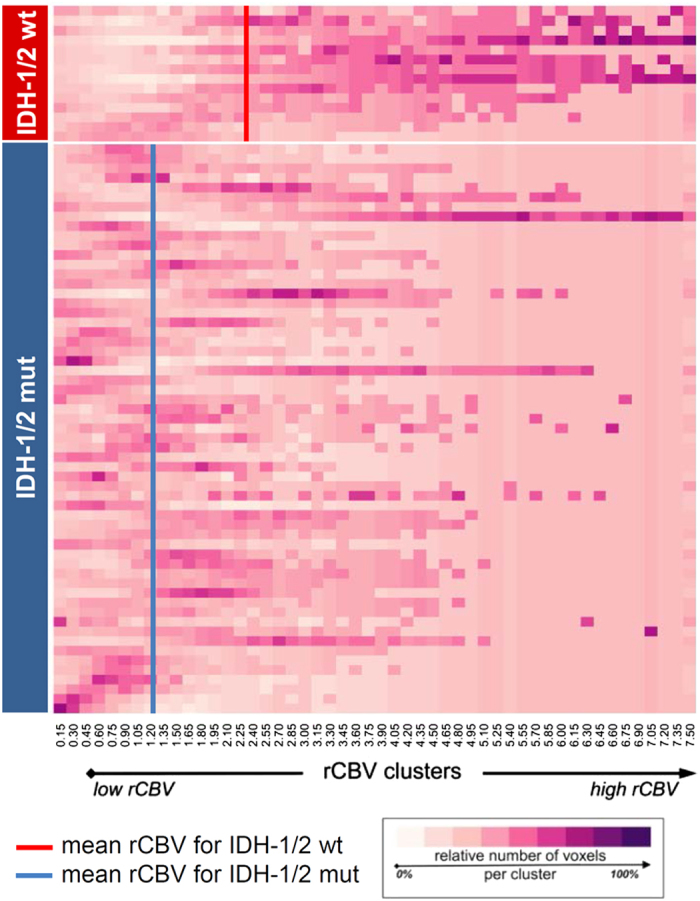
The color of the voxel corresponds to the relative number of rCBV-voxels in each cluster and goes from white (low frequency) to purple (high frequency). Overall, IDH-1/2 wild-type tumors (top of the heat-map) clustered at a significantly higher rCBV as compared to IDH-1/2 mutant tumors (bottom of the heat-map) (see Table 2 for statistical results).
Figure 3.

Pre-treatment MRI (FLAIR and corresponding rCBV images) of six representative patients with IDH-1/2 mutant (a–c) and wild-type (d–f) gliomas. Histogram analysis demonstrates the distribution of rCBV-voxels, with IDH-1/2 mutant tumors clustering at substantially lower values as compared to their wild-type counterpart.
Table 2. rCBV histogram analysis for differentiating IDH -1/2 mutant and wildtype tumors.
| Percentile | Mann-Whitney U test | Logistic regression | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| p-value | z-score | OR | (95% CI) | p-value | AUC | (95% CI) | Correctly classified | PPV | NPV | |
| 5 | 0.00 | 3.46 | 0.07 | (0.01–0.94) | 0.05 | 66.7% | (50.2%–83.2%) | 82.2% | 82.9% | 66.7% |
| 10 | 0.05 | 1.93 | 0.08 | (0.01–0.55) | 0.01 | 71.6% | (55.8%–87.3%) | 83.6% | 84.1% | 75.0% |
| 15 | 0.01 | 2.49 | 0.08 | (0.01–0.43) | 0.00 | 74.8% | (60.1%–89.6%) | 82.2% | 83.8% | 60.0% |
| 25 | 0.00 | 2.87 | 0.09 | (0.02–0.41) | 0.00 | 79.9% | (67.5%–92.3%) | 82.2% | 83.8% | 60.0% |
| 50 | 0.00 | 4.11 | 0.17 | (0.04–0.76) | 0.02 | 85.5% | (75.2%–95.8%) | 83.6% | 86.2% | 62.5% |
| 75 | 0.00 | 4.40 | 0.23 | (0.06–0.88) | 0.03 | 88.0% | (77.2%–98.9%) | 86.3% | 87.7% | 75.0% |
| 90 | 0.00 | 4.89 | 0.31 | (0.13–0.78) | 0.01 | 92.2% | (84.3%–100.0%) | 87.7% | 89.1% | 77.8% |
| 95 | 0.00 | 4.88 | 0.40 | (0.22–0.75) | 0.00 | 92.2% | (84.8%–99.7%) | 86.3% | 88.9% | 70.0% |
| 99 | 0.00 | 4.72 | 0.56 | (0.42–0.76) | 0.00 | 90.8% | (83.9%–97.7%) | 82.2% | 85.9% | 55.6% |
Abbreviations: AUC = Area under the curve; CI = confidence interval; NPV = negative predictive value; OR = Odds ratio; PPV = positive predictive value.
Discussion
The recent discovery that genes encoding IDH-1/2 are recurrently mutated in several cancers, which induce a neomorphic enzyme activity that results in the production of the oncometabolite (R)-2HG, unveiled a fascinating and complex biology behind the dysregulation and functional consequences of metabolites in cancers29. However, the resulting biological effects of IDH mutation and (R)-2HG accumulation remain controversial, exemplified in the case of HIF1A signaling, a major oncogenic pathway in malignant glioma. While some studies demonstrated that IDH mutation increases HIF1A levels 30,31, several others reported opposite results20,21,22: Koivunen et al. found EGLN activation in response to (R)-2HG accumulation, in turn leading to increased HIF1A degradation20. More recently, many HIF1A target genes were shown to be lower expressed in IDH mutant gliomas32. In line with the latter reports, the results from the present study show that IDH mutant tumors are characterized by an increased expression of EGLN prolyl 4-hydroxylases that mark HIF1A for polyubiquitylation and proteasomal degradation, leading to decreased HIF1A activation and downstream inhibition of hypoxia, vasculo- and angiogenesis related signaling. These signaling pathways are a prerequisite for aggressive tumor behavior which is underscored by the relative indolent course of low-grade and anaplastic gliomas carrying IDH mutations, as compared to their wild-type counterpart which usually carry a dismal prognosis similar to those of glioblastoma33. Most importantly, our semi-automatic radiogenomic imaging approach showed that these differential molecular signatures between IDH mutant and wild-type tumors translate into distinct phenotypes which are non-invasively predictable with rCBV imaging in a clinical setting. The prognostic value of rCBV imaging has already been shown before by Law et al.34, who reported that lower rCBV translates into significantly prolonged time to progression in 189 glioma patients. While these authors did not account for IDH, it seems reasonable to assume that this finding indeed reflects IDH status, given that in their study the proportion of tumors with high rCBV considerably increases with tumor grade, as does the percentage of IDH wild type tumors in general35.
Despite the straightforward results of the present study, we however have to acknowledge several limitations. First, differential mRNA expression and genotype/imaging phenotype analyses were performed in two independent cohorts (The Cancer Genome Atlas (TCGA) and local dataset), thus we were not able to directly define an mRNA expression correlate for the analyzed rCBV samples, and instead used the IDH mutation status as a connector between the TCGA and local dataset. Second, we were not able to determine whether decreased angiogenesis in IDH mutant tumors is solely a consequence of increased expression of EGLN prolyl 4-hydroxylases and subsequent decreased HIF1A activation or whether epigenetic modifications in IDH mutant tumors that result in reorganization of the methylome and gene expression may have also had implications on angiogenesis.
In conclusion (Fig. 4), these findings (1) show that IDH mutation status is associated with a distinct angiogenesis transcriptome signature which is non-invasively predictable with rCBV imaging and (2) highlight the potential future of radiogenomics (i.e. the correlation between cancer imaging and genomic features) towards a more accurate diagnostic workup of brain tumors.
Figure 4. Synopsis of how IDH mutation status is linked to angiogenesis and alteration in rCBV on DSC-MRI in human glioma based on this hypothesis generating study.

In brief, mutations in cancer-associated IDH acquire neoactivity producing 2-hydroxyglutarate (2HG). (R)-2HG potentiates EglN activity that mark the hypoxia inducible factor HIF1A for polyubiquitylation and proteasomal degradation, leading to decreased HIF1A activation in IDH mutant tumors, compared with their wild-type counterparts. This results in a distinct transcriptome signature induced by upregulation of hypoxia, vasculo- and angiogenesis related signaling pathways in IDH wild-type tumors, which is non-invasively detectable with rCBV imaging.
Methods
Analysis of differential mRNA expression (TCGA dataset)
RNA sequencing raw data (mapped to genes) and curated IDH-1/2 mutation data were downloaded from TCGA data portal on October 15th, 2014. In total, RNA sequencing and curated mutation data was available for 288 unique samples with low-grade diffuse (WHO °II) or anaplastic (WHO °III) gliomas (detailed sample information is provided in Supplementary Table 1) which were then analyzed using Bioconductor 3.036, a package implemented in R 3.1.2 (R Foundation for Statistical Computing, Vienna, Austria)37. First, normalization and differential gene expression analysis of RNA sequencing counts was performed using the edgeR package38, which assumes a negative binomial distribution of count data, filtering lowly expressed transcripts.
To study relative pathway activity on the level of individual samples, we used the GSVA package25. Using a nonparametric approach, GSVA transforms a gene-by-sample gene expression matrix into a gene set–by-sample gene set enrichment matrix, facilitating the identification of differentially activated genesets for each sample. GSVA was performed on 17 gene sets related to hypoxia, vasculo- and angiogenesis (a detailed listing is provided in Supplementary Table 2) identified through the Molecular Signatures Database (MSigDB - www.broadinstitute.org/gsea/msigdb)39 and literature research. Next, empirical Bayes moderated t statistics40 were calculated, and gene sets with FDR-q < 0.05 were considered to have significantly differential activity between samples with mutant and wild-type IDH-1/2 tumors. Of these gene sets, individual GSVA enrichment scores were plotted as a heatmap with the gplots package.
Furthermore, the differential upstream biological causes and probable downstream effects between mutant and wild-type IDH-1/2 tumors were analyzed with the proprietary Ingenuity Pathway Analysis (IPA, QIAGEN, Redwood City, CA, USA) using a fold change filter of |1.5| and FDR-q < 0.05. Briefly, the software calculates both an overlap p value (based on Fisher’s exact test) and an activation z score, which is based on the expression state of activating and inhibiting genes, for manually curated upstream regulators and downstream biological functions. Only results with both an FDR-q < 0.05 and a z score > |2| were considered significant.
Analysis of MRI data (local dataset)
The local ethics committee of the University of Heidelberg approved retrospective data evaluation, and the requirement for patient informed consent was waived (ethics approval number: S-320/2012). Data evaluation was performed in accordance with relevant guidelines and regulations. Pre-treatment MRI scans of all adult patients (age ≥18 years) with newly diagnosed, pathologically confirmed low-grade diffuse (WHO grade II) or anaplastic (WHO grade III) gliomas in the period of July 2009 and April 2015 were screened. Out of these, dynamic susceptibility contrast-enhanced (DSC) and conventional MRI was available for 73 individual patients (detailed sample information is provided in Supplementary Table 1), which were then selected for subsequent analysis.
Images were acquired in the routine clinical workup using a 3 Tesla MR system (Magnetom Verio/Trio TIM, Siemens Healthcare, Erlangen, Germany) with a 12-channel head-matrix coil. Acquisition of the DSC-MRI sequence was performed as described previously41. In brief, prior dynamic imaging, a 0.1 mmol/kg prebolus dose of gadoterate meglumine (Gd-DOTA, DOTAREM, Guerbet, France) was administered to diminish T1 effects that might result from agent extravasation. DSC-PWI was obtained with a T2*-weighted gradient-echo EPI sequence during the bolus injection of a standard dose (0.1 mmol/kg) of intravenous gadoterate meglumine. Twenty-six to 28 slices with a thickness of 5 mm were acquired with spectral fat suppression (TE = 36 ms, TR = 2220 ms, FA = 90°, field of view = 240 mm × 240 mm, image matrix = 128 × 128). In total, 50 to 75 dynamic measurements were performed. Fluid attenuated inversion recovery (FLAIR) images were acquired with the following parameters: TI = 2400 ms; TE = 85 ms; TR = 8500 ms; section thickness, 5 mm; interslice gap, 5%.
Post-processing of DSC-MRI and FLAIR data was performed with dedicated software (Olea Sphere v 2.3, Olea Medical, La Ciotat, France). First, a rigid-body registration was applied to the DSC-MRI data to correct for motion artifacts. An arterial-input function (AIF) was determined automatically using cluster analysis techniques42 and deconvolution of the AIF was performed using a time-insensitive block-circulant singular value decomposition (cSVD)43. Whole-brain relative CBV (rCBV) maps were generated by using an established tracer kinetic model applied to the first-pass data, with rCBV values computed pixel-by-pixel as the area under the concentration time curve (AUC) divided by the AUC of the AIF44. Mathematical correction of the contrast agent leakage from the intravascular to extracellular space was performed with the method suggested by Boxerman et al.45. Next, the FLAIR-hyperintense tumor was outlined according to a semi-automated region-growing segmentation method, which examines neighboring voxels of an initial seed point/voxel and determines whether the voxel neighbors should be added to the ROI. Automatic iteration of this process was performed on each slice of the FLAIR-images until the hyperintense FLAIR abnormality (excluding those resulting from obvious leukariosis) was included in the ROI. Automatic co-registration of rCBV maps with corresponding FLAIR images which included the previously segmented FLAIR ROIs was performed prior voxel-wise extraction of rCBV values from the segmented tumor. The extracted rCBV voxel values from individual patients were then distributed into 50 equally spaced clusters (width of 0.15) for rCBV-values in the range of 0.0 to 7.5. Finally, the relative number of rCBV-voxels in each cluster were plotted in R as a heatmap with the gplots package. Separate extraction and analysis of rCBV voxels from the contrast-enhancing tumor parts was not performed since this was only present in a small minority of patients included in the present study.
The rCBV histogram distribution was further analyzed to evaluate whether rCBV is different between patients IDH-1/2 mutant and wild-type tumors. We evaluated the significance of several percentiles (5th, 10th, 25th, 50st, 75th, 90st, 95th, 99th) from the rCBV histogram distribution with a Mann-Whitney U test. Furthermore, leave-one-out cross-validated logistic regression analysis was performed to assess the significance of each rCBV percentile for predicting IDH-1/2 mutation status (using a cut-off of the predicted probability of 0.5). P-values < 0.05 were considered significant.
IDH mutation status assessment (local dataset)
IDH-1/2 mutation screening was performed for all evaluated patients (n=73) with immunohistochemistry (IHC) for IDH1-R132H and DNA sequencing for IHC-negative cases. Tissue was provided by the Department of Neuropathology, Institute of Pathology, University of Heidelberg Medical Center, Germany in accordance with local ethical approval. All experiments were performed in accordance with relevant guidelines and regulations.
IHC of formalin-fixed paraffin-embedded tumor tissue for IDH1-R132H was performed as described previously46. In brief, sections cut to 3 μm were incubated and processed on a Ventana BenchMark XT® immunostainer (Ventana Medical Systems, Tucson, AZ, USA). Antibodies were anti-human IDH1-R132H (H09, Dianova, Hamburg, Germany) The Ventana staining procedure included pretreatment with cell conditioner 2 (pH 6) for 60 min, followed by incubation with primary antibody at 37 °C for 32 min. Incubation was followed by Ventana standard signal amplification, UltraWash, counterstaining with one drop of hematoxylin for 4 min and one drop of bluing reagent for 4 min. For visualization, ultraView™ Universal DAB Detection Kit (Ventana Medical Systems) was used. IDH1-R132H staining was evaluated as described previously47.
DNA sequencing for IHC-negative cases was performed as described previously1. In brief, DNA-extraction from paraffin-embedded tumor was performed tissue using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany). A fragment of 129-bp length spanning the catalytic domain of IDH1 including codon 132 was amplified using sense primer IDH1f CGGTCTTCAGAGAAGCCATT and antisense primer IDH1r GCAAAATCACATTATTGCCAAC, and a fragment of 150 bp length spanning the sequence encoding the catalytic domain of IDH2 including codon 172 was amplified using the sense primer IDH2f AGCCCATCATCTGCAAAAAC and antisense primer IDH2r CTAGGCGAGGAGCTCCAGT. Sequences were determined using a semiautomated sequencer (ABI 3100 Genetic Analyzer, Applied Biosystems, Foster City, CA) and the Sequence Pilot version 3.1 (JSI-Medisys, Kippenheim, Germany).
Additional Information
How to cite this article: Kickingereder, P. et al. IDH mutation status is associated with a distinct hypoxia/angiogenesis transcriptome signature which is non-invasively predictable with rCBV imaging in human glioma. Sci. Rep. 5, 16238; doi: 10.1038/srep16238 (2015).
Supplementary Material
Acknowledgments
The results published here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov. PK is a fellow of the Medical Faculty Heidelberg Postdoc-Program. FS is a fellow of the Medical Faculty Heidelberg Postdoc-Program.
Footnotes
Author Contributions P.K. designed the overall study, P.K. and B.W. performed analysis of the TCGA dataset, P.K. and A.R. performed analysis of the local MRI. dataset, F.S. and A.v.D. performed IDH mutation status assessment of the local dataset, P.K. performed statistical analysis, P.K. and B.W. wrote large parts of the manuscript, A.R., F.S., A.v.D., S.H., W.W. and M.B. contributed to the manuscript.
References
- Hartmann C. et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118, 469–474, doi: 10.1007/s00401-009-0561-9 (2009). [DOI] [PubMed] [Google Scholar]
- Nobusawa S., Watanabe T., Kleihues P. & Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 15, 6002–6007, doi: 10.1158/1078-0432.CCR-09-0715 (2009). [DOI] [PubMed] [Google Scholar]
- Lai A. et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol 29, 4482–4490, doi: 10.1200/JCO.2010.33.8715 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483, doi: 10.1038/nature10866 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30, doi: 10.1016/j.ccr.2010.12.014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C. et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120, 707–718, doi: 10.1007/s00401-010-0781-z (2010). [DOI] [PubMed] [Google Scholar]
- Wick W. et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27, 5874 - 5880 (2009). [DOI] [PubMed] [Google Scholar]
- Leu S. et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro Oncol 15, 469–479, doi: 10.1093/neuonc/nos317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis E. R. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361, 1058–1066, doi: 10.1056/NEJMoa0903840 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns R. A. et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 119, 1901–1903, doi: 10.1182/blood-2011-11-391748 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amary M. F. et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 224, 334–343, doi: 10.1002/path.2913 (2011). [DOI] [PubMed] [Google Scholar]
- Wang P. et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 32, 3091–3100, doi: 10.1038/onc.2012.315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744, doi: 10.1038/nature08617 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R. et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO reports 12, 463–469, doi: 10.1038/embor.2011.43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478, doi: 10.1038/nature10860 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa M. E. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567, doi: 10.1016/j.ccr.2010.11.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oermann E. K., Wu J., Guan K. L. & Xiong Y. Alterations of metabolic genes and metabolites in cancer. Semin Cell Dev Biol 23, 370–380, doi: 10.1016/j.semcdb.2012.01.013 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer N., Zasada C., Kempa S. & Cramer T. The growing complexity of HIF-1alpha’s role in tumorigenesis: DNA repair and beyond. Oncogene 32, 3569–3576, doi: 10.1038/onc.2012.510 (2013). [DOI] [PubMed] [Google Scholar]
- Maxwell P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275, doi: 10.1038/20459 (1999). [DOI] [PubMed] [Google Scholar]
- Koivunen P. et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488, doi: 10.1038/nature10898 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman J. A. et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625, doi: 10.1126/science.1231677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D., Ma S., Xiong Y. & Guan K. L. R-2-hydroxyglutarate as the key effector of IDH mutations promoting oncogenesis. Cancer Cell 23, 274–276, doi: 10.1016/j.ccr.2013.03.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A. & Harris A. L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491, 364–373, doi: 10.1038/nature11706 (2012). [DOI] [PubMed] [Google Scholar]
- del Peso L. et al. The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J Biol Chem 278, 48690–48695, doi: 10.1074/jbc.M308862200 (2003). [DOI] [PubMed] [Google Scholar]
- Hanzelmann S., Castelo R. & Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics 14, 7, doi: 10.1186/1471-2105-14-7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia A. C. Jr. et al. MR cerebral blood volume maps correlated with vascular endothelial growth factor expression and tumor grade in nonenhancing gliomas. AJNR Am J Neuroradiol 26, 777–783 (2005). [PMC free article] [PubMed] [Google Scholar]
- Ginat D. T., Mangla R., Yeaney G., Schaefer P. W. & Wang H. Correlation between dynamic contrast-enhanced perfusion MRI relative cerebral blood volume and vascular endothelial growth factor expression in meningiomas. Acad radiol 19, 986–990, doi: 10.1016/j.acra.2012.04.006 (2012). [DOI] [PubMed] [Google Scholar]
- Jain R. et al. Correlation of perfusion parameters with genes related to angiogenesis regulation in glioblastoma: a feasibility study. AJNR Am J Neuroradiol 33, 1343–1348, doi: 10.3174/ajnr.A2956 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns R. A. & Mak T. W. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer discovery 3, 730–741, doi: 10.1158/2159-8290.CD-13-0083 (2013). [DOI] [PubMed] [Google Scholar]
- Zhao S. et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324, 261–265, doi: 10.1126/science.1170944 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. et al. Glioma derived isocitrate dehydrogenase-2 mutations induced up-regulation of HIF-1alpha and beta-catenin signaling: possible impact on glioma cell metastasis and chemo-resistance. Int J Biochem Cell Biol. 44, 770–775, doi: 10.1016/j.biocel.2012.01.017 (2012). [DOI] [PubMed] [Google Scholar]
- Chesnelong C. et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol 16, 686–695, doi: 10.1093/neuonc/not243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N.et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 372, 2481-2498, doi: 10.1056/NEJMoa1402121 (2015). [DOI] [PMC free article] [PubMed]
- Law M. et al. Gliomas: predicting time to progression or survival with cerebral blood volume measurements at dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging. Radiology 247, 490–498, doi: 10.1148/radiol.2472070898 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H. et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773, doi: 10.1056/NEJMoa0808710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80, doi: 10.1186/gb-2004-5-10-r80 (2004). [DOI] [PMC free article] [PubMed]
- R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2014).
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Molec. Biol. 3, Article3, doi: 10.2202/1544-6115.1027 (2004). [DOI] [PubMed] [Google Scholar]
- Kickingereder P. et al. Relative cerebral blood volume is a potential predictive imaging biomarker of bevacizumab efficacy in recurrent glioblastoma. Neuro Oncol., 2015. Aug; 17(8): 1139–47. doi: 10.1093/neuonc/nov028. Epub 2015 Mar 9. (http://www.ncbi.nlm.nih.gov/pubmed/25754089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouridsen K., Christensen S., Gyldensted L. & Ostergaard L. Automatic selection of arterial input function using cluster analysis. Magn Reson Med. 55, 524–531, doi: 10.1002/mrm.20759 (2006). [DOI] [PubMed] [Google Scholar]
- Wu O. et al. Tracer arrival timing-insensitive technique for estimating flow in MR perfusion-weighted imaging using singular value decomposition with a block-circulant deconvolution matrix. Magn Reson Med. 50, 164–174, doi: 10.1002/mrm.10522 (2003). [DOI] [PubMed] [Google Scholar]
- Ostergaard L., Weisskoff R. M., Chesler D. A., Gyldensted C. & Rosen B. R. High resolution measurement of cerebral blood flow using intravascular tracer bolus passages. Part I: Mathematical approach and statistical analysis. Magn Reson Med. 36, 715–725 (1996). [DOI] [PubMed] [Google Scholar]
- Boxerman J., Schmainda K. & Weisskoff R. Relative cerebral blood volume maps corrected for contrast agent extravasation significantly correlate with glioma tumor grade, whereas uncorrected maps do not. AJNR Am J Neuroradiol 27, 859–867 (2006). [PMC free article] [PubMed] [Google Scholar]
- Sahm F. et al. Farewell to oligoastrocytoma: in situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol 128, 551–559, doi: 10.1007/s00401-014-1326-7 (2014). [DOI] [PubMed] [Google Scholar]
- Capper D. et al. Application of mutant IDH1 antibody to differentiate diffuse glioma from nonneoplastic central nervous system lesions and therapy-induced changes. Am J Surg Pathol. 34, 1199–1204, doi: 10.1097/PAS.0b013e3181e7740d (2010). [DOI] [PubMed] [Google Scholar]
- Losman J. A. & Kaelin W. G. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013 Apr 15; 27(8): 836–852. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3650222/. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.