

Abstract
Increasing evidence reveals that deregulation of miRNAs contributes to carcinogenesis of the human non-small cell lung cancer (NSCLC). Our study discovered that the expression of miR-449a was markedly decreased in NSCLC cells with high metastatic capacity and tissues of positive lymph node metastasis. Moreover, our results showed that miR-449a could act as a tumor suppressor by inhibiting the invasion of NSCLC cells in vitro and in vivo. Mechanistically, miR-449a inhibited the expression of MAP2K1 by direct targeting its 3’UTR, and regulated the activity of MEK1/ERK1/2/c-Jun pathway through an auto-regulatory feedback loop. Furthermore, the histone methylation mediated the decreased expression of miR-449a through SUZ12. Taken together, the novel connection between miR-449a and MAP2K1 demonstrated here provided a new, potential therapeutic target for the treatment of non-small cell lung cancer.
Keywords: Non-small cell lung cancer (NSCLC), miR-449a, MAP2K1, invasion
Introduction
Non-small cell lung cancer (NSCLC), including adenocarcinoma (ADC), squamous cell carcinoma (SCC), adenosquamous cell carcinoma (ASC) and large cell carcinoma (LCC), accounts for 80-85% of all lung cancer cases [1,2]. The overall 5-year survival rate for NSCLC patients remains at approximately 15%, despite recent advances in surgery, radiotherapy and chemotherapy [2]. Therefore, thorough understanding of the molecular mechanisms underlying NSCLC development and progression is of great therapeutic value. Non-coding small RNAs have been implicated to play important roles in NSCLC pathogenesis [3,4], which may potentially serve as novel NSCLC diagnostic markers and therapeutic targets.
Mature miRNAs are short, non-coding RNAs of approximately 22 nucleotides that regulate gene expression at post-transcriptional level by targeting the 3’-UTR region of mRNA and leading to their translational inhibition or deregulation [5]. MiRNAs regulate various biological processes, including cell differentiation, proliferation, apoptosis, stress resistance, fat metabolism, and development. Their dysregulation have been shown to associate with multiple human diseases such as cancer, either as oncogenes or tumor suppressors [6]. The role of miRNAs in tumorigenesis and tumor progression, includes both tumor cell proliferation and metastasis [5,7-10]. For example, miR-10b promotes breast tumor metastasis, while miR-335 and miR-126 suppress the cancer metastasis [11,12]. Therefore, miRNAs could potentially act as the next generation of therapeutic targets for malignant tumors [13].
The miR-449a has been mapped to the first intron of CDC20B gene, which located on human chromosome 5q11.2, a region identified as a susceptibility locus in a variety of malignancies, including lung cancer. Accumulating evidence has demonstrated that the expression of miR-449a is frequently decreased in several types of malignant tumors, for instance those of prostate, gastric and bladder [14-16]. In these types of cancer, miR-449a induces cell cycle arrest, apoptosis and senescence by regulating critical cell cycle motors or apoptosis inhibitors such as histone deacetylase-1 (HDAC-1), CDK6, CDC25A or sirtuin1 (SIRT1) [14-17]. However, the role of miR-449a in lung cancer development remains unclear.
In the current study, we identified that miR-449a was differentially expressed between NL9980 and L9981 cells, both commonly used NSCLC cell lines with distinct metastatic capacity- through microRNA chip analyses. Further characterization of miR-449a in other NSCLC cell lines found that miR-449a functions as a metastasis suppressor. Over-expression of miR-449a can suppress NSCLC cell invasion and migration in vitro and in vivo. MiR-449a mediates metastasis suppressing activity by modulating SUZ12 expression. Through bioinformatics analysis, we identified MAP2K1 (MEK1) as a direct target of miR-449a. Furthermore, we found that MAPK signaling pathway is involved in miR-449a-regulated NSCLC metastasis, in which AP-1 can directly regulate miR-449a expression by a negative feedback loop. Finally, we found that miR-449a expression in lung cancer cells is epigenetically inactivated by the oncogenic trimethylation of histone H3Lys27 (H3K27me3), which can be reversed by epigenetic drug treatment targeting histone modifications. These findings reveal a feedback mechanism to regulate miR-449a expression and suggest that miR-449a may represent a novel therapeutic target for NSCLC metastasis.
Materials and methods
Cell culture and transfection
The human lung cancer cell lines NL9980 and L9981 were established from the same human lung large cell carcinoma cell line, WCQH-9801. NL9980 cells have much higher metastatic potential than L9981 cells. L9981-luc, 95C and 95D were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA) medium supplemented with 10% fetal bovine serum (FBS, GIBCO), 100 IU/ml penicillin and 100 IU/ml streptomycin in humidified 5% CO2 at 37°C. Transfection of miRNA/siRNA was carried out using Lipofectamine 2000 (Initrogen, CA, USA) according to the manufacturer’s recommendation. The mature hsa-miR-449a mimic/inhibitor, non-specific control, were synthesized by GenePharma (Shanghai, China). Sequences are listed in Supplementary Table 1.
Patient sample preparation
A total of 30 cases of NSCLC were obtained from the 1st January 2006 to the 31st December 2009 at the General Hospital of Tianjin Medical University. None of the 30 patients had received radiation therapy or chemotherapy before surgery. Tissue samples were stored in liquid nitrogen before use. The TNM staging system of the UICC (1997) was used to classify the specimens. The study has been approved by hospital ethical committee. Clinicopathological information of the patients about age, histological type, stage and lymph node metastasis was obtained from patient records and summarized in Table 1.
Table 1.
Correlation between miR-449a expression and clinicopathological features of non-small cell lung cancer
| Factor | No. of cases | MiR-449a Expression | χ2 value | P value | |
|---|---|---|---|---|---|
|
| |||||
| lower | higher | ||||
| Age | |||||
| <62 | 11 | 6 | 5 | ||
| ≥62 | 19 | 9 | 10 | 0.144 | 0.705 |
| Gender | |||||
| Man | 23 | 13 | 10 | ||
| Woman | 7 | 2 | 5 | 1.677 | 0.195 |
| Smoking history | |||||
| Smoker | 18 | 13 | 5 | ||
| Non smoker | 12 | 2 | 10 | 8.889 | 0.003* |
| Histology | |||||
| Sq | 16 | 9 | 7 | ||
| Ad | 13 | 5 | 8 | ||
| Other | 1 | 1 | 0 | 1.870 | 0.469 |
| Stage | |||||
| I+II | 9 | 2 | 7 | ||
| III+IV | 21 | 13 | 8 | 3.968 | 0.046* |
| Lymph node status | |||||
| Negative | 5 | 0 | 5 | ||
| Positive | 25 | 15 | 10 | 6.000 | 0.042* |
Significant difference.
χ2 test was used to evaluate the correlation of mir-449a expression and clinical pathologic parameters.
MiRNA microarray
Differentially expressed miRNAs were identified using Agilent miRNA chip (Agilent Technologies Inc. CA, USA) in primary lung cancer tissues and matched metastasis lymph nodes, and one pair of lung cancer cell lines with different metastasis ability, respectively. MiRNA was isolated using the PreAnalytiX PAXgene miRNA isolation kit (Qiagen) according to the manufacturer’s protocol. Hybridization, scanning, and data processing were performed by Genetimes Technology Inc. (Shanghai, China).
Plasmid construction and stable transfection in L9981-luc cells
The eukaryotic expression vector pcDNA3.1 (+) (Invitrogen) was used to construct the miRNAs expression plasmid. Genomic sequence of the human miR-449a gene was cloned from human genomic DNA. PCR product was digested by Xho I and BamH I, and then cloned into pcDNA3.1 (+) vector and named as pcDNA3.1-miR-449a. L9981-luc cells were stably transfected pcDNA3.1-miR-449a in the presence of Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions and placed under Hygromycin B (400 µg/ml) selection for 4 weeks. Surviving colonies were isolated and named as L9981-luc-miR-449a.
Target prediction
The target genes of miR-449a were predicted using three Bioinformatics algorithms: miRanda (microrna.org), PicTar (4-way) and TargetScans. The MAK2K1 was chosen to construct dual-Luciferase reporter vectors.
Real-time quantitative PCR
Quantitative RT-PCR was carried out using SYBR○RPremix Ex TaqTM (Code DRR041A, Takara). Total RNA isolated using the mirVana Kit (Applied Biosystems, CA) was subsequently reverse transcribed to cDNA according to the manufacturer’s protocol (Takara). The U6 and GAPDH were used as internal controls, respectively. All primer sequences were listed in Table 2. The reactions were placed in a 96-well plate (ABI) or 384-well plate using a pre-heated real-time PCR instrument (ABI 7500) or (ABI 7500HT), respectively. Program of the real-time instrument was cosisted of 10 sec at 95°C followed by 40 cycles of 95°C for 5 s and 60°C for 34 s. Three independent experiments were performed to analyze relative gene expressions and each sample was tested in triplicate. Ct values were used to calculate the expression of RNA levels. The amount of target gene expression (2-ΔΔCt) was normalized using the endogenous GAPDH or U6 reference, the amount of target gene in the control sample was set as the calibrator at 1.0.
Table 2.
Primers sequence of quantitative real-time PCR
| Gene | Sense primer (5’→3’) | Antisense primer (5’→3’) |
|---|---|---|
| U6 | TGCGGGTGCTCGCTTCGGCAGC | CCAGTGCAGGGTCCGAGGT |
| GAPDH | GCACCGTCAAGGCTGAGAAC | TGGTGAAGACGCCAGTGGA |
| miR-449a | TGCGGTGGCAGTGTATTGTTAGC | CCAGTGCAGGGTCCGAGGT |
| MAP2K1 | GAATTCATGCCCAAGAAGAAGCCGACGCCCAT | CTCGAGTTAGACGCCAGCAGCATGGGTTGG |
Plasmid construction and Luciferase reporter assay
The full-length 3’untranslated region (3’UTR) of MAP2K1 was amplified from human genomic DNA, and the PCR product was cloned into pMIR-GLOTM Luciferase vector (pMIR, Invetrogen) downstream of the firefly luciferase coding region. The construct was named as pMIR-MAP2K1-wt. Mutations of miR-449a binding sites were introduced by site-directed mutagenesis and the resulted plasmid was called pMIR-MAP2K1-Mut. MAP2K1 overexpression was introduced by pCMV-MAP2K1 expression plasmid. Primer sequences used in construction were list in Table 2. All the sequences of resulted reporter plasmids were verified by sequence analysis. Luciferase assays were conducted using 1×104 L9981 cells plated in a 48-well plate. Co-transfection was performed using 200ng pMIR-MAP2K1-wt, pMIR-MAP2K1-mut or pMIR-GLOTM empty vector and either 80 ng miR-449a expression vector or pCDNA 3.1(+) empty vector. Forty-eight hours after transfection, the cells were harvested and assayed for both firefly and renilla luciferase using the Dual-luciferase glow assay (Promega, Madison, WI). All transfection experiments were conducted in triplicate.
ChIP assay
The cells were crosslinked and processed according to the Chromatin Immunoprecipitation (ChIP) Assay Kit (Invitrogen) protocol. Antibodies to H3K9me3 (Invitrogen) or H3K27me3 (Invitrogen) were used at 10 μg per immunoprecipitation reaction respectively. ChIP results were analyzed by real-time PCR.
Western blot analysis
L9981 cells were transfected with 100nmol miR-449a mimics or miR-449a inhibitor in six-well plate, the cells were washed twice with PBS after 48 h, then scraped off and harvested. Subsequently cells were lysed in RIPA buffer (contain 1‰ PMSF protease inhibitors) at 4°C for 30 min and centrifuged at 12,000 rpm for 15 min, the protein concentrations were quantified using the BCA protein assay (Pierce, Rockford, IL). Samples with 50 μg total protein were loaded on a 10% SDS-PAGE gel and transferred onto a NC (nitrocellulose) membrane. The membrane was incubated for 60 min in PBS containing 0.1% Tween-20 and 5% skim milk to block nonspecific binding, followed by incubation at 4°C overnight with antibodies against human MEK1, c-Jun, p-c-Jun, ERK1/2, p-ERK1/2 and MMP2 (Cell Signal Technology). The membrane was washed three times in PBS with 0.1% Tween-20 for 10 min and then incubated for 1 h with HRP-conjugated secondary antibody (Booster Biological Technology, Wuhan, China). The membrane was then washed thoroughly in PBS containing 0.1% Tween-20; blots were detected using ECL substrate following manufacturer’s recommendation. β-actin was used as an endogenous protein for normalization.
Cell invasion assay
Transwell assays were used to measure cell invasion capability. L9981 and A549 cells were transfected with miR-449a mimics or NC, NL9980 cell was transfected with miR-449a inhibitor or NC, respectively. Sixteen hours later, transfected cells were trypsinized and resuspended, 1.0×104 cells in 200 μl RPMI-1640 medium were placed into the upper chambers (8-mm pore size; Millipore). The lower chambers were filled with 600 μl complete medium with 10% FBS. After incubation for 48 h at 37°C, non-invading cells were removed from the top of the chamber with a cotton swab. The rescued assay, L9981 and A549 cell was transfected with miR-449a or co-transfected with miR-449a and pCMV-MAP2K1, NL9980 cell was transfected with anti-miR-449a or co-transfected with anti-miR-449a and siMAP2K1. 16 hours later, cells were processed as the described invasion assay. The invaded cells on the lower surface of the inserts were fixed and stained with 0.1% crystal violet, and five random fields for each insert were counted at 200× magnifications. Transwell assays were conducted in duplicate in two separate experiments.
In vivo metastasis assay
For the in vivo metastasis assays, the stable cell line L9981-luc-miR-449a and control cell line L9981-luc-pcDNA3.1 were collected and suspended in 0.2 ml PBS for each mouse (four in each group, 6-8 weeks age), and the cells were injected into left side of the posterior flank of nude mouse. Thirty minutes after cell injection, luciferase substrate was added at a dose of 150 mg/kg and live images of the mouse were obtained using an IVIS200 (Xenogene, USA). These data were classified as Day 0. Luciferase activity was measured every 6 days using the same protocol. Tumor growth and metastasis were measured periodically. After sacrifice, the lung and liver of the mice were dissected and aligned on culture plates in order to detect metastatic fluorescent foci on the surface. The areas of fluorescence were captured and a metastatic value using arbitrary units was calculated using Image-Pro plus software (Media Cybernetics, Bethesda, MD, USA).
Statistical analysis
The SPSS 13.0 software (IBM SPSS Inc., Chicago, IL, USA) was applied to complete data processing. Each experiment was repeated at least three times. Statistical significance was assessed by comparing mean values (± SD) using Student’s t test for independent groups, χ 2 test was used to evaluate the correlation of mir-449a expression and clinical pathologic parameters. Results were considered statistically significant when satisfied for P<0.05 (*) and P<0.01 (**).
Results
MiR-449a is downregulated in lung cancer cell and tumor tissues and its expression is inversely correlated with lung cancer metastasis
Although it has been reported that a number of miRNAs were implicated in NSCLC metastasis, additional novel miRNAs might be involved in this process due to the complexity of NSCLC metastasis. We analyzed the miRNA expression profile of lung cancer cell lines with high or low metastatic capacity, L9981 and NL9980, by miRNA microarray analysis. A total of 10 miRNAs were identified to be down-regulated in L9981 compared with NL9980 (Figure 1A). MiR-371-5p, miR-449a, miR-483-5p, miR-34a, miR-338-3p, miR-1224-5p, and miR-500 showed the lowest expression, among which the role of miR-449a in lung cancer metastasis remains unknown. qRT-PCR confirmed that miR-449a expression was significantly decreased in two lung cancer cell lines with highly metastatic capacity, L9981 and 95D, compared with lowly metastatic capacity cell line NL9980 and 95C. The expression of miR-449a was also downregulated in lung cancer cell line A549 and YTMLC9 (Figure 1B).
Figure 1.
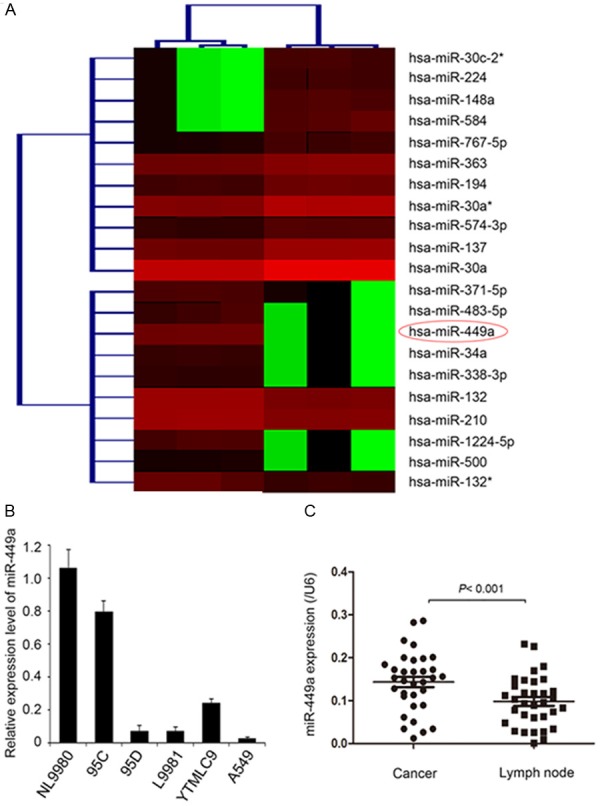
Expression of miR-449a was negatively correlated with metastasis of lung cancer. A. MicroRNA chips show the significant downregulation of miR-449a in L9981 cells which have strong metastatic ability. B. Detection of miR-449a expression in lung cancer cells with different metastasis ability using real-time PCR. C. Metastatic lymph node tissues showed lower expression of miR-449a compared with cancer tissues in 30 cases of matched clinical specimens. Statistical significance is indicated as: P<0.001, Student’s t test.
Furthermore, we found that the expression of miR-449a was significantly decreased in metastatic lymph nodes compared with primary tumor tissues of NSCLC (P<0.001, Figure 1C). To gain insight into the detailed biological role of miR-449a in human lung cancer progression, we analyzed the association between miR-449a expression and tumor clinicopathological features in 30 patients with NSCLC. The result showed that downregulation of miR-449a was significantly associated with smoking history (P=0.003), clinical stage (P=0.046), and lymph node metastasis (P=0.042). However, no significant correlation was observed between miR-449a expression and age, gender or tumor histology (Table 1). This result showed that miR-449a downregulation is correlated with increased lung cancer metastasis, suggesting that miR-449a might inhibit migration and invasion of lung cancer cells.
MiR-449a inhibits migration and invasion of NSCLC cells in vitro and in vivo
To test the role of miR-449a on the ability of lung cancer cell invasion, we increased miR-449a expression in L9981 and A549 cells by transfecting with miR-449a mimics. Mimics-dependent restoration of miR-449a expression in both cell types inhibited cell invasion by transwell assays (P=0.001 or P=0.003, Figure 2A). Consistently, the silence of miR-449a by miR-449a inhibitor in NL9980 cells promoted cell invasion (P=0.005, Figure 2A). To confirm the tumor suppressor function of miR-449a in vivo, L9981-luc cells stably expressing miR-449a (named L9981-luc-pcDNA-miR-449a) were subcutaneously implanted into nude mice to generate a xenograft model. Comparing with vehicle control, the fluorescence intensity of both primary tumor and metastatic nodules in the lung and liver was dramatically decreased in miR-449a-overexpressed group (Figure 2B). In addition, the volume of primary tumors was also significantly decreased in miR-449a-overexpressed group than control group (P=0.026, Figure 2C). As expected, miR-449 expression in the tumors was dramatically elevated in L9981-luc-pcDNA-miR-449a group comparing to the control, measured by real-time quantitative PCR (P=0.002, Figure 2D). These results demonstrated that miR-449a inhibited the invasive capability of lung cancer cells in vitro and in vivo.
Figure 2.

Demonstrate the relationship between miR-449a and lung cancer cell metastasis in cells and animals. (A) The invasion capability of L9981, A549, and NL9980 cells with indicated transfections were detected by transwell assay. The data is shown as mean ± SD. Student’s t test, **P<0.01. (B) The primary tumor site, lung and liver metastasis of L9981-luc-pcDNA-miR-449a and L9981-luc-pcDNA cells were detected by in vivo metastasis assay in mice. (C) The tumor size in xenograft experiments was shown. The data is shown as mean ± SD. Student’s t test, *P<0.05. (D) The miR-449a expression was detected by real-time quantitative PCR assays in each tumor as shown in (D). **P<0.01, Student’s t test.
MiR-449a inhibits MEK1/ERK1/2/c-Jun pathway in lung cancer cells
To investigate the underlying molecular mechanism by which miR-449a inhibits the migration and invasion of lung cancer cells, we identified the potential targets of miR-449a using several computational methods including TargetSan, PicTar (4-way) and miRanda. MAP2K1, one of the predicted potential targets of miR-449a, was selected as the candidate for further study due to its critical roles in cancer cell metastasis. To further determine whether miR-449a could directly associate with the conserved binding site in the 3’-untranslated region (3’UTR) of MAP2K1 mRNA, we constructed a reporter plasmid harboring the wild-type or mutant binding site sequence in MAP2K1 3’-UTR, which located downstream of luciferase reporter gene (Figure 3A and 3B). In L9981 cells transfected with wild-type reporter plasmid, miR-449a significantly decreased the luciferase activity compared with control (P=0.007, Figure 3C). However, in L9981 cells transfected with mutant binding site sequence, the luciferase activity was not affected by miR-449a (Figure 3C). Furthermore, overexpression of miR-449a in L9981 (P=0.003) and A549 (P=0.000) cells markedly inhibited the expression of MAP2K1 at both mRNA and protein level. In consistent with this finding, the mRNA and protein level of MAP2K1 were significantly increased by the downregulation of miR-449a in NL9980 (P=0.001) cells (Figure 3D and 3E). These results demonstrated that the expression of MAP2K1 is regulated by miR-449a through directly targeting its 3’UTR in lung cancer cells.
Figure 3.

MiR-449a could downregulate p-ERK, c-Jun, p-c-Jun and MMP2 through its target gene MAP2K1. A. Figures explained MAP2K1 3’ UTR and miR-449a complementary sequences. B. Construction of reporter gene plasmids and point mutation plasmids of MAP2K1 3’ UTR. C. Reporter assay results demonstrated can decrease the activity of wild-type gene plasmid MAP2K1 3’UTR report, but has no effect on mutant plasmids. The data shown as mean ± SD. Student’s t test, **P<0.01. D and E. L9981 and A549 was transfected with miR-449a and NL9980 was transfected with anti-miR-449a, then testing MAP2K1 expression in mRNA and protein. The data shown as the means ± SD. Student’s t test, **P<0.01. F. To detect the expression of MAP2K1, ERK1/2, p-ERK1/2, c-Jun, p-c-Jun and MMP2 in L9981 transfected with miR-449a, NL9980 transfected with anti-miR-449a and MEK inhibitor U0126. G. The invasion ability of L9981, A549, and NL9980 cells with indicated transfections were detected by transwell assays. The data shown as the means ± SD. Student’s t test, **P<0.01.
As the MEK1/ERK1/2/c-Jun pathway play a critical role in cell migration and invasion, we further investigated whether MEK1/ERK1/2/c-Jun pathway was manipulated by miR-449a through targeting MAP2K1. We found that overexpression of miR-449a in L9981 cells reduced the protein level of MAP2K1, p-ERK1/2, c-Jun, p-c-Jun, and MMP2 (Figure 3F). Moreover, inhibition of miR-449a in NL9980 cells increased the level of MAP2K1, p-ERK1/2, c-Jun, p-c-Jun, and MMP2. In addition, MEK1 inhibitor (U0126) could eliminate the upregulation of p-ERK1/2, c-Jun, p-c-Jun, and MMP2 by miR-449a. In order to verify that MAP2K1 mediates the role of miR-449a in suppressing NSCLC invasion, the rescued assay was performed. We discovered that the invasion ability of A549 and L9981 cells was restored by simultaneous overexpression of miR-449a and MAP2K1, compared with overexpression of miR-449a only. When both miR-449a and MAP2K1 were knocked down, the invasion ability of the cells was inhibited as miR-449a knockdown (Figure 3G). These results indicated that miR-449a regulated the activity of MEK1/ERK1/2/c-Jun pathway.
MiR-449a regulates MEK1/ERK1/2/c-Jun through an auto-regulatory feedback loop
MiR-449a was known to restrain E2F1 activity and cell proliferation through a negative feedback loop [18]. Accordingly, in this study, we investigated whether miR-449a could also utilize a negative feedback loop mechanism to inhibit MEK1/ERK1/2/c-Jun pathway, therefore limiting cell invasion and metastasis in lung cancer. The results showed that knockdown of MAP2K1 (MEK1) decreased miR-449a level in A549 and L9981 cells, whereas MAP2K1 overexpression increased miR-449a level in NL9980 cells (Figure 4A). The promoter sequence of miR-449a was analyzed using the TFSEARCH software online, and three AP-1 binding sites were identified at core promoter region of miR-449a (Figure 4B), which implicated that AP-2 might regulate the transcription of miR-449a. Therefore, the sequence flanking the core promoter region of miR-449a including wild type or mutant binding sites of AP-1 were subcloned into luciferase reporter plasmid (Pro-AP1 and Pro-AP1mut1, 2, 3) (Figure 4C). Luciferase reporter assays showed that overexpression of MAP2K1 resulted in an increased luciferase activity in NL9980 cells transfected with plasmid with wildtype AP1-2 binding site, compared with control, whereas, the mutant binding site of AP1-2 abrogated the activity of MAP2K1 (Figure 4C). Furthermore, the increased level of miR-449a induced by MAP2K1 overexpression was disrupted by c-Jun knockdown in NL9980 (Figure 4D). Consistently, MAP2K1 kn-ockdown eliminated miR-449a upregulation induced by c-Jun overexpression in A549 and L9981 cells (Figure 4E and 4F). Taken together, these results indicated that the miR-449a transcription was promoted by MAP2K1-activated c-Jun, which provided an auto-regulatory negative feedback circuit in which MAP2K1 activated c-Jun, subsequently promoted the transcription of miR-449a, further increased expression of MAP2K1 and the activation of MEK1/ERK1/2/c-jun pathway, which contributed to the promotion of metastatic ability of lung cancer cells.
Figure 4.

MAP2K1 controlled negative feedback regulation of miR-449a expression through c-Jun. A. miR-449a expression was detected by real-time PCR after MAP2K1 overexpression in NL9980 or knockdown in L9981 and A549. B. Three AP-1 binding sites in miR-449a promoter. C. Point mutation report gene test discovered only AP1-2 can affect MAP2K1 regulation of miR-449a promoter. D. Knockdown of c-Jun expression in NL9980 blocked MAP2K1 over-expression induced miR-449a upregulation. E and F. Over-expression of c-Jun inhibited miR-449a downregulation mediated by MAP2K1knockdown in L9981 and A549. The data is shown as the means ± SD. Student’s t test, **P<0.01.
Histone methylation mediates the decreased expression of miR-449a by SUZ12
MiR-449a was found to be consistently and markedly downregulated in highly metastatic lung cancer cell lines and metastatic lymph nodes [19], but the underlying mechanism was unknown. Previous studies revealed that miR-449a expression could be epigenetically repressed by histone modification in hepatocellular carcinoma cells [20]. Therefore, we investigated whether the repression of miR-449a expression was also regulated by histone methylation in lung cancer cells. MiR-449a is known to be mapped to the first intron of CDC20 on chromosome 5 and shares a common promoter with CDC20 (Figure 5A). ChIP assay was performed to analyze the histone methylation status of CDC20 promoter using two antibodies against specific histone modifications: H3K9me3 and H3K27me3, which were generally associated with chromatin with transcriptional activity. The results showed that the level of H3K27me3 was significantly higher in L9981 (P=0.005) and A549 (P=0.007) cells with upregulated miR-449a level than that in NL9980 cells with downregulated miR-449a, however, the level of H3K9me3 was not significantly altered among these three cell lines (Figure 5B). SUZ12 is an essential component of Polycomb Repressive Complex2 (PRC2), which modifies the transcription by affecting the methylation of histone and DNA (Pasini D et al). We also investigated whether SUZ12 might regulate the H3K27me3 level of CDC20 promoter. The expression of SUZ12 were markedly inhibited by siRNA in L9981 and A549 cells (Figure 5C), and the level of H3K27me3 was consequently reduced by SUZ12 siRNA in L9981 cells, compared with siRNA control (P=0.037, Figure 5D). In addition, the results showed that the expression of miR-449a were increased by SUZ12 siRNA in both L9981 (P=0.002) and A549 (P=0.001) cells (Figure 5E). These data suggested that the altered expression of miR-449a in lung cancer cells was regulated by histone methylation probably mediated by SUZ12.
Figure 5.
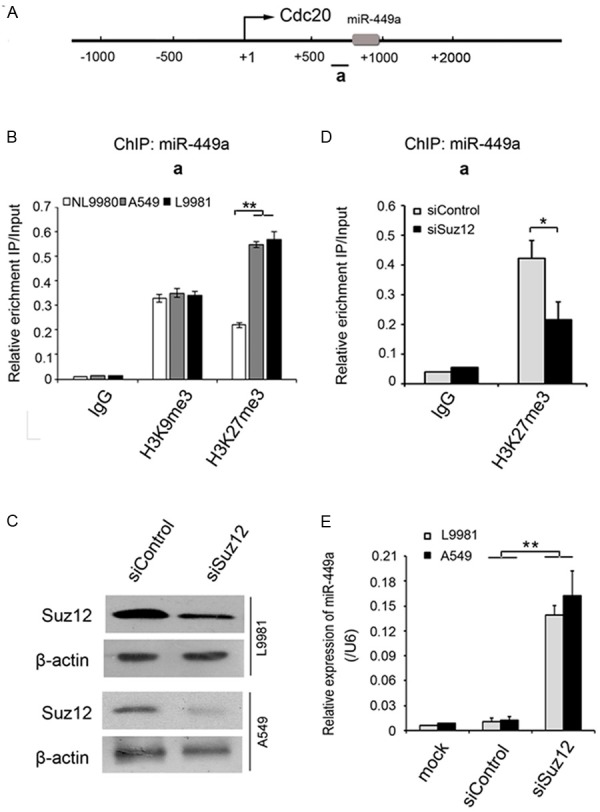
The histone H3K27me3 level of miR-449a promoter, which is mediated by SUZ12, correlates with metastasis capability in lung cancer cell lines. A. MiR-449a is located in the CDC20 gene. B. The histone H3K27me3 but not H3K9me3 level at miR-449a promoter is significantly higher in highly metastatic lung cancer cell line L9981 and A549, than low metastatic lung cancer cell line NL9980, which can be down-regulated by SUZ12 siRNA. C. The western blot assay confirmed SUZ12 expression interference. D. The histone H3K27me3 marks at miR-449a promoter was reduced after histone methyltransferase was disturbed in L9981 cells. E. MiR-449a expression was remarkably increased along with SUZ12 interference in L9981 and A549 cells. The data is shown as the means ± SD. Student’s t test, **P<0.01.
Discussion
Invasion and metastasis are the leading causes of cancer-related morbidity and mortality, especially for NSCLC. In NSCLC cases, the high recurrence rate often confounds the long term survival of patients after curative resection, which is mainly due to the spread of lymphatic metastasis. Therefore, it is critical to identify metastatic factors and better understand the underlying molecular mechanisms involved in the progression of invasion and metastasis. Recent findings from investigations of human cancer support a fundamental role of miRNA in human cancer invasion and metastasis [21,22], and studies to identify miRNA target genes have identified novel cancer invasion and metastasis suppressor genes, demonstrating that miRNAs are valuable tools for better understanding of cancer biology. Therefore, miRNAs started a novel avenue to uncover the molecular mechanism of cancer invasion and metastasis and to develop potential therapeutics against human cancer. Herein, we performed miRNA profiling of a pair of lung cancer cell lines, L9981 and NL9980, which had high and low metastatic ability, respectively, to identify miRNAs that might be involved in cancer invasion and metastasis.
In the present study, 21 miRNAs was found differentially expressed between L9981 and NL9980: 11 of which, including miR-224, -137, -363 etc., were validated to be up-regulated; and 10 of which, including miR-449a, -34a, -500 etc. were validated to be down-regulated in L9981. Among the miRNAs, miR-449a was taken for further characterization for several reasons. Firstly, the function of miR-449a is almost unknown. Secondly, patients with low miR-449a expression were more likely than high expression patients to have advanced disease (P=0.046) and lymph node metastasis (P=0.042). Finally, the level of miR-449a expression had an almost 10-fold difference between L9981 and NL9980 cells (between 95C and 95D). Consistent with our findings, a recent study showed miR-449a inhibits cell proliferation and is downregulated in gastric cancer [23]. All of these suggested a potential cancer invasion and metastasis suppressor role of miR-449a. In support of this notion, its suppressive activity to NSCLC invasion and metastasis was investigated in vitro and in vivo.
The fundamental function of miRNAs is to regulate target genes by direct target mRNA degradation or protein synthesis inhibition, depending on the degree of complementarity with the target 3’UTR [24]. Recent studies show that miRNAs can act as inhibitors of human cancer metastasis and invasion by targeting some oncogenes. It has been reported that the miR-409 suppresses tumor cell invasion and metastasis by directly targeting radix in gastric cancer [21]. MiR-218 inhibits the invasion and metastasis of gastric cancer by targeting the Robol receptor [25]. MiR-34a inhibits migration and invasion of colon cancer cells via targeting Fra-1 [26]. However, few studies have focused on the mechanisms of miR-449a-regulated NSCLC invasion and metastasis. To explore the molecular mechanism underlying miR-449a function, we use bioinformatics analysis to search for its target genes, especially those involved in cancer migration and/or invasion. MAP2K1 (MEK1) was detected by Target-Scan program. Results from luciferase reporter assays support that MAP2K1 is the direct target of miR-449a. MAP2K1 expression was also found suppressed by miR-449a mainly through translational inhibition. Therefore, MAP2K1 mRNA and protein levels were highly expressed in L9981 compared with NL9980.
MAPK is activated by specific MAPK kinase (MAPKK) through the phosphorylation of specific threonine and tyrosine residues (Thr-X-Tyr). MAPKK is also activated by MAPKK kinase via similar mechanisms [27,28]. These kinases constitute MAP kinase cascades. In mammalian cells, at least four MAPKs, including extracellular signal-regulated kinases (ERKs) [29], c-Jun N-terminal kinase/stress-activated protein kinases (JNK/SAPKs) [30], p38 [31], and ERK5/big MAP kinase have been identified [32].
We hypothesized that miR-449a inhibits the invasion and metastasis of NSCLC cells by regulating MEK1/ERK1/2/c-Jun signal pathway. Several salient findings reported here support this hypothesis. Firstly, forced miR-449a expression inhibited the expression of MAP2K1 (MEK1) in L9981 cells, concomitant with decreased p-ERK1/2, c-Jun, p-c-Jun, and MMP2. Secondly, inhibition of miR-449a in NL9980 cells enhanced the expression of MAP2K1, p-ERK1/2, c-Jun, p-c-Jun, and MMP2. Thirdly, up-regulation of p-ERK1/2, c-Jun, p-c-Jun, and MMP2 was inhibited after adding MEK1 inhibitor (U0126) in NL9980 cells. Moreover, miR-449a was recently shown to be directly transactivated by E2F1. It is very interesting that miR-449a is placed at a key node in a feedback circuit in which E2F1 transactivates the transcription of miR-449 that in turn targets and inhibits CDC25A and CDK6. Reduced levels of CDC25A and CDK6 result in the dephosphorylation of pRB and subsequently inhibits the release of E2F1 [33]. Whether miR-449a regulates MEK1/ERK1/2/c-Jun signal pathway through a feedback circuit was investigated. Our studies demonstrated that MEK1 up-regulated the transcription of miR-449 by c-Jun through MEK1/ERK1/2/c-Jun signal cascade. In turn, miR-449a inhibited MEK1 protein expression by directly targeting MAP2K1 (MEK1), suggesting a distinct negative feedback loop regulating the lung cancer invasion and metastasis. Collectively, these findings demonstrated that miR-449a inhibits the invasion and metastasis of lung cancer cells by regulating MEK1/ERK1/2/c-Jun pathway through an auto-regulatory negative feedback circuit.
SUZ12 could function as an oncogene by promoting proliferation and metastasis in human tumor, including ovarian cancer, breast cancer, and mantle cell lymphoma [34-37]. We confirmed the altered expression of miR-449a was regulated by histone methylation probably mediated by SUZ12 in lung cancer cells. Our study suggests that miR-449a is a novel tumor invasion and metastasis suppressor in lung cancer. Moreover, the present study demonstrates that MAP2K1 (MEK1) is a miR-449a target gene that is required for lung cancer cell invasion and metastasis. Finally, miR-449a inhibits the invasion and metastasis abilities of lung cancer cells by regulating MEK1/ERK1/2/c-Jun pathway through an auto-regulatory negative feedback circuit.
Furthermore, MEK1 can also be targeted by other miRNAs besides miR-449a. For example, a recent report indicates that both miR-1826 and miR-424 can suppress MEK1 expression by interacting with the same miR-449a binding site located in the MEK1 3’UTR [38,39], suggesting that multiple miRNAs may have an additive or synergetic effect on regulation of gene expression, which may also result in an additive or synergetic effect on inhibition/promotion of tumor invasion and metastasis. On the other hand, it is very likely that miR-449a may also regulate other genes simultaneously to inhibit NSCLC invasion and metastasis. For example, miR-449a also targets EFNB1. A recent study demonstrates that up-regulated expression of EFNB1 C-terminal peptide can suppress the dissemination of gastric cancer [40]. In addition to these three targets identified in this study, miR-449a may possess other targets yet to be identified. Therefore, the observed miR-449a-mediated inhibition of NSCLC invasion and metastasis is likely due to simultaneously targeting multiple genes.
Acknowledgements
This study was partly supported by the grants from National Natural Science Foundation of China (No. 81000950), National 863 Program (No. 2012AA02A201, No. 2012AA02A502), and National 973 Program (No. 2010CB529405).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Spira A, Ettinger DS. Multidisciplinary management of lung cancer. N Engl J Med. 2004;350:379–392. doi: 10.1056/NEJMra035536. [DOI] [PubMed] [Google Scholar]
- 3.Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y, Zhu X, Chen B, Wu J, Li M. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res. 2013;73:5402–5415. doi: 10.1158/0008-5472.CAN-13-0297. [DOI] [PubMed] [Google Scholar]
- 4.Yongchun Z, Linwei T, Xicai W, Lianhua Y, Guangqiang Z, Ming Y, Guanjian L, Yujie L, Yunchao H. MicroRNA-195 inhibits non-small cell lung cancer cell proliferation, migration and invasion by targeting MYB. Cancer Lett. 2014;347:65–74. doi: 10.1016/j.canlet.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188–6196. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- 7.Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krutzfeldt J, Poy MN, Stoffel M. Strategies to determine the biological function of microRNAs. Nat Genet. 2006;38(Suppl):S14–19. doi: 10.1038/ng1799. [DOI] [PubMed] [Google Scholar]
- 9.Cho WC. OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer. 2007;6:60. doi: 10.1186/1476-4598-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, Li Y, Lai M. The microRNA network and tumor metastasis. Oncogene. 2010;29:937–948. doi: 10.1038/onc.2009.406. [DOI] [PubMed] [Google Scholar]
- 11.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 12.Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy SD, Gajula RP, Pakala SB, Kumar R. MicroRNAs and cancer therapy: The next wave or here to stay. Cancer Biol Ther. 2010;9:479–482. doi: 10.4161/cbt.9.7.11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noonan EJ, Place RF, Pookot D, Basak S, Whitson JM, Hirata H, Giardina C, Dahiya R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene. 2009;28:1714–1724. doi: 10.1038/onc.2009.19. [DOI] [PubMed] [Google Scholar]
- 15.Bou KT, Futoma-Kazmierczak E, Jacobsen A, Krogh A, Bardram L, Hother C, Gronbaek K, Federspiel B, Lund AH, Friis-Hansen L. miR-449 inhibits cell proliferation and is down-regulated in gastric cancer. Mol Cancer. 2011;10:29. doi: 10.1186/1476-4598-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Lin YW, Mao YQ, Wu J, Liu YF, Zheng XY, Xie LP. MicroRNA-449a acts as a tumor suppressor in human bladder cancer through the regulation of pocket proteins. Cancer Lett. 2012;320:40–47. doi: 10.1016/j.canlet.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 17.Lize M, Klimke A, Dobbelstein M. MicroRNA-449 in cell fate determination. Cell Cycle. 2011;10:2874–2882. doi: 10.4161/cc.10.17.17181. [DOI] [PubMed] [Google Scholar]
- 18.Lize M, Pilarski S, Dobbelstein M. E2F1-inducible microRNA 449a/b suppresses cell proliferation and promotes apoptosis. Cell Death Differ. 2010;17:452–458. doi: 10.1038/cdd.2009.188. [DOI] [PubMed] [Google Scholar]
- 19.Luo W, Huang B, Li Z, Li H, Sun L, Zhang Q, Qiu X, Wang E. MicroRNA-449a is downregulated in non-small cell lung cancer and inhibits migration and invasion by targeting c-Met. PLoS One. 2013;8:e64759. doi: 10.1371/journal.pone.0064759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buurman R, Gurlevik E, Schaffer V, Eilers M, Sandbothe M, Kreipe H, Wilkens L, Schlegelberger B, Kuhnel F, Skawran B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA- 449 in hepatocellular carcinoma cells. Gastroenterology. 2012;143:811–820. e1–15. doi: 10.1053/j.gastro.2012.05.033. [DOI] [PubMed] [Google Scholar]
- 21.Zheng B, Liang L, Huang S, Zha R, Liu L, Jia D, Tian Q, Wang Q, Wang C, Long Z, Zhou Y, Cao X, Du C, Shi Y, He X. MicroRNA-409 suppresses tumour cell invasion and metastasis by directly targeting radixin in gastric cancers. Oncogene. 2012;31:4509–4516. doi: 10.1038/onc.2011.581. [DOI] [PubMed] [Google Scholar]
- 22.Huang Q, Gumireddy K, Schrier M, le SC, Nagel R, Nair S, Egan DA, Li A, Huang G, Klein-Szanto AJ, Gimotty PA, Katsaros D, Coukos G, Zhang L, Pure E, Agami R. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008;10:202–210. doi: 10.1038/ncb1681. [DOI] [PubMed] [Google Scholar]
- 23.Bou KT, Futoma-Kazmierczak E, Jacobsen A, Krogh A, Bardram L, Hother C, Gronbaek K, Federspiel B, Lund AH, Friis-Hansen L. miR-449 inhibits cell proliferation and is down-regulated in gastric cancer. Mol Cancer. 2011;10:29. doi: 10.1186/1476-4598-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regu- lation in animal cells. Curr Opin Cell Biol. 2009;21:452–460. doi: 10.1016/j.ceb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Tie J, Pan Y, Zhao L, Wu K, Liu J, Sun S, Guo X, Wang B, Gang Y, Zhang Y, Li Q, Qiao T, Zhao Q, Nie Y, Fan D. MiR-218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet. 2010;6:e1000879. doi: 10.1371/journal.pgen.1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu J, Wu G, Lv L, Ren YF, Zhang XJ, Xue YF, Li G, Lu X, Sun Z, Tang KF. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis. 2012;33:519–528. doi: 10.1093/carcin/bgr304. [DOI] [PubMed] [Google Scholar]
- 27.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 28.Zettergren JG, Peterson LL, Wuepper KD. Keratolinin: the soluble substrate of epidermal transglutaminase from human and bovine tissue. Proc Natl Acad Sci U S A. 1984;81:238–242. doi: 10.1073/pnas.81.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cobb MH, Robbins DJ, Boulton TG. ERKs, extracellular signal-regulated MAP-2 kinases. Curr Opin Cell Biol. 1991;3:1025–1032. doi: 10.1016/0955-0674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 30.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 31.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 32.Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Feng M, Jiang X, Wu Z, Li Z, Aau M, Yu Q. miR-449a and miR-449b are direct transcriptional targets of E2F1 and negatively regulate pRb-E2F1 activity through a feedback loop by targeting CDK6 and CDC25A. Genes Dev. 2009;23:2388–2393. doi: 10.1101/gad.1819009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb- mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39:761–772. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Cai Q, Wu H, Vathipadiekal V, Dobbin ZC, Li T, Hua X, Landen CN, Birrer MJ, Sanchez-Beato M, Zhang R. SUZ12 promotes human epithelial ovarian cancer by suppressing apoptosis via silencing HRK. Mol Cancer Res. 2012;10:1462–1472. doi: 10.1158/1541-7786.MCR-12-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Perez D, Sanchez E, Maestre L, Suela J, Vargiu P, Di LL, Martinez N, Alves J, Piris MA, Sanchez-Beato M. Deregulated expression of the polycomb-group protein SUZ12 target genes characterizes mantle cell lymphoma. Am J Pathol. 2010;177:930–942. doi: 10.2353/ajpath.2010.090769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasini D, Bracken AP, Jensen MR, Lazzerini DE, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirata H, Hinoda Y, Ueno K, Nakajima K, Ishii N, Dahiya R. MicroRNA-1826 directly targets beta-catenin (CTNNB1) and MEK1 (MAP2K1) in VHL-inactivated renal cancer. Carcinogenesis. 2012;33:501–508. doi: 10.1093/carcin/bgr302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakashima T, Jinnin M, Etoh T, Fukushima S, Masuguchi S, Maruo K, Inoue Y, Ishihara T, Ihn H. Down-regulation of mir-424 contributes to the abnormal angiogenesis via MEK1 and cyclin E1 in senile hemangioma: its implications to therapy. PLoS One. 2010;5:e14334. doi: 10.1371/journal.pone.0014334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka M, Kamata R, Yanagihara K, Sakai R. Suppression of gastric cancer dissemination by ephrin-B1-derived peptide. Cancer Sci. 2010;101:87–93. doi: 10.1111/j.1349-7006.2009.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.