

Abstract
Gastric Carcinoma is one of the most common cancers in the world. A large number of differentially expressed genes have been identified as being associated with gastric cancer progression, however, little is known about the underlying regulatory mechanisms. To address this problem, we developed a differential networking approach that is characterized by including a nascent methodology, differential coexpression analysis (DCEA), and two novel quantitative methods for differential regulation analysis. We first applied DCEA to a gene expression dataset of gastric normal mucosa, adenoma and carcinoma samples to identify gene interconnection changes during cancer progression, based on which we inferred normal, adenoma, and carcinoma-specific gene regulation networks by using linear regression model. It was observed that cancer genes and drug targets were enriched in each network. To investigate the dynamic changes of gene regulation during carcinogenesis, we then designed two quantitative methods to prioritize differentially regulated genes (DRGs) and gene pairs or links (DRLs) between adjacent stages. It was found that known cancer genes and drug targets are significantly higher ranked. The top 4% normal vs. adenoma DRGs (36 genes) and top 6% adenoma vs. carcinoma DRGs (56 genes) proved to be worthy of further investigation to explore their association with gastric cancer. Out of the 16 DRGs involved in two top-10 DRG lists of normal vs. adenoma and adenoma vs. carcinoma comparisons, 15 have been reported to be gastric cancer or cancer related. Based on our inferred differential networking information and known signaling pathways, we generated testable hypotheses on the roles of GATA6, ESRRG and their signaling pathways in gastric carcinogenesis. Compared with established approaches which build genome-scale GRNs, or sub-networks around differentially expressed genes, the present one proved to be better at enriching cancer genes and drug targets, and prioritizing disease-related genes on the dataset we considered. We propose this extendable differential networking framework as a promising way to gain insights into gene regulatory mechanisms underlying cancer progression and other phenotypic changes.
Keywords: Gastric cancer (GC), gene regulation network (GRN), differential network analysis, differential coexpression analysis (DCEA), differentially regulated genes (DRGs), carcinogenesis, gene regulatory mechanisms
Introduction
Gastric Carcinoma (GC) is one of the most common and lethal tumor in human, which is characterized by unlimited proliferation, a high degree of metastasis, and relatively poor prognosis [1]. During the last decade, quite a series of high-throughput studies, including genetic variation study [2-5], genome-wide association study (GWAS) [6], gene expression profiling and analysis [7-9], epigenetic variation study [10], and very recent integrative genomic analysis [11-14], have substantially contributed to the comprehensive understanding of GC. A large number of genes have been identified as being associated with gastric cancer progression, however, the underlying dysregulation mechanisms of gastric carcinogenesis remain poorly understood.
Cancer is believed to involve dysregulation of multiple fundamental cell processes such as proliferation, differentiation, migration, apoptosis, etc. Gene regulatory network (GRN) modelling has be widely used to obtain novel insights into the pathogenesis of complex diseases [15]. In recent years, numerous reverse engineering approaches have been developed to infer GRNs from gene expression data, including Boolean model [16], Bayesian model [17], relevance model [18], differential equation model [19], etc. In order to reduce the false positive rate, prior knowledge such as transcription factor (TF)-target regulatory relationships and miRNA-target regulatory relationships, could also be integrated into modelling framework, leading to reverse and forward integrated approaches [20,21]. These expression data-based methods theoretically enable cellular context-specific, or conditional GRNs to be constructed, which are the basis for exploring transcriptomic behavior under certain conditions. However, for the sake of the limitation of available data for specific subjects, in a quite long period of time, people had to infer conceptual GRNs based on gene expression profiles across various cellular contexts, for example, building a carcinoma GRN based on expression profiles of 60 National Cancer Institute cell lines (NCI60) in our previous work [21]. In this case, there was no chance to decipher differential regulatory relationships between tumor types, let alone the gene regulation dynamics of a certain tumor. It is only recently that the accumulation of transcriptome data and the improvement of computational strategies have allowed researchers to infer conditional GRNs [22-30], and greatly inspired the newly emerging theme “differential networking” [31-33]. It has been widely accepted that differential network analysis helps to identify specific regulatory relationships that are dysfunctional in a given disease state, which is essential for the elucidation of the pathophysiological processes.
Network analysis offers the possibility to comprehensively understand biology, however, it dramatically increases the computational complexity. In order to reduce the computational burden and generate testable hypotheses, the search space needs to be narrowed down. A commonly used strategy is to build a subnetwork around a given set of genes, for example, previously reported disease-related genes [22-24]. In this way, a large number of relations which are not directly associated with the subject of interest are pre-filtered out. However, this strategy highly depends on prior knowledge and may lose the chance to find out novel regulators or regulatory relationships that have not yet been documented as critical. Therefore, researchers recently set out to build subnetworks around differentially expressed genes [25-27], which proved to be capable of discovering context related genes.
In the transcriptome analysis domain, differential co-expression analysis (DCEA) is emerging as a prospective complement to differential expression analysis (DEA) [31]. Rather than calculating expression level changes of individual genes between two phenotypes (e.g., normal and cancer), DCEA looks at changes in gene co-expression patterns, and thus offers hints about the disrupted regulatory relationships or abnormal regulations specific to the phenotype of interest (in this case, cancer) [34-36]. Following this sense, we speculated that the subnetwork around differentially coexpressed genes (DCGs), instead of differentially expressed genes (DEGs), may lead to more insightful findings on regulatory mechanisms of phenotypic changes.
In this work, we first identified a set of differentially co-expressed genes or gene pairs from gene expression data of gastric normal mucosa, adenoma and carcinoma samples, by using differential coexpression analysis methods we developed previously [34,35], and then inferred stage-specific GRNs among these genes with reverse-forward integrated modelling method [21]. The topological properties and functional relevance of the three GRNs specific for normal, adenoma and carcinoma were analyzed, and the enrichment of known cancer genes and drug targets was observed in these stage-specific GRNs. We then implemented two quantitative methods to prioritize differentially regulated genes (DRGs) and links (DRLs) between adjacent stages (normal vs. adenoma; adenoma vs. carcinoma). It was found that known cancer genes and drug targets are significantly higher ranked, and most of the genes in two top-10 DRG lists have been reported to be GC related (~62%), or cancer relevant (~93%). Furthermore, the top 4% normal vs. adenoma DRGs (36 genes) and top 6% adenoma vs. carcinoma DRGs (56 genes) are worthy of further investigation to determine their association with gastric cancer. The differential networking information around these genes offers insightful clues to explore differential regulation mechanisms underlying gastric carcinogenesis. Finally, we compared our strategy with the previously established ones which model GRNs at the whole genome level, or model subnetworks around differentially expressed genes. Our strategy turns out to outperform the others in terms of enriching cancer genes and drug targets, and prioritizing disease-related genes. This study represents a complete framework for exploring differential regulation mechanisms underlying phenotypic changes, and the application to gastric cancer demonstrates its potential in cancer research.
Materials and methods
Gene expression datasets, cancer genes and drug targets
The normalized gene expression profile of gastric carcinoma GSE24375 [7] was downloaded from Gene Expression Omnibus (GEO) and all measurements were log2 transformed. The dataset involved eight patient-matched gastric normal mucosa, adenoma and carcinoma samples and two additional carcinoma samples. Probe sets with more than 20% missing values were discarded, while probe sets with less missing values were filled up with KNN method. After probe sets filtering, 18468 probe sets were mapped to Gene Symbols based on their platform annotations and 12658 unique genes were obtained.
We also downloaded the mRNA expression dataset of stomach adenocarcinoma (STAD) from The Cancer Genome Atlas (TCGA) Data Protal (https://tcga-data.nci.nih.gov/tcga/), which contains sequenced 28 matched tumor-normal pairs with IlluminaHiseq platform. After discarding genes with more than 20% missing values, we got 19211 RPKM normalized and log2 transformed unique genes.
A total of 486 cancer genes and 2093 drug targets were downloaded from Cancer Gene Census (http://cancer.sanger.ac.uk/cancergenome/projects/census/) [37] and DrugBank (http://www.drugbank.ca/) [38], respectively, out of which 347 cancer genes and 1654 drug targets were covered by the 12658 unique genes in the processed GSE24375 dataset.
Identification of DCGs, DCLs and DEGs
Differentially coexpressed genes (DCGs) and differentially coexpressed links (DCLs) were identified with differential coexpression analysis (DCEA) algorithms developed in our previous studies [34,35]. Specifically, DCGs with q-value less than 0.05 were picked out by using DCp method in DCGL package, while DCLs were identified by modified LFC model incorporated in DCe method in DCGL package [35]. Differentially expressed genes (DEGs) were selected by limma method with p-value less than 0.05 [39].
Characterization of network topology
For a network G(V,E) with N nodes and M edges, the degree of a node is defined as the number of connections or edges the node has to other nodes. If the network is directed, the in-degree is the number of inward edges, and the out-degree is the number of outward edges.
Betweenness [40] which measures how frequently a node locates on all shortest paths between two other nodes is defined as following:
Bet (v) = ∑ s ≠ t ≠ v ≠ Є V (σst(v)/σst) (Equation 1)
Where σst is the number of shortest paths linking vertex s and vertex t; σst(v) is the number of shortest paths that must pass vertex v. Nodes with high betweenness are bottle-necks for information transmission.
Closeness [40] is defined as the average shortest paths from vertex v to all other reachable vertices except itself. The measure of Closeness centrality of node v is defined as following:
Cls (v) = ∑ t Є V\v dist (v, t)/(N - 1) (Equation 2)
Where dist(v,t) means the shortest path distance between node v and node t. Closeness is the mean distance between a node and all other nodes in the network indicating its reachable ability.
Clustering Coefficient [40] is a count of the probability that any two nodes are linked together if they have a neighbor in common. Let vertex j and vertex t be two neighbors of vertex v and e(j,t) be the edge between them. The clustering coefficient of vertex v is defined as following:
CC (v) = (2|{e (j, t)})/kv(kv - 1) (Equation 3)
Where kv is the number of neighbors of node v; |{e (j, t)}| is the number of edges existing among kv neighbors.
Modelling of gene regulatory network
The multivariant linear regression model has been successfully utilized to infer gene regulatory relationships from gene expression data [20,21,41]. In this work, still by using the linear regression model, we constructed stage-specific gene regulatory networks based on forward predicted TF-target relationships and gene expression profiles. Using the protocol described in our previous work [20], a set of candidate TF-target regulatory relationships were generated based on the information from UCSC (http://genome.ucsc.edu/), including 214,607 regulatory relationships involving 215 human TFs and 16,835 targets. For a specific target g which has m regulators, its expression level (log2 expression value), Ag, is modeled by the following multivariate linear regression equation (Equation 4):
Ag = Atf1 * b1 + Atfi * bi + ... + Atfm * bm + interception + err (Equation 4)
Atfi is the mRNA levels (log2 expression value) of regulator i, while bi is the to-be-estimated regulation efficacy of regulator TFi which regulates gene g. Interception is a constant across all genes, and err, assumed to follow a normal distribution with a zero mean, represents the variation of gene g’s mRNA level that cannot be interpreted by its regulators. In brief, the expression level of a particular gene g is modeled by all of its candidate regulators, and the eventual regulators and their regulation efficacies are determined by the stepwise linear regression.
In the candidate TF-target relationships, a target can be regulated by more than 50 TFs (WG_targets, Figure S1), which is inconsistent with the common sense that a gene is rarely regulated by over 20 regulators [42]. Therefore, when we inferred gene regulatory networks at the whole genome level, we conducted a pre-filtering step to filter out those insignificant regulation relationships as we did previously [21]. However, since the differentially coexpressed or differentially expressed genes in GSE24375 are averagely regulated by only two TFs according to the candidate relationships, with max number of 15 (Figure S1), the pre-filtering step was ignored when modelling from DCGs or DEGs.
Prioritization of differentially regulated genes and links
We first proposed a quantitative metric (Equation 6) to measure the differential regulation of a gene across conditions. For a specific gene i (TF or target), which has N neighbors in condition A and M neighbors in condition B, the regulation efficacy between gene ii and its neighbors calculated by linear regression model in two conditions are X=(Xi1,...,Xik,....Xin), Y=(Yi1,...,Yik,....Yim), the differential regulation (DR) of gene i is defined as following:
DRi = √(∑j = 1 n (Xij - Yij)2/n) (Equation 5)
Xik and Yik are regulation efficacies between gene i and k in GRN A and B, respectively. Since gene ii could have different neighbor sets in two GRNs, when a neighbor gene exists only in one GRN, we assume its counterpart regulation efficacy in another GRN to be zero. DR measure captures the average regulation changes of a gene between two GRNs.
We then implemented modified LFC model [35] to screen gene pairs which display significant changes in regulation efficacy between two GRNs, say, differentially regulated gene pairs or links (DCLs). Similar to our previous work [35], for gene pairs (links) which have both positive or both negative regulation efficacy in two GRNs, we categorized them into bins according to their maximum regulatory efficiency, and within each bin, we selected 10% links with highest log fold changes to fit a curve y=a+(b/x), in which y is the log ratio of regulation efficacy and x is the maximum regulation efficacy. Links lying above the fitted curve were regarded as differentially regulated links (DRLs). For links which have positive regulation efficacy in one GRN and negative regulation efficacy in another GRN, we exchanged x and y to fit y=a+(b/x) curve, and picked out the links lying above the curve as DRLs.
Results
Construction of stage-specific gene regulatory networks
Gene expression profile of gastric cancer progression was downloaded from the Gene Expression Omnibus (GEO), where eight patient-matched gastric normal mucosa, adenomas and carcinomas, and two additional carcinomas were examined (GSE24375) [7]. We first applied DCGL package [35] to extract differentially coexpressed genes (DCGs) and differentially coexpressed gene pairs, or links (DCLs) between adjacent stages, resulting in a total of 309 DCGs and 18407 DCLs for normal vs. adenoma, 237 DCGs and 22719 DCLs for adenoma vs. carcinoma. All DCGs and the gene pairs in the DCLs which involved at least one DCG were included in the following network construction procedure. In total, two gene sets including 1793 and 2066 genes were obtained, corresponding to normal vs. adenoma and adenoma vs. carcinoma, respectively. According to the philosophy of differential coexpression analysis (DCEA), these genes were potentially relevant to differential regulation (DR) during phenotypic changes [31,34,36], and thus named as DR-relevant genes for short. The two DR-relevant gene sets were merged into a union set containing a total of 2524 genes. Based on the stage-specific expression data of the union gene set, we separately built three stage-specific GRNs with comparable sizes, corresponding to normal, adenoma and carcinoma, by using stepwise linear regression method [20,21]. It should be noted that due to the usage of DCEA, the regulatory relationships which keep stable during cancer progression tend to be ignored by our model, that is, the resultant network would enrich differential regulatory relationships. The network statistics are included in Table 1. Out of 28 TFs in the starting 2524 DR-relevant genes, 22 are retained in all the three GRNs, and the other six do not exist in any of the three. We propose these 22 TFs to be relevant to differential regulation underlying carcinogenesis, while the other six probably do not have significant regulatory relationships with their targets during cancer progression. It is interesting that the adenoma network has the most links (1173) or regulatory relationships among the three, about 33.9% more than the normal network (876), and the carcinoma network has the least links (816) (Table 1). The detailed topological comparison of the three networks will be described in the next section.
Table 1.
Cancer genes and drug targets enrichment of GRNs
| Subtypes | Stage-specific GRNs | No. of genes | No. of links | No. of cancer genes (p-value) | No. of drug targets (p-value) |
|---|---|---|---|---|---|
| DCG_GRNs | Normal | 521 | 878 | 26 (0.0034) | 84 (0.039) |
| Adenoma | 766 | 1173 | 32 (0.0159) | 133 (0.0005) | |
| Carcinoma | 575 | 816 | 29 (0.0015) | 106 (0.0002) | |
| DEG_GRNs | Normal | 621 | 1087 | 28 (0.001) | 90 (0.307) |
| Adenoma | 706 | 1184 | 26 (0.12) | 119 (0.0098) | |
| Carcinoma | 604 | 955 | 25 (0.039) | 95 (0.058) | |
| WG_GRNs | Normal | 3344 | 4063 | 136 (0.43) | 456 (0.256) |
| Adenoma | 4072 | 5498 | 149 (0.489) | 558 (0.142) | |
| Carcinoma | 4099 | 5428 | 156 (0.92) | 573 (0.037) |
The enrichment significance (p-value) was calculated by Fisher’s Exact Test. Three types of GRNs, DCG-GRNs, DEG-GRNs and WG-GRNs, were included in this table. The latter two, DEG-GRNs and WG-GRNs, were described in the section of “Method comparison”.
To globally understand the functional relevance of the three stage-specific networks, we performed functional enrichment analysis of the genes in each network in Gene Ontology (GO) by using DAVID [43]. It was found that the terms of “positive regulation of cell differentiation”, “positive regulation of apoptosis”, “positive regulation of cell death” and “negative regulation of cell migration” were exclusively enriched in normal GRN, the terms of “negative regulation of protein ubiquitination” and “negative regulation of ubiquitin-protein ligase activity during mitotic cell cycle” were exclusively enriched in adenoma GRN, while the terms of “negative regulation of apoptosis”, “growth factor activity” and “negative regulation of cell death” were exclusively enriched in carcinoma GRN. These observations are consistent with our basic understanding of cancer progression. Furthermore, we estimated the enrichment of known cancer genes and drug targets in the three networks by Fisher’s Exact Test. As shown in Table 1, the three DCG-GRNs (normal, adenoma and carcinoma) were all enriched with known cancer genes and therapeutic targets. It is suggested that our stage-specific GRNs have the potential to highlight crucial cancer-related regulation relationships, thus proving the rationality of our modelling strategy.
Global topological comparison of stage-specific grns
In order to investigate the dynamic changes of gene regulation in gastric cancer progression, we first assessed the topological differences between stage-specific GRNs. In-degree (In-Deg), out-degree (Out-Deg), betweenness (Bet), clustering coefficient (CC) and closeness (Cls) of each gene in three networks and those of three networks were calculated, and their differences between adjacent stages were tested by Wilcoxon rank-sum test (Table 2). For a certain node in a network, Deg reflects its connectedness, with In-Deg and Out-Deg corresponding to inward and outward links respectively; Bet represents its bridging character in connecting two other nodes; CC measures the closeness of its neighbors while Cls measures its closeness to all other nodes in the network. Generally speaking, these parameters describe the centrality of a node from diverse angles, and their counterparts for a network reflect the network compactness from both global and local perspectives [44].
Table 2.
Topological comparison of adjacent stage-specific GRNs
| Stages | Out-Deg | In-Deg | Bet | CC | Cls |
|---|---|---|---|---|---|
| Normal_GRN* | 39.9 | 1.68 | 0.83 | 0.036 | 3.70E-06 |
| Normal vs. Adenoma (p-value) | 0.638 | 0.0167 | 0.72 | 2.87E-08 | < 2.2e-16 |
| Adenoma_GRN* | 53.3 | 1.53 | 0.78 | 0.163 | 1.70E-06 |
| Adenoma vs. Carcinoma (p-value) | 0.24 | 0.24 | 0.38 | 0.033 | < 2.2e-16 |
| Cancer_GRN* | 37.09 | 1.42 | 0.709 | 0.1428 | 3.04E-06 |
Means the mean value of a topological measure of a GRN.
Topological difference significance (p-value) was calculated by Wilcoxon rank-sum test.
Since the network In-Deg, CC and Cls significantly change in both comparisons, normal vs. adenoma and adenoma vs. carcinoma, we conclude that the compactness of gene regulation significantly alters as gastric cancer progresses. Noticing that the adenoma network has the highest CC value, the lowest Cls, the highest Out-Deg, and the most regulatory relationships (1173) among the three networks (Table 1), we propose that adenoma seems to be an intermediate stage with a more compact network topology compared with both normal and carcinoma. This is at least partly resulted from quite a batch of transient regulatory relationships which occur specifically at the stage of adenoma. These transient regulations enable the GRN to achieve more efficiency in spreading information among nodes, which probably contribute to the promotion of cellular proliferation in the early stage of carcinogenesis.
Since the network Out-Deg didn’t display any significant difference for both comparisons (Table 2), we turned to check the Out-Deg values of every TFs in the networks. As shown in Figure 1A, the Out-Deg values of the 22 TFs show similar patterns across the three GRNs. We furthermore defined a measure, targets diversity of a regulator (TDR), to examine how the TF targets changed between adjacent GRNs. For TFi, the TDR is as following (Equation 6):
Figure 1.
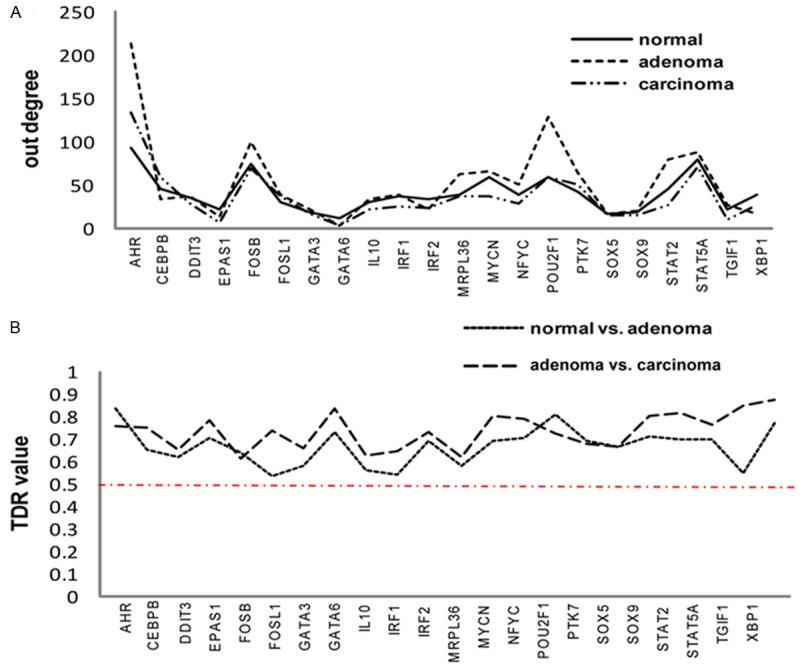
Distribution of Out-degree and TDR values of TFs in three stage-specific GRNs. A. Out-degree distribution of 22 TFs involved in three stage-specific GRNs. B. Distribution of TDR values of 22 TFs in normal vs. adenoma and adenoma vs. carcinoma comparisons.
TDR (i) = 1 - ∩ (tg1, tg2)/∪ (tg1, tg2) (Equation 6)
Where tg1 and tg2 are targets of TFi in GRN1 and GRN2, respectively. The TDR value reaches its maximum value, 1, when gene set tg1 and gene set tg2 do not overlap at all. While in this case, the Out-Deg of TFi could still be stable across GRNs, which would blur the distinction of TFi between adjacent stages in terms of its regulation functions. According to Figure 1B, more than 50% targets regulated by a certain TF change between adjacent GRNs, although the Out-Deg of the TF keeps stable across stages, which is consistent with previous report [45]. This also coincides with our inference that the 22 TFs involved in our GRNs would be relevant to differential regulation underlying gastric carcinogenesis.
Classification of target genes and regulatory relationships in terms of regulation change between stages
As found above, the regulators involved in the three stage-specific GRNs are the same while the targets regulated by a certain TF remarkably change between adjacent GRNs. We therefore examined the change of regulation in detail, both qualitatively and quantitatively. For a certain comparison, normal vs. adenoma or adenoma vs. carcinoma, the targets and regulatory relationships (links) in each network could be classified into “overlapped” and “stage-specific”. The numbers of “overlapped” targets (Figure 2A) or links (Figure 2B), and “stage-specific” targets (Figure 2A) or links (Figure 2B), are shown as Venn diagrams. In this step, the link weight, or regulation efficacy, was not taken into consideration.
Figure 2.
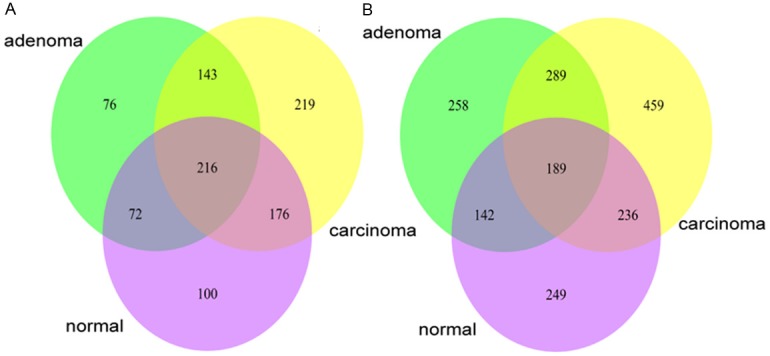
Venn diagrams for targets (A) and regulatory relationships (B) involved in three stage-specific GRNs. (A) illustrates the target numbers. (B) illustrates the number of gene regulatory relationships (links).
Still for a certain comparison, we further classified the overlapped targets in Figure 2A into “common” group and “varied” group (Table 3). Targets in “common” group are regulated by identical TFs in two GRNs, while targets in “varied” group are regulated by diverse TFs. Meanwhile, the overlapped links were also grouped as “common” and “varied”. If the change of the regulation efficacy of a link is equal to or smaller than the average value, it is “common”, otherwise it is “varied”. Since targets and links in “stage-specific” group and “varied” group involve regulation changes more than those in “common” group, they were combined to be a “differential” group as shown in Table 3. The ”differential” targets or links represent over 80% of total targets or links in two comparisons, indicating that our stage-specific GRNs are indeed enriched with differential regulatory relationships as expected.
Table 3.
Classification of targets and links contained in two adjacent GRNs
| Comparison | Groups | No. of targets (%) | No. of links (%) |
|---|---|---|---|
| Normal vs. Adenoma | Common | 132 (15.2%) | 285 (18.1%) |
| Differential (varied and stage-specific) | 770 (84.8%) | 1228 (81.9%) | |
| Adenoma vs. Carcinoma | Common | 145 (15.7%) | 241 (15.4%) |
| Differential (varied and stage-specific) | 781 (84.3%) | 1323 (84.5%) |
“Differential” means targets or links with substantial changes between two stage-specific GRNs; “common” means targets or links with only slight or no changes between two stage-specific GRNs.
In order to evaluate the power of our grouping strategy, we then analyzed the enrichment of cancer genes and drug targets in four groups of targets, and found that two “differential” target groups were enriched with cancer genes and drug targets, whereas two “common” target groups were not enriched with cancer genes or drug targets (Table 4, Table S1). It demonstrates that by combining DCEA strategy and linear regression modelling method, we efficiently narrowed down the search space and obtained stage-specific networks enriching subject relevant genes.
Table 4.
Enrichment analysis of cancer genes and drug targets in common and differential targets
| Comparison | Groups | No. of targets | No. of Control Genes | No. of cancer genes (p-value) | No. of drug targets (p-value) |
|---|---|---|---|---|---|
| Normal vs. Adenoma | Common | 132 | 12526 (12658-132) | 4 (0.785) | 26 (0.027) |
| Differential | 770 | 11888 (12658-770) | 33 (0.012) | 134 (0.0003) | |
| Adenoma vs. Carcinoma | Common | 145 | 12513 (12658-145) | 6 (0.297) | 25 (0.137) |
| Differential | 781 | 11877 (12658-781) | 32 (0.023) | 148 (1.67e-6) |
The significance (p-value) of the enrichment of cancer genes or drug targets in a targets group was calculated by Fisher’s Exact Test. The 12658 genes assayed by Affymetrix Microarray GPL10982 were taken as the respondents population. The calculation of the p values of every “common” or “differential” groups were displayed in Table S1.
Prioritization of differentially regulated genes and links
A key issue in differential regulation analysis is how to quantitatively analyze dynamic changes of gene regulation. To address this problem, we developed two methods to measure the differential regulation of a specific gene or a gene pair across GRNs, based on which we could prioritize differentially regulated genes (DRGs) and differentially regulated gene pairs or links (DRLs). The differential regulation (DR) of a gene is defined with Equation 5 in Materials and Methods, which captures the average regulation change between a gene and its GRN neighbors during phenotypic changes. In this way, genes involved in two adjacent GRNs could be ranked by their DRs in a descending order. Additionally, differentially regulated links (DRLs) between adjacent GRNs could be obtained by utilizing an modified LFC model [35], and ranked by their absolute changes of regulation efficacy in a descending order. It is our assumption that genes or gene pairs with higher ranks in the DRG or DRL lists play more important roles in gastric cancer progression.
To estimate the power of DR ranking, we carried out perturbation tests in which the gene order of DRG lists was randomly perturbed for 5000 times. Cancer genes or drug targets were then separately mapped to the 5000 random lists, forming an empirical null distribution of rank sums. It was found that cancer genes and drug targets were significantly higher ranked in normal vs. adenoma DRG list compared with those ranked in random lists, with p values of 0.026 and 0.023, respectively. For adenoma vs. carcinoma, p values of cancer genes and drug targets were 0.06 and 0.03. These results verified the effectiveness of DR measure in prioritizing disease-related genes. Coinciding with our basic understanding that transcription factors (TFs) play crucial roles in the proliferation and differentiation of cells, TFs were overrepresented at the extreme top of two DRGs lists with p values of 1.88e-14 for normal vs. adenoma and 3.85e-13 for adenoma vs. carcinoma.
In order to further test the DR measure in predicting disease genes, we calculated the percentage of known cancer genes in top x% of DRG list (x=1, 2, 3,…., 100). According to Figure 3, top 4% DRGs for normal vs. adenoma and top 6% DRGs for adenoma vs. carcinoma contained the highest percentage of cancer genes. We then randomly selected 4% and 6% of genes from two DRG lists and calculated the percentage of cancer genes in the random set for 5000 times to generate empirical null distributions for two DRG lists. For normal vs. adenoma DRGs, the percentage of cancer genes in the top 4% DRGs is 11%, while the average percentage of cancer genes in random gene sets is about 4%. For adenoma vs. carcinoma DRGs, the percentage of cancer genes in the top 6% DRGs is 5%, while the average percentage of cancer genes in random gene sets is 2%. The statistical significances (p values) of the top 4% normal vs. adenoma DRGs and top 6% adenoma vs. carcinoma DRGs in prioritizing cancer genes are 0.012 and 0.067, respectively. After doing a literature review in PubMed, we found that more than 50% genes contained in top 4% normal vs. adenoma DRGs and top 6% adenoma vs. carcinoma DRGs were reported to be GC related (Table S2). We therefore propose that our top 4%, 36 DRGs for normal vs. adenoma and top 6%, 56 DRGs for adenoma vs. carcinoma are worthy of further investigation to determine their association with gastric cancer. It is noted that the top 3% genes from adenoma vs carcinoma have no cancer genes. We speculate they probably include promising novel cancer genes relevant to gastric cancer. Compared to the transition from normal to cancer, or normal to adenoma, the transition from adenoma to carcinoma have been studied less frequently, which might be a reason that there are no cancer genes in the top 3% genes from adenoma vs carcinoma.
Figure 3.
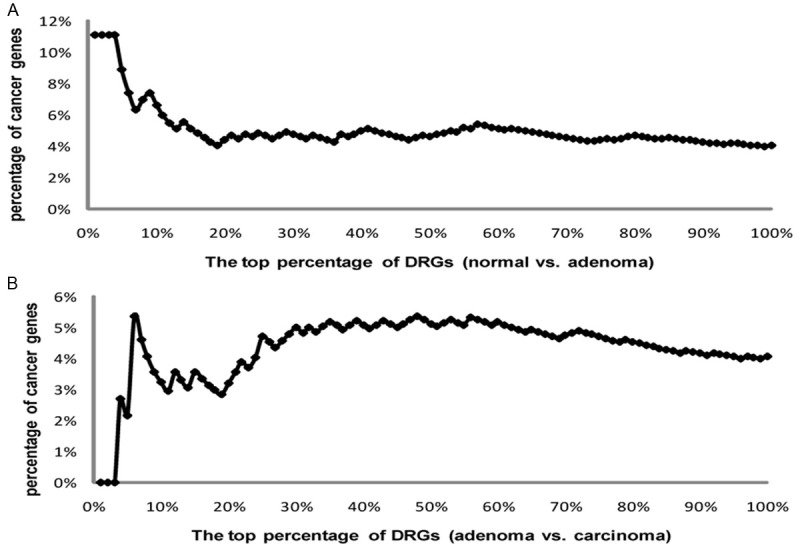
The distribution of the percentages of cancer genes in two DRG lists. A. The percentages of cancer genes in normal vs. adenoma DRG list. B. The percentages of cancer genes in adenoma vs. carcinoma DRG list.
We also examined the performance of DRL ranking by checking the rank of cancer genes and drug targets. When a gene was involved in more than one DRLs, we took the highest rank as the unique one of the gene. Similarly, perturbation tests indicate that cancer genes and drug targets are significantly higher ranked in normal vs. adenoma DRG list compared with those ranked in perturbed random DRL lists, with p values of 0.02 and 0.07. The p values of cancer genes and drug targets in adenoma vs. carcinoma DRL list are 0.08 and 0.06. Although these p values are greater than those for DRG lists, and three of them are even not significant at the 0.05 level, the DRL rank could still be useful in prioritizing regulation relationships around a certain DRG.
Table 5 lists the top ten genes in two DRG lists corresponding to normal vs. adenoma and adenoma vs. carcinoma. Among the 16 genes in Table 5, ten (ESRRG [46], LIMS1 [47,48], CEBPB [49], AHR [50], POU2F1 [51], TGIF1 [52], GATA3 [53], GATA6 [54], SOX9 [55], HEPH [56]) have been reported to be associated with gastric carcinoma (GC); another five genes (IRF2 [57], RGS3 [58], MRPL36 [59], TRIB1 [60] and FOSB [61] are cancer related, all of which are transcriptionally regulated by known GC related genes according to FANTOM database [62], including IRF1, NFKB1, MYC and SP1; the left one gene (RIC8B) is probably GC related since it was transcriptionally regulated by GC genes in our networks, such as STAT5A. Additionally, among the 16 top ranked genes, ESRRG [46], IRF2 [57] and CEBPB [49] are known cancer drug targets in CancerResource database [63], and they might be promising targets for gastric cancer therapy.
Table 5.
The top ten ranked genes in DRG lists and DRL lists
| DRG lists | DRL lists | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Normal vs. Adenoma | Adenoma vs. Carcinoma | Normal vs. Adenoma | Adenoma vs. Carcinoma | ||||||
|
| |||||||||
| Genes | Rank | Genes | Rank | TF | Target | Rank | TF | Target | Rank |
| LIMS1 | 1 | ESRRG | 1 | GATA3 | LIMS1 | 1 | IRF2 | ESRRG | 1 |
| FOSB | 2 | LIMS1 | 2 | SOX9 | LIMS1 | 2 | CEBPB | ESRRG | 2 |
| MRPL36 | 3 | IRF2 | 3 | FOSB | RIC8B | 3 | FOSB | ESRRG | 3 |
| GATA3 | 4 | RGS3 | 4 | MRPL36 | MME | 4 | POU2F1 | ESRRG | 4 |
| RGS3 | 5 | FOSB | 5 | MRPL36 | HEPH | 5 | CEBPB | RGS3 | 5 |
| TRIB1 | 6 | CEBPB | 6 | FOSB | PDGFRB | 6 | IRF1 | ESRRG | 6 |
| GATA6 | 7 | TRIB1 | 7 | GATA3 | FABP2 | 7 | CEBPB | DMKN | 7 |
| SOX9 | 8 | AHR | 8 | FOSB | ID1 | 8 | FOSB | ID1 | 8 |
| HEPH | 9 | POU2F1 | 9 | TGIF1 | PFN1 | 9 | STAT2 | ID1 | 9 |
| RIC8B | 10 | TGIF1 | 10 | STAT5A | RGS3 | 10 | TGIF1 | NAV2 | 10 |
The genes are sorted by the DR values. Genes in bold refer to GC-related genes; genes in italic refer to cancer-related genes.
Table 5 also lists the top ten ranked gene pairs in two DRLs lists corresponding to normal vs. adenoma and adenoma vs. carcinoma in comparison. Among the 23 genes in the two top ten ranked DRLs (Table 5), 12 are GC related genes (ESRRG [46], LIMS1 [47,48], CEBPB [49], POU2F1 [51], TGIF1 [52], GATA3 [53], SOX9 [55], HEPH [56], IRF1 [64], ID1 [65], PDGFRB [66] , STAT5A [67]); 8 are cancer related genes (IRF2 [57], RGS3 [58], MRPL36 [59], FOSB [61], MME [68], STAT2 [69], FABP2 [70], PFN1 [71]); the left 3 are probably GC related genes (RIC8B, NAV2, DMKN) since they are regulated by GC genes in our GRNs as seen in Table 5. Additionally, ID1, PDGFRB, STAT5A, STAT2, MME and IRF1 are known cancer drug targets in CancerResource database [63].
We then focused on the top ranked DRGs and their surrounding top ranked DRLs to generate hypotheses on the regulation mechanisms of gastric carcinogenesis. For example, GATA6, one of the top-10 DRGs in normal vs. adenoma comparison, has been reported to play a key role in controlling cell apoptosis and cell cycle of gastric cancer, while the dynamic mechanisms of gene regulation during cancer progression remains unclear [54]. In our differential networking analysis, GATA6 forms DRLs with LIMS1 and FMOD in normal vs. adenoma, and with PTTG1 in adenoma vs. carcinoma (Figure 4A). As shown in Figure 4A, the negative regulation of LIMS1 by GATA6 decreased sharply from normal to adenoma, and was even lost in carcinoma. LIMS1 was reported to promote the tumorigenesis of gastric tumor cells by physically interacting with ILK [48] and inhibit apoptosis in fibrosarcoma cell through the suppression of BIM [72]. The decrease of negative regulation of LIMS1 by GATA6 in adenoma and carcinoma stage may lead to the over-expression of LIMS1, which further induces tumorigenesis and inhibits apoptosis as plotted in Figure 4B. The over-expression of LIMS1 and down-expressin of GATA6 in carcinoma stage was verified by data from TCGA, as well as the present GSE24375 (Figure S2). Similarly, the negative regulation of FMOD by GATA6 disappeared from normal to adenoma (Figure 4A). FMOD is not a known GC gene, but it was reported to be up-regulated in chronic lymphocytic leukaemia (CLL) cells and the silencing of FMOD resulted in the apoptosis of CLL cells [73]. The loss of negative regulation of FMOD by GATA6 in adenoma and carcinoma stage may cause the over-expression of FMOD, which inhibits apoptosis and thus promotes tumorigenesis (Figure 4B). The over-expression of FMOD in adenoma and carcinoma stage was observed in both GSE24375 and TCGA dataset (Figure S2). As for PTTG1, the positive regulation of PTTG1 by GATA6 was reversed to be negative from normal to adenoma, while the change was slight, and then disappeared in carcinoma stage; however, the expression level of PTTG1 was found to continuously rise in GSE24375 from normal to adenoma and to carcinoma, which is consistent with the observation in TCGA data (Figure S2). PTTG1, promoting cancer by inhibiting p53, was reported to be over-expressed in gastric carcinoma and correlated to lymph node metastasis of gastric carcinoma [74]. It seems that GATA6 exerts its PTTG1-mediated cancerigenic function mainly at the late stage (Figure 4B). In addition, we noticed that the positive regulation of CAPN1 by GATA6 in normal stage disappeared in adenoma and reappeared in carcinoma (Figure 4A). Although CAPN1 does not fall in the top 4% or top 6% DRGs, and the link from GATA6 to CAPN1 is not a DRL either, CAPN1 was reported to promote the cell apoptosis through the activation of P53 (KEGG pathway ID: hsa04210). We therefore proposed that the loss of positive regulation of CAPN1 by GATA6 may lead to the inhibition of P53 and thus the suppression of apoptosis, which may induce the progression of normal to adenoma (Figure 4B). In this sense, decreased expression of CAPN1 gene might be a potential early stage biomarker for gastric cancer. The down-expression of CAPN1 in adenoma and carcinoma stage was observed in both GSE24375 and TCGA data (Figure S2). Recently, a whole genome sequencing study of gastric carcinoma found a non-synonymous mutation (YH323Y deletion) located in the transcriptional activation domain (residue 147-381) of GATA6 protein [12]. To decipher the potential functional impact of the YH323Y deletion, we generated a three-dimensional protein structure model of GATA6 by using the software FR-t5-M [75]. As shown in Figure 4C, although the normal (left) and mutation (right) GATA6 proteins have similar topology, the YH323H deletion remarkably affects the local region so that a pair of antiparallel beta sheets disappear in the mutated GATA6 protein. We speculate that the YH323H deletion might affect the function of GATA6 through altering its conformation in the DNA binding domain. The temporal molecular events underlying gastric cancer progression involving GATA6 and its down-stream signaling pathway are worth further experimental exploration.
Figure 4.
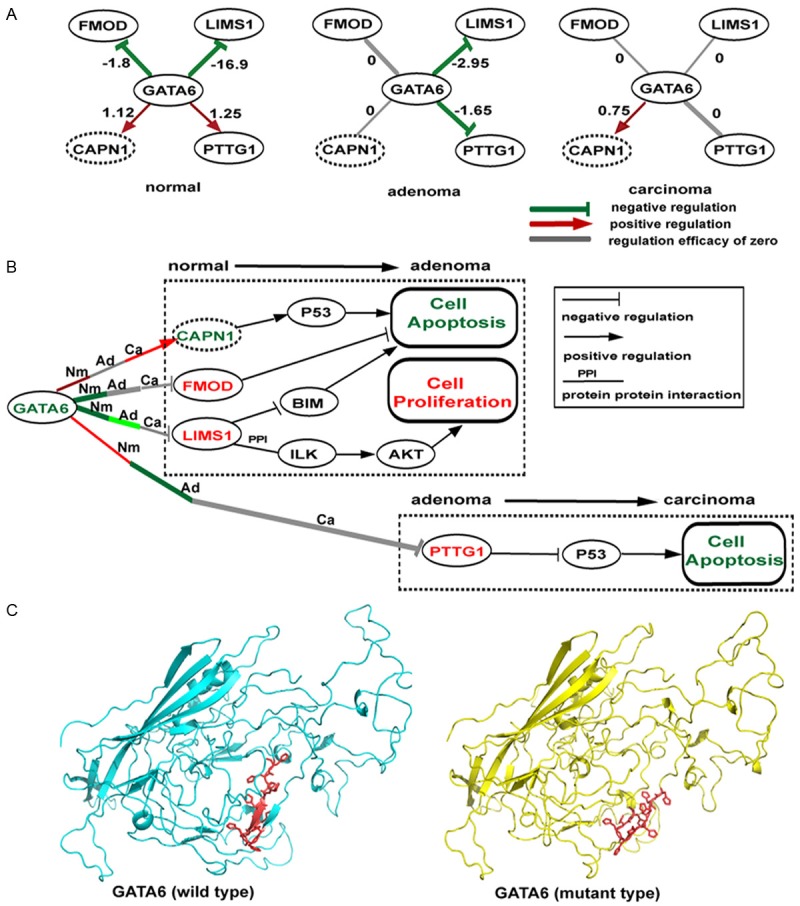
The proposed dysregulation mechanisms around GATA6. A. GATA6-centered subnetworks out of the three GRNs corresponding to normal, adenoma and carcinoma. GATA6 is a TF, and the other nodes are its targets. Nodes in solid-line ovals represent DRLs related targets; nodes in dotted-line ovals are unrelated to DRLs. Links in red, green and grey represent positive, negative and absent regulation relationships calculated with dataset GES24375; links in bold are DRL related. Numbers on the links indicate the regulation efficacies. B. The proposed mechanism by which GATA6 induces gastric adenoma. Links in red, green and grey still represent positive, negative and absent regulation relationships at normal (Nm), adenoma (Ad) and carcinoma (Ca) stage calculated with dataset GES24375. The darker the color, the larger the absolute regulation efficacy. Links in black are gene-gene interconnections obtained from literatures. The color of gene symbol, red or green, represents up- or down-expression in stage transition according to dataset GES24375 and TCGA data. Boxes indicate biological processes, with red color referring to activation and green color referring to inhibition. C. Homology models of normal and mutation GATA6 protein are represented in cyan cartoon and yellow cartoon, respectively. The mutated local region (residue 323-332) is shown in red.
Another interesting example is ESRRG, which is ranked top one in our adenoma vs. carcinoma DRG list (Table 5), and has been reported to be one of five gastric prognostic signature genes [46]. Although ESRRG is not a TF in our GRNs, it is a nuclear receptor, and functions as a transcription activator in the absence of bound ligand. ESRRG was also reported to promote the development of breast cancer by activating HIF1A [76] and DNMT1 [77]. Since ESRRG is the top one DRG for adenoma vs. carcinoma comparison, while ranks 656 (top 72%) for normal vs. adenoma, we propose that ESRRG may stimulate gastric carcinogenesis at relatively late stage. According to our differential network analysis, CEBPB, IRF2, FOSB, IRF1, and POU2F1 form 5 DRLs with ESRRG respectively (Figure 5A), which are all top ten ranked in adenoma vs. carcinoma DRL list (Table 5). From adenoma to carcinoma, CEBPB, IRF2 and FOSB elevated their positive regulation to ESRRG, while IRF1 and POU2F1 increased their inhibition to ESRRG. In all, the positive regulation of ESRRG was enhanced in carcinoma stage, and its over-expression was indeed observed in both GSE24375 and TCGA data (Figure S3). Among ESRRG’s regulators, CEBPB is a known GC related gene, whose over-expression was correlated with metastasis [49]; IRF2 was a cancer gene, and it was reported to promote cell growth and inhibit cell apoptosis in pancreatic cancer [57]; FOSB is a cofactor of AP1 and functions as a driver of skin cancer development [78]. The over-expressions of CEBPB, IRF2 and FOSB in carcinoma stage were also observed in both GSE24375 and TCGA data (Figure S3). Therefore, we speculated that the positive regulators, CEBPB, IRF2 and FOSB may promote the gastric carcinogenesis by activating ESRRG (Figure 5B). IRF1, a known suppressor of gastric carcinoma [79], was observed to enhance its inhibition to ESRRG from adenoma to carcinoma according to our networks, while in this case, it failed to reverse the carcinogenesis process (Figure 5B). The down-expression of IRF1 in carcinoma stage was observed in both GSE24375 and TCGA data (Figure S3). POU2F1, a known GC related gene, increases cell proliferation by activating ERK signalling, and its over-expression correlates with gastric poor prognosis [51]; however, POU2F1 was found to negatively regulate ESRRG in our carcinoma GRN, suggesting that POU2F1 should have alternative down-stream signaling pathways which effectively promote gastric carcinogenesis.
Figure 5.
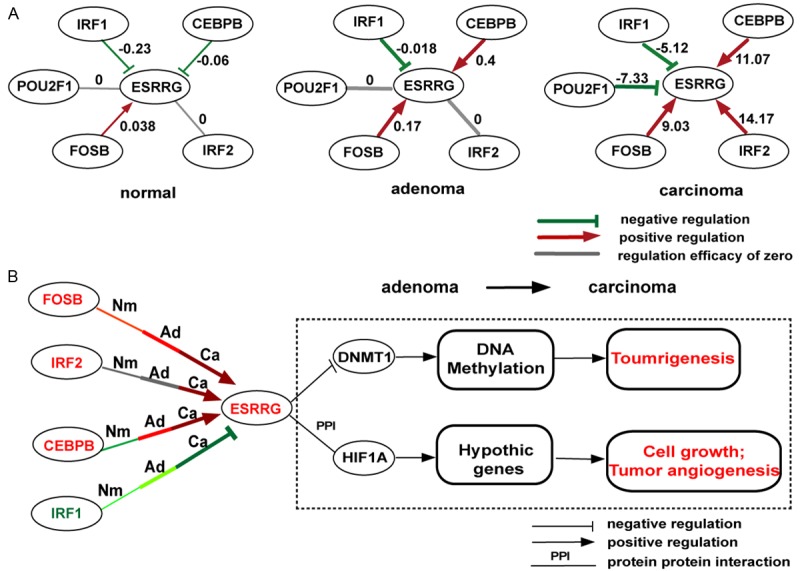
The proposed dysregulation mechanisms around ESRRG. A. ESRRG-centered subnetworks out of the three GRNs corresponding to normal, adenoma and carcinoma. ESRRG is a target, and the other nodes are its regulators. Nodes in solid-line ovals represent DRL related targets; nodes in dotted-line ovals are unrelated to DRLs. Links in red, green and grey represent positive, negative and absent regulation relationships calculated with dataset GES24375; Links in bold are DRL related. Numbers on the links indicate the regulation efficacies. B. The proposed mechanisms by which ESRRG induces gastric carcinoma. Links in red, green and grey still represent positive, negative and absent regulation relationships at normal (Nm), adenoma (Ad) and carcinoma (Ca) stage calculated with dataset GES24375. The darker the color, the larger the absolute regulation efficacy. Links in black are gene-gene interconnections obtained from literatures. The color of gene symbol, red or green, represents up- or down-expression in stage transition according to dataset GES24375 and TCGA data. Boxes indicate biological processes, with red color referring to activation and green color referring to inhibition.
Method comparison
As described in Introduction, to reduce the computational complexity, researchers built subnetworks around previously reported disease-related genes [22-24], or differentially expressed genes [25-27], which enabled the discovery of context related genes. In order to evaluate the power of our DCEA-based differential network modelling and analysis method in prioritizing disease related genes, we compared DCEA-based method with differential expression analysis based method and whole genome method, with three types of networks shortly named as DCG-GRNs, DEG-GRNs and WG-GRNs, respectively.
To infer DEG-GRNs, we first utilized limma method [39] to obtain two DEG lists (P<0.05) for normal vs. adenoma and adenoma vs. carcinoma comparisons, including 1822 and 994 genes respectively. We then utilized stratified sampling method to extract DEGs from the two DEG lists in equal proportion, resulting in a final list with the gene number equal to the number of DR-relevent genes used to infer DCG-GRNs, 2524. For WG-GRN modelling, all 12658 genes (whole genome, WG for short) contained in the GSE24375 gene expression profiles were all used. The statistics of three types of stage-specific networks, DCG-GRNs, DEG-GRNs and WG-GRNs, are listed in Table 1. Functional enrichment analysis of network genes shows that DCG-GRNs enrich cancer genes and drug targets more significantly than DEG-GRNs and WG-GRNs (Table 1), suggesting that GRN modelling started from DR-relevant genes can effectively highlight context relevant genes compared with DEGs based modelling, as well as whole genome modelling. The insignificant enrichment of cancer genes and drug targets in WG-GRNs implies the necessity of seed genes selection in reducing not only the computational complexity but also the background noise.
Similar to the above section, we also estimated the differential regulation extent (DRs) of genes in DEG-GRNs and WG-GRNs. In order to compare the power of DRG lists from DCG-GRNs, DEG-GRNs and WG-GRNs in prioritizing cancer genes, we calculated the percentages of cancer genes and drug targets in their top 5%, 10%, 15%....80% DRG lists, respectively. Wilcoxon-test of the percentages of cancer genes and drug targets in DRG lists (Table 6) suggests that DRG lists for DCG-GRNs are more efficient than DRG lists of DEG-GRNs and WG-GRNs in prioritizing disease genes.
Table 6.
Comparison of DRG lists of DCG-GRNs, DEG-GRNs and WG-GRNs
| DRG list | Gene sets | DCG-GRNs vs. DEG-GRNs (p-value) | DCG-GRNs vs. WG-GRNs (p-value) |
|---|---|---|---|
| Normal vs. Adenoma | Cancer Genes | 0.005 | 0.00029 |
| Drug Targets | 0.00024 | 0.00042 | |
| Adenoma vs. Carcinoma | Cancer Genes | 0.013 | 0.0123 |
| Drug Targets | 0.00087 | 0.0122 |
p-value: significance of Wilcoxon test of percentages of cancer genes and drug targets in different DRG lists.
Discussion
Differential network analysis, aiming to identify different networking, is a new emerging area in systems biology. For gene regulation network, the biological idea underlying differential networking is differential regulation. As is well known, gene regulation is cellular context-specific and dynamic in nature. That is, regulatory components are differently activated or inhibited under varying conditions, and the topology of the underlying networks changes accordingly, which is considered causal to phenotypic changes. Since cancer has a nature of multistep, inferring stage-specific GRN and finding out the dysregulation mechanisms during carcinogenesis with the aid of differential network analysis is worthwhile, yet challenging, for both computational and experimental biologists. In this work, we proposed a novel framework for differential network modelling and analysis, and applied it to a gastric cancer dataset.
In the current efforts in carcinogenesis studies, lots of attention has been paid on differential expression. Differentially expressed functional-relevant genes are always selected to explain regulation mechanisms underlying pathogenesis. The two terms, differentially expressed genes and differentially regulated genes, are even regarded as the same in quite a lot of literatures. When performing network modelling, in order to reduce the computational complexity, some researchers built gene regulatory subnetworks around differentially expressed genes, and did manage to identify potential drug targets or context related genes [26,27]. However, it has been well accepted that cancers originates from genomic changes in genes regulating cell growth and differentiation, followed by the dysregulation of cell signaling transduction, which causes abnormal expression of a large number of genes, and eventual over-activation of cell proliferation [80]. That is to say, causal signals would be submerged within differentially expressed genes, and in this sense, differentially expressed genes are more likely to be the consequences of differential regulation mechanisms, rather than the causes of phenotypic changes. More importantly, causal factors are not necessarily differentially expressed. For example, if a mutation occurs to DNA binding domain or activation domain of a TF, the TF could no longer activate its target genes, even though it keeps its original expression level. Taking p53 as an example, it has been well established that p53 pathway could be disrupted through a point mutation that inactivates its capacity to bind specifically to its cognate recognition sequence [81]. In this case, the expression correlation of the TF with its targets is disrupted in the abnormal state, which could be captured by a new emerging strategy, differential coexpression analysis (DCEA) [31,34,35]. DCEA was designed to explore gene interconnection changes, instead of expression level changes, and has been considered more promising in identifying differential regulation mechanisms of phenotypic changes than differential expression analysis [31]. Therefore, in the present work, we built stage-specific subnetworks around differentially coexpressed genes (DCGs) identified with our previously developed DCEA methods [34,35], instead of around differentially expressed genes as previous studies did [25-27]. Benefiting from the use of DCEA, the search space of gene regulatory network modelling was successfully narrowed down to the genes most relevant to differential regulation, which was verified by the observation of the enrichment of cancer genes and drug targets in our stage-specific networks (Table 1), and especially in the “differential” groups of target genes (Tables 3, 4). The comparison of DCEA-based modelling method with differential expression analysis based modelling and whole genome modelling methods proved that DCG-GRNs highlighted context relevant genes more effectively than DEG-GRNs and WG-GRNs did (Table 6).
The final goal of differential regulatory network analysis is to explore the dynamic changes of gene regulation and identify differential regulatory relationships. It is assumed that differential regulatory relationships play crucial roles in carcinogenesis, and the involved genes have the potential to be drug targets. The more significant the difference is, the more promising the genes would be. However, network comparison is always a challenge in systems biology, especially when the networks are large and complex. In previous differential network analysis studies, some of them ignored the quantitative information in the original GRNs, and constructed qualitative differential network by removing common interactions/regulations of conditional networks [23,24,26,29]; some semi-quantitatively compared small-scale networks under varying conditions [22,25,27,28,30]. Generally speaking, all these efforts greatly reduced the calculation complexity by discarding quantitative information or inferring subnetworks around a given set of genes, while they did not make full use of regulation strength information involved in GRNs. In our design, by integrating DCEA to linear regression modelling method, we first obtained stage-specific differential regulation-enriched networks with links weighted by regulation efficacy. After that, by implementing DR measure (Equation 5) and the modified LFC model [34], the change in regulation strength of a certain gene with its neighbors in the GRN, and the change in regulation strength of a certain link could be estimated and ranked. In this way, all genes and links in the regulatory networks could be prioritized based on the extent of differential regulation. Furthermore, the modelling and anlysis pipeline is not limited by known disease-related genes, and therefore enables the discovery of novel regulators or regulatory relationships that have not yet been associated to the disease of interest. By using the present pipeline, we identified 36 DRGs for normal to adenoma transition and 56 DRGs for adenoma to carcinoma transition, out of which more than 50% have been reported to be GC related (Table S1). The rest genes are therefore worthy of further experimental investigation to determine their association with gastric cancer.
Among the 16 differentially regulated genes (DRGs) involved in the two top-10 DRG lists corresponding to normal vs. adenoma and adenoma vs. carcinoma comparison, 10 have been reported to be gastric cancer, and 5 to be cancer related. We noticed that out of the 16 DRGs, only one gene, TRIB1, was differentially expressed between normal and adenoma (adjusted P=0.03) in GSE24375, and no one was differentially expressed between adenoma and carcinoma. This strongly supports the necessity of DCEA-based methods in discovering disease related genes. To make it clear, we also specially tested the power of DEGs in prioritizing cancer genes. Two DEG sets discovered in normal vs. adenoma and adenoma vs. carcinoma comparison by limma method were separately ranked by their p values in an ascending order. The two gene lists were then cut to the same length as the corresponding DRG lists. Perturbation tests showed that cancer genes were not significantly higher ranked in both normal vs. adenoma DEG list (p value =0.11) and adenoma vs. carcinoma DEG list (p value =0.48). It is our belief that the differential networking information involved in DRGs and DRLs, together with prior knowledge on transcriptional regulation and the changes in absolute expression level, help to elucidate the regulatory mechanisms of phenotypic changes, which starts from disruption or switch of regulation relationships, results in changes of gene expression and thus functional alterations of crucial processes.
It is widely accepted that carcinogenesis is a multistep process, especially for intestinal gastric carcinoma, progressing through the stages of chronic gastritis, atrophy, intestinal metaplasia, adenoma, and finally gastric carcinoma [82]. The study of sequential changes of GRNs from normal to premalignant lesions and to cancers would provide insights into the global landscape of the dynamics of carcinogenesis. Recently, a colon cancer research of state-specific gene-gene interaction networks with respect to normal, adenoma and cancer found that the gene and link numbers, the mean degree and the mean clustering coefficient increased in the progression from normal to adenoma and to cancer [44]; however, our differential networks analysis of gastric cancer GRNs with respect to normal, adenoma and carcinoma found that the network size and complexity expanded from normal to adenoma and then shrank from adenoma to carcinoma. This up and down trend was also observed in our DEG based GRNs and whole genome GRNs (Table 1). According to our observation, adenoma seems to be an intermediate stage with most compact network topology compared with normal and carcinoma. Quite a lot of regulatory relationships transiently occur at the stage of adenoma, and then disappear when adenoma develop into carcinoma. Specifically, the target number of a TF (so called out-degree) increased from 39.9 (normal) to 53.3 (adenoma), and then decreased to 37.09 (carcinoma). After checking the transient regulations specific for adenoma, we found that Wnt/β-catenin pathway are more active in gastric adenoma than both normal and carcinoma, which is consistent with some previous reports [83,84]. In our GRNs, AXIN2 is negatively regulated by GATA3 specifically in adenoma stage, which could lead to the accumulation of β-catenin, and thus the activation of Wnt signaling pathway [83]. Furthermore, we identified the Wnt pathway related DRLs for normal vs. adenoma and adenoma vs. carcinoma, and found that the regulation efficacy related to Wnt pathway averagely increased from normal to adenoma (0.48), and then decreased from adenoma to carcinoma (-0.31), quantitatively proving the activation of Wnt pathway in adenoma stage. It seems that at the early stage of carcinogenesis, a number of transient regulations accumulate, which accelerate the celluar proliferation by affecting some crucial signal pathways, and eventually result in a drastic transition to carcinoma. This is consistent with the speculation that the progression of cancer may involve a critical transition [85]. Our observation is also coherent with some previous reports. A transcriptomic study of colorectal carcinogenesis found that majority of gene expression changes occur in the transition from normal to adenoma rather than from adenoma to carcinoma [86]. A DNA methylation study of gastric carcinogenesis suggested that accumulation of DNA methylation occur during the early stages of carcinogenesis and predetermines the future cancer type [87]. At this point, the dynamic changes from normal to adenoma seem to be more worthy of investigation although the phenotype has not deteriorated to the worst.
In all, the present framework combines our previously developed differential coexpression analysis (DCEA) strategy [34,35], reverse-forward integrated GRN modelling method [20,21] and novel quantitative methods for prioritization of differential regulation, to implement differential network modelling and analysis. DCEA enriches differential regulations, forward engineering strategy reduces the false positive rate, reverse engineering strategy utilizes the temporary information from expression data, and the methods for quantifying differential regulation fully exploit the quantitative information in the model. As expected, all these factors efficiently facilitate the differential network analysis with reasonable time consuming, and help to generate insightful hints on dysfucntional regulation mechanisms underlying carcinogenesis. In the case study, based on the quite limited sample size, we obtained valuable observations worthy of further exploration. We believe that if applied to data with a larger sample size, the performance of our approach will be enhanced significantly. The last but not least, the present framework could be easily applied to other clinical topics, and other phenotypic changes associated with development, aging, or other case-control settings. Four years ago, de la Fuente reviewed as follows, “Although we still have not reached a stage where the elucidation of differential regulatory networks is commonly feasible, recent advances have described the first steps towards this goal - the identification of differential coexpression networks.” With the accumulation of whole genome expression data, and the improvement of computational algorithms, it is time to decipher the dysfunctional regulators and their relevant signaling pathway through efficient differential network analysis.
Acknowledgements
This work was supported by the grants from the National “973” Key Basic Research Development Program (2012CB316501 and 2013CB910801), the National Natural Science Foundation of China (31171268, 81272279, and 91229106), the Program of International S&T Cooperation (2014DFB30020), and the the Fundamental Research Program of Shanghai Municipal Commission of Science and Technology (14DZ1951300 and 14DZ2252000).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Figueiredo C, Garcia-Gonzalez MA, Machado JC. Molecular pathogenesis of gastric cancer. Helicobacter. 2013;18(Suppl 1):28–33. doi: 10.1111/hel.12083. [DOI] [PubMed] [Google Scholar]
- 2.Buffart TE, Carvalho B, van Grieken NC, van Wieringen WN, Tijssen M, Kranenbarg EM, Verheul HM, Grabsch HI, Ylstra B, van de Velde CJ, Meijer GA. Losses of chromosome 5q and 14q are associated with favorable clinical outcome of patients with gastric cancer. Oncologist. 2012;17:653–662. doi: 10.1634/theoncologist.2010-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holbrook JD, Parker JS, Gallagher KT, Halsey WS, Hughes AM, Weigman VJ, Lebowitz PF, Kumar R. Deep sequencing of gastric carcinoma reveals somatic mutations relevant to personalized medicine. J Transl Med. 2011;9:119. doi: 10.1186/1479-5876-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song HR, Kim HN, Kweon SS, Choi JS, Shim HJ, Cho SH, Chung IJ, Park YK, Kim SH, Choi YD, Joo KW, Shin MH. Genetic variations in the PRKAA1 and ZBTB20 genes and gastric cancer susceptibility in a Korean population. Mol Carcinog. 2013;52(Suppl 1):E155–60. doi: 10.1002/mc.22063. [DOI] [PubMed] [Google Scholar]
- 5.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, Lee SP, Ho SL, Chan AK, Cheng GH, Roberts PC, Rejto PA, Gibson NW, Pocalyko DJ, Mao M, Xu J, Leung SY. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, Hu Z, Wu C, Dai J, Li H, Dong J, Wang M, Miao X, Zhou Y, Lu F, Zhang H, Hu L, Jiang Y, Li Z, Chu M, Ma H, Chen J, Jin G, Tan W, Wu T, Zhang Z, Lin D, Shen H. A genome-wide association study identifies new susceptibility loci for non-cardia gastric cancer at 3q13.31 and 5p13.1. Nat Genet. 2011;43:1215–1218. doi: 10.1038/ng.978. [DOI] [PubMed] [Google Scholar]
- 7.Kim H, Eun JW, Lee H, Nam SW, Rhee H, Koh KH. Gene expression changes in patientmatched gastric normal mucosa, adenomas, and carcinomas. Exp Mol Pathol. 2011;90:201–209. doi: 10.1016/j.yexmp.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Wu XM, Shao XQ, Meng XX, Zhang XN, Zhu L, Liu SX, Lin J, Xiao HS. Genome-wide analysis of microRNA and mRNA expression signatures in hydroxycamptothecin-resistant gastric cancer cells. Acta Pharmacol Sin. 2011;32:259–269. doi: 10.1038/aps.2010.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao WJ, Wu HL, He BS, Zhang YS, Zhang ZY. Analysis of long non-coding RNA expression profiles in gastric cancer. World J Gastroenterol. 2013;19:3658–3664. doi: 10.3748/wjg.v19.i23.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gopal G, Raja UM, Shirley S, Rajalekshmi KR, Rajkumar T. SOSTDC1 down-regulation of expression involves CpG methylation and is a potential prognostic marker in gastric cancer. Cancer Genet. 2013;206:174–182. doi: 10.1016/j.cancergen.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Fan B, Dachrut S, Coral H, Yuen ST, Chu KM, Law S, Zhang L, Ji J, Leung SY, Chen X. Integration of DNA copy number alterations and transcriptional expression analysis in human gastric cancer. PLoS One. 2012;7:e29824. doi: 10.1371/journal.pone.0029824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, Ho SL, Chan AK, Man JL, Foglizzo V, Ng MK, Ching YP, Cheng GH, Xie T, Fernandez J, Li VS, Clevers H, Rejto PA, Mao M, Leung SY. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, McCleland M, Stawiski EW, Gnad F, Mayba O, Haverty PM, Durinck S, Chen YJ, Klijn C, Jhunjhunwala S, Lawrence M, Liu H, Wan Y, Chopra V, Yaylaoglu MB, Yuan W, Ha C, Gilbert HN, Reeder J, Pau G, Stinson J, Stern HM, Manning G, Wu TD, Neve RM, de Sauvage FJ, Modrusan Z, Seshagiri S, Firestein R, Zhang Z. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat Commun. 2014;5:3830. doi: 10.1038/ncomms4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kreeger PK, Lauffenburger DA. Cancer systems biology: a network modeling perspective. Carcinogenesis. 2010;31:2–8. doi: 10.1093/carcin/bgp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao Y. A tutorial on analysis and simulation of boolean gene regulatory network models. Curr Genomics. 2009;10:511–525. doi: 10.2174/138920209789208237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan ZS, Collins L, Kasabov N. Bayesian learning of sparse gene regulatory networks. Biosystems. 2007;87:299–306. doi: 10.1016/j.biosystems.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 18.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vu TT, Vohradsky J. Nonlinear differential equation model for quantification of transcriptional regulation applied to microarray data of Saccharomyces cerevisiae. Nucleic Acids Res. 2007;35:279–287. doi: 10.1093/nar/gkl1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu K, Yu H, Hua YJ, Li YY, Liu L, Xie L, Li YX. Combinatorial network of primary and secondary microRNA-driven regulatory mechanisms. Nucleic Acids Res. 2009;37:5969–5980. doi: 10.1093/nar/gkp638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H, Tu K, Wang YJ, Mao JZ, Xie L, Li YY, Li YX. Combinatorial network of transcriptional regulation and microRNA regulation in human cancer. BMC Syst Biol. 2012;6:61. doi: 10.1186/1752-0509-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang B, Li H, Riggins RB, Zhan M, Xuan J, Zhang Z, Hoffman EP, Clarke R, Wang Y. Differential dependency network analysis to identify condition-specific topological changes in biological networks. Bioinformatics. 2009;25:526–532. doi: 10.1093/bioinformatics/btn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moignard V, Macaulay IC, Swiers G, Buettner F, Schutte J, Calero-Nieto FJ, Kinston S, Joshi A, Hannah R, Theis FJ, Jacobsen SE, de Bruijn MF, Gottgens B. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat Cell Biol. 2013;15:363–372. doi: 10.1038/ncb2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK, Regev A. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–468. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gill R, Datta S. A statistical framework for differential network analysis from microarray data. BMC Bioinformatics. 2010;11:95. doi: 10.1186/1471-2105-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madhamshettiwar PB, Maetschke SR, Davis MJ, Reverter A, Ragan MA. Gene regulatory network inference: evaluation and application to ovarian cancer allows the prioritization of drug targets. Genome Med. 2012;4:41. doi: 10.1186/gm340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma C, Xin M, Feldmann KA, Wang X. Machine learning-based differential network analysis: a study of stress-responsive transcriptomes in Arabidopsis. Plant Cell. 2014;26:520–537. doi: 10.1105/tpc.113.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng L, Yu J, Huang T, Jia H, Dong Q, He F, Yuan W, Qin L, Li Y, Xie L. Differential combinatorial regulatory network analysis related to venous metastasis of hepatocellular carcinoma. BMC Genomics. 2012;13(Suppl 8):S14. doi: 10.1186/1471-2164-13-S8-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Araki R, Seno S, Takenaka Y, Matsuda H. An estimation method for a cellular-state-specific gene regulatory network along tree-structured gene expression profiles. Gene. 2013;518:17–25. doi: 10.1016/j.gene.2012.11.090. [DOI] [PubMed] [Google Scholar]
- 30.Gambardella G, Moretti MN, de Cegli R, Cardone L, Peron A, di Bernardo D. Differential network analysis for the identification of condition-specific pathway activity and regulation. Bioinformatics. 2013;29:1776–1785. doi: 10.1093/bioinformatics/btt290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de la Fuente A. From ‘differential expression’ to ‘differential networking’ - identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010;26:326–333. doi: 10.1016/j.tig.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gill R, Datta S. Differential network analysis in human cancer research. Curr Pharm Des. 2014;20:4–10. doi: 10.2174/138161282001140113122316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu H, Liu BH, Ye ZQ, Li C, Li YX, Li YY. Linkbased quantitative methods to identify differentially coexpressed genes and gene pairs. BMC Bioinformatics. 2011;12:315. doi: 10.1186/1471-2105-12-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu BH, Yu H, Tu K, Li C, Li YX, Li YY. DCGL: an R package for identifying differentially coexpressed genes and links from gene expression microarray data. Bioinformatics. 2010;26:2637–2638. doi: 10.1093/bioinformatics/btq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Yu H, Liu BH, Zhao Z, Liu L, Ma LX, Li YX, Li YY. DCGL v2.0: an R package for unveiling differential regulation from differential co-expression. PLoS One. 2013;8:e79729. doi: 10.1371/journal.pone.0079729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 40.McDermott JE, Diamond DL, Corley C, Rasmussen AL, Katze MG, Waters KM. Topological analysis of protein co-abundance networks identifies novel host targets important for HCV infection and pathogenesis. BMC Syst Biol. 2012;6:28. doi: 10.1186/1752-0509-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He F, Balling R, Zeng AP. Reverse engineering and verification of gene networks: principles, assumptions, and limitations of present methods and future perspectives. J Biotechnol. 2009;144:190–203. doi: 10.1016/j.jbiotec.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 42.Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 44.Chung FH, Lee HH, Lee HC. ToP: A trend-of-disease-progression procedure works well for identifying cancer genes from multi-state cohort gene expression data for human colorectal cancer. PLoS One. 2013;8:e65683. doi: 10.1371/journal.pone.0065683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, Gerstein M. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature. 2004;431:308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- 46.Yin Y, Zhuo W, Zhao Y, Chen S, Li J, Wang L, Zhou T, Si JM. Converting a microarray signature into a diagnostic test: a trial of custom 74 gene array for clarification and prediction the prognosis of gastric cancer. PLoS One. 2013;8:e81561. doi: 10.1371/journal.pone.0081561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim SK, Jang HR, Kim JH, Noh SM, Song KS, Kim MR, Kim SY, Yeom YI, Kim NS, Yoo HS, Kim YS. The epigenetic silencing of LIMS2 in gastric cancer and its inhibitory effect on cell migration. Biochem Biophys Res Commun. 2006;349:1032–1040. doi: 10.1016/j.bbrc.2006.08.128. [DOI] [PubMed] [Google Scholar]
- 48.Zhu ZL, Yan BY, Zhang Y, Yang YH, Wang ZM, Zhang HZ, Wang MW, Zhang XH, Sun XF. PINCH expression and its clinicopathological significance in gastric adenocarcinoma. Dis Markers. 2012;33:171–178. doi: 10.3233/DMA-2012-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du YX, Zhang LH, Wang XH, Xing XF, Cheng XJ, Du H, Hu Y, Li YA, Zhu YB, Jia YN, Lin Y, Ji JF. [Expression of CCAAT/enhancer binding protein beta in human gastric carcinoma and its clinical significance] . Zhonghua Wei Chang Wai Ke Za Zhi. 2013;16:179–182. [PubMed] [Google Scholar]
- 50.Lai DW, Liu SH, Karlsson AI, Lee WJ, Wang KB, Chen YC, Shen CC, Wu SM, Liu CY, Tien HR, Peng YC, Jan YJ, Chao TH, Lan KH, Arbiser JL, Sheu ML. The novel Aryl hydrocarbon receptor inhibitor biseugenol inhibits gastric tumor growth and peritoneal dissemination. Oncotarget. 2014;5:7788–7804. doi: 10.18632/oncotarget.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian J, Kong X, Deng N, Tan P, Chen H, Wang J, Li Z, Hu Y, Zou W, Xu J, Fang JY. OCT1 is a determinant of synbindin-related ERK signalling with independent prognostic significance in gastric cancer. Gut. 2015;64:37–48. doi: 10.1136/gutjnl-2013-306584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu ZL, Wen JF, Xiao DS, Zhen H, Fu CY. Effects of transforming growth interacting factor on biological behaviors of gastric carcinoma cells. World J Gastroenterol. 2005;11:84–88. doi: 10.3748/wjg.v11.i1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keshari RP, Wang W, Zhang Y, Wang DD, Li YF, Yuan SQ, Qiu HB, Huang CY, Chen YM, Xia JC, Zhou ZW. Decreased expression of the GATA3 gene is associated with poor prognosis in primary gastric adenocarcinoma. PLoS One. 2014;9:e87195. doi: 10.1371/journal.pone.0087195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sulahian R, Casey F, Shen J, Qian ZR, Shin H, Ogino S, Weir BA, Vazquez F, Liu XS, Hahn WC, Bass AJ, Chan V, Shivdasani RA. An integrative analysis reveals functional targets of GATA6 transcriptional regulation in gastric cancer. Oncogene. 2014;33:5637–48. doi: 10.1038/onc.2013.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi YJ, Song JH, Yoon JH, Choi WS, Nam SW, Lee JY, Park WS. Aberrant expression of SOX9 is associated with gastrokine 1 inactivation in gastric cancers. Gastric Cancer. 2014;17:247–254. doi: 10.1007/s10120-013-0277-3. [DOI] [PubMed] [Google Scholar]
- 56.Hinoi T, Gesina G, Akyol A, Kuick R, Hanash S, Giordano TJ, Gruber SB, Fearon ER. CDX2-regulated expression of iron transport protein hephaestin in intestinal and colonic epithelium. Gastroenterology. 2005;128:946–961. doi: 10.1053/j.gastro.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 57.Sakai T, Mashima H, Yamada Y, Goto T, Sato W, Dohmen T, Kamada K, Yoshioka M, Uchinami H, Yamamoto Y, Ohnishi H. The roles of interferon regulatory factors 1 and 2 in the progression of human pancreatic cancer. Pancreas. 2014;43:909–916. doi: 10.1097/MPA.0000000000000116. [DOI] [PubMed] [Google Scholar]
- 58.Tatenhorst L, Senner V, Puttmann S, Paulus W. Regulators of G-protein signaling 3 and 4 (RGS3, RGS4) are associated with glioma cell motility. J Neuropathol Exp Neurol. 2004;63:210–222. doi: 10.1093/jnen/63.3.210. [DOI] [PubMed] [Google Scholar]
- 59.Piao L, Li Y, Kim SJ, Byun HS, Huang SM, Hwang SK, Yang KJ, Park KA, Won M, Hong J, Hur GM, Seok JH, Shong M, Cho MH, Brazil DP, Hemmings BA, Park J. Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res. 2009;69:3397–3404. doi: 10.1158/0008-5472.CAN-08-3235. [DOI] [PubMed] [Google Scholar]
- 60.Mashima T, Soma-Nagae T, Migita T, Kinoshita R, Iwamoto A, Yuasa T, Yonese J, Ishikawa Y, Seimiya H. TRIB1 Supports Prostate Tumorigenesis and Tumor-Propagating Cell Survival by Regulation of Endoplasmic Reticulum Chaperone Expression. Cancer Res. 2014;74:4888–4897. doi: 10.1158/0008-5472.CAN-13-3718. [DOI] [PubMed] [Google Scholar]
- 61.Kataoka F, Tsuda H, Arao T, Nishimura S, Tanaka H, Nomura H, Chiyoda T, Hirasawa A, Akahane T, Nishio H, Nishio K, Aoki D. EGRI and FOSB gene expressions in cancer stroma are independent prognostic indicators for epithelial ovarian cancer receiving standard therapy. Genes Chromosomes Cancer. 2012;51:300–312. doi: 10.1002/gcc.21916. [DOI] [PubMed] [Google Scholar]
- 62.Sulahian R, Casey F, Shen J, Qian ZR, Shin H, Ogino S, Weir BA, Vazquez F, Liu XS, Hahn WC, Bass AJ, Chan V, Shivdasani RA. An integrative analysis reveals functional targets of GATA6 transcriptional regulation in gastric cancer. Oncogene. 2014;33:5637–5648. doi: 10.1038/onc.2013.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahmed J, Meinel T, Dunkel M, Murgueitio MS, Adams R, Blasse C, Eckert A, Preissner S, Preissner R. CancerResource: a comprehensive database of cancer-relevant proteins and compound interactions supported by experimental knowledge. Nucleic Acids Res. 2011;39:D960–967. doi: 10.1093/nar/gkq910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu X, Ru J, Zhang J, Zhu LH, Liu M, Li X, Tang H. miR-23a targets interferon regulatory factor 1 and modulates cellular proliferation and paclitaxel-induced apoptosis in gastric adenocarcinoma cells. PLoS One. 2013;8:e64707. doi: 10.1371/journal.pone.0064707. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Yang G, Zhang Y, Xiong J, Wu J, Yang C, Huang H, Zhu Z. Downregulation of Id1 by small interfering RNA in gastric cancer inhibits cell growth via the Akt pathway. Mol Med Rep. 2012;5:1075–1079. doi: 10.3892/mmr.2012.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzuki S, Dobashi Y, Hatakeyama Y, Tajiri R, Fujimura T, Heldin CH, Ooi A. Clinicopathological significance of platelet-derived growth factor (PDGF)-B and vascular endothelial growth factor-A expression, PDGF receptor-beta phosphorylation, and microvessel density in gastric cancer. BMC Cancer. 2010;10:659. doi: 10.1186/1471-2407-10-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seidl C, Port M, Gilbertz KP, Morgenstern A, Bruchertseifer F, Schwaiger M, Roper B, Senekowitsch-Schmidtke R, Abend M. 213Biinduced death of HSC45-M2 gastric cancer cells is characterized by G2 arrest and up-regulation of genes known to prevent apoptosis but induce necrosis and mitotic catastrophe. Mol Cancer Ther. 2007;6:2346–2359. doi: 10.1158/1535-7163.MCT-07-0132. [DOI] [PubMed] [Google Scholar]
- 68.Boberg DR, Batistela MS, Pecharki M, Ribeiro EM, Cavalli IJ, Lima RS, Urban CA, Furtado-Alle L, Souza RL. Copy number variation in ACHE/EPHB4 (7q22) and in BCHE/MME (3q26) genes in sporadic breast cancer. Chem Biol Interact. 2013;203:344–347. doi: 10.1016/j.cbi.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 69.Liang Z, Gao LH, Cao LJ, Feng DY, Cao Y, Luo QZ, Yu P, Li M. Detection of STAT2 in early stage of cervical premalignancy and in cervical cancer. Asian Pac J Trop Med. 2012;5:738–742. doi: 10.1016/S1995-7645(12)60117-5. [DOI] [PubMed] [Google Scholar]
- 70.Kato I, Land S, Barnholtz-Sloan J, Severson RK. APOE and FABP2 Polymorphisms and History of Myocardial Infarction, Stroke, Diabetes, and Gallbladder Disease. Cholesterol. 2011;2011:896360. doi: 10.1155/2011/896360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yao W, Ji S, Qin Y, Yang J, Xu J, Zhang B, Xu W, Liu J, Shi S, Liu L, Liu C, Long J, Ni Q, Li M, Yu X. Profilin-1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3-HIF1alpha axis. Mol Cancer. 2014;13:187. doi: 10.1186/1476-4598-13-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen K, Tu Y, Zhang Y, Blair HC, Zhang L, Wu C. PINCH-1 regulates the ERK-Bim pathway and contributes to apoptosis resistance in cancer cells. J Biol Chem. 2008;283:2508–2517. doi: 10.1074/jbc.M707307200. [DOI] [PubMed] [Google Scholar]
- 73.Lee YH, Schiemann WP. Fibromodulin suppresses nuclear factor-kappaB activity by inducing the delayed degradation of IKBA via a JNK-dependent pathway coupled to fibroblast apoptosis. J Biol Chem. 2011;286:6414–6422. doi: 10.1074/jbc.M110.168682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Severin J, Waterhouse AM, Kawaji H, Lassmann T, van Nimwegen E, Balwierz PJ, de Hoon MJ, Hume DA, Carninci P, Hayashizaki Y, Suzuki H, Daub CO, Forrest AR. FANTOM4 EdgeExpressDB: an integrated database of promoters, genes, microRNAs, expression dynamics and regulatory interactions. Genome Biol. 2009;10:R39. doi: 10.1186/gb-2009-10-4-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dai W, Song T, Wang X, Jin X, Deng L, Wu A, Jiang T. Improvement in low-homology template-based modeling by employing a model evaluation method with focus on topology. PLoS One. 2014;9:e89935. doi: 10.1371/journal.pone.0089935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ao A, Wang H, Kamarajugadda S, Lu J. Involvement of estrogen-related receptors in transcriptional response to hypoxia and growth of solid tumors. Proc Natl Acad Sci U S A. 2008;105:7821–7826. doi: 10.1073/pnas.0711677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y, Wang L. Nuclear receptor SHP inhibition of Dnmt1 expression via ERRgamma. FEBS Lett. 2011;585:1269–1275. doi: 10.1016/j.febslet.2011.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Budczies J, Brockmoller SF, Muller BM, Barupal DK, Richter-Ehrenstein C, Kleine-Tebbe A, Griffin JL, Oresic M, Dietel M, Denkert C, Fiehn O. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: alterations in glutamine and beta-alanine metabolism. J Proteomics. 2013;94:279–288. doi: 10.1016/j.jprot.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 79.Nozawa H, Oda E, Ueda S, Tamura G, Maesawa C, Muto T, Taniguchi T, Tanaka N. Functionally inactivating point mutation in the tumor-suppressor IRF-1 gene identified in human gastric cancer. Int J Cancer. 1998;77:522–527. doi: 10.1002/(sici)1097-0215(19980812)77:4<522::aid-ijc8>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 80.Yan LH, Wei WY, Cao WL, Zhang XS, Xie YB, Xiao Q. Overexpression of E2F1 in human gastric carcinoma is involved in anti-cancer drug resistance. BMC Cancer. 2014;14:904. doi: 10.1186/1471-2407-14-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 82.Correa P. Human gastric carcinogenesis: A multistep and multifactorial process-First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- 83.Wang J, Ray PS, Sim MS, Zhou XZ, Lu KP, Lee AV, Lin X, Bagaria SP, Giuliano AE, Cui X. FOXC1 regulates the functions of human basal-like breast cancer cells by activating NF-kappaB signaling. Oncogene. 2012;31:4798–4802. doi: 10.1038/onc.2011.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hidaka Y, Mitomi H, Saito T, Takahashi M, Lee SY, Matsumoto K, Yao T, Watanabe S. Alteration in the Wnt/beta-catenin signaling pathway in gastric neoplasias of fundic gland (chief cell predominant) type. Hum Pathol. 2013;44:2438–2448. doi: 10.1016/j.humpath.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 85.Chen L, Liu R, Liu ZP, Li M, Aihara K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci Rep. 2012;2:342. doi: 10.1038/srep00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Regalo G, Resende C, Wen X, Gomes B, Duraes C, Seruca R, Carneiro F, Machado JC. C/EBP alpha expression is associated with homeostasis of the gastric epithelium and with gastric carcinogenesis. Lab Invest. 2010;90:1132–1139. doi: 10.1038/labinvest.2010.79. [DOI] [PubMed] [Google Scholar]
- 87.Kaneda A, Matsusaka K, Sakai E, Funata S. DNA methylation accumulation and its predetermination of future cancer phenotypes. J Biochem. 2014;156:63–72. doi: 10.1093/jb/mvu038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.