

Abstract
The CIP2A gene is an oncogene associated with solid and hematologic malignancies [1]. CIP2A protein is an oncoprotein and a potential cancer therapy target [2]. Literature shows that CIP2A inhibits the tumor suppressor protein PP2A [3] which downregulates phophorylation of AKT, a hallmark of cancers [4] and stabilizes the proto-oncogene, c-MYC in tumor cells [5], the comprehensive action of CIP2A and its functional interaction(s) with other oncoproteins and tumor suppressors is not clearly established. Recently we tried to put forward a contextual mode-of-action of CIP2A protein in a review which proposed that CIP2A influences oncogenesis via an “oncogenic nexus” [1]. In this review we critically evaluated the potential relevance of the mode-of-action of the “oncogenic nexus” of CIP2A in breast carcinogenesis and appraised the role of this nexus in different PAM50 luminal A, PAM50 luminal B, PAM50 HER2-enriched and PAM50 basal BC. This review has a novel approach. Here we have not only compiled and discussed the latest developments in this field but also presented data obtained from c-BioPortal and STRING10 in order to substantiate our view regarding the mode-of-action of the “oncogenic nexus” of CIP2A. We functionally correlated alterations of genes pertaining to the “oncogenic nexus” of CIP2A with protein-protein interactions between the different components of the nexus including (1) subunits of PP2A, (2) multiple transcription factors including MYC oncogene and (3) components of the PI3K-mTOR and the MAPK-ERK oncogenic pathways. Using these proteins as “input” to STRING10 we studied the association, Action view, at the highest Confidence level. OncoPrints (c-BioPortal) showed alterations (%) of regulatory subunits genes of PP2A (PPP2R1A and PPP2R1B) along with alterations of CIP2A in breast invasive carcinoma (TCGA, Nature 2012 & TCGA, Provisional). Similar genetic alterations of PP2A were also observed in samples of breast tumors at our Avera Research Institute, SD. In an attempt to critically evaluate the role of “oncogenic nexus” of CIP2A in subtypes of BC, we used PPP2R1A and PPP2R1B as “inputs” into the STRING10 and obtained their predicted association (Action view) in respect to CIP2A. The outcome of this exercise has been discussed in the light of the literature in the BC research in the context of “oncogenic nexus” of CIP2A. In summary, herein we review the progress in our understanding of how CIP2A regulates oncogenic transformations of breast cells via PP2A-CIP2A “oncogenic nexus” and how we can prospect the clinical relevance of CIP2A in the context of BC.
Keywords: CIP2A, PP2A, breast tumors, prognosis, c-MYC, biomarkers
Introduction
Cancer is a complex and diverse group of disease characterized by the immortalization of differentiated cells, reactivation of stem cells and invasion of cells through different embryonic layers leading to unharnessed growth and metastasis of a tumor mass [6]. Tumorigenesis is initiated by a single and/or sequential multiple triggers/events/hits. Studies have established that triggers are almost always genetic in nature naming cancer as a genetic disease. Genetic events that trigger these oncogenic transformations, which bring a diverse range of changes in the context of different cancer types have so far demonstrated that upregulation of certain discrete common pathways including, (1) mutagenic activation of oncogenic pathways (RAS pathway, PI3K pathway and telomerase activation) as a result of growth factor receptor mutation/overexpression or activating mutations of several components of the respective pathway(s), (2) mutagenic inactivation of several tumor suppressor proteins (e.g. PP2A, p53, retinoblastoma Rb protein, and PTEN) and (3) mutagenic or epigenetic or stability dependent activation of oncogenic transcription factors in the transformed cells (e.g. c-MYC). Although broadly classified, these categories of oncogenic changes, once initiated are highly influential and interactive in orchestrating complete and enduring changes in the genetic and protein makeup of the cells, an “oncogenic nexus”, towards the clonal growth and metastasis of the tumor. The sequence of oncogenic events which are causally associated with the growth and metastasis of a tumor although unique to each patient, the course of growth, drug/radiation response and the development of resistance to drug/radiation can be attributed to a comparable consequences of genetic alterations in either their oncogene(s)/tumor suppressor(s) genes/oncogenic transcription factors which collectively constitute each patient’s “oncogenic nexus”. This review describes the involvement of the “oncogenic nexus” of CIP2A and presents the modus operandi of the “oncogenic nexus” of CIP2A [1] in breast carcinigenesis with special reference to different subtypes of breast cancers and evaluates the clinical relevance of PP2A-CIP2A “oncogenic nexus” in the context of targeted therapies.
“Oncogenic nexus” of CIP2A in breast cancers: how does it work?
CIP2A is an onco-protein [1,3,7]. KIAA1524 (Protein CIP2A; Cancerous inhibitor of PP2A; P90 Autoantigen; FLJ12850; MGC163436; p90) is a protein-coding gene for CIP2A protein. In 2002 Soo Hoo and group reported cloning and characterization of this novel 90 kDa ‘companion’ auto-antigen of p62 overexpressed in hepatocellular carcinoma [8]. Two alternatively spliced human isoforms of KIAA1524 have been described. The protein product of KIAA1524, CIP2A (referred as CIP2A hereafter) is an endogenous physiological inhibitor of tumor suppressor PP2A phosphatase. CIP2A oncoprotein (905 amino acids; 102185 Dalton) stabilizes MYC protein in human malignancies, promotes anchorage-independent cell growth and is involved in tumor formation. CIP2A is rarely present (low levels) in non-transformed/non-malignant adult cells. In a typically transformed cell where CIP2A is present in abundance, it is sub-cellularly localized as an integral part of plasma membrane and concentrated around the peri-nuclear region [8]. Using confocal microscopy, Peng and group were able to confirm their previous finding that p90/CIP2A was predominately localized in the cytoplasm in lung cancer specimens, which was consistent with their IHC data [9]. At the cellular level CIP2A is an integral protein which functions via protein binding.
CIP2A promotes tumorigenesis and associates with cancer progression in breast [10]. Yu and group studied the expression and regulatory effects of CIP2A protein in breast cancer and the correlation between CIP2A protein expression and the prognosis of breast cancer [11]. In determining the relationship between CIP2A protein and clinico-pathological parameters of breast cancer they reported that (1) CIP2A expression has linear correlation with lymph node metastasis as well as distant metastasis, tumor size, histological grade, triple-negative breast cancer, and TNM stage, (2) CIP2A expression also significantly related to chemotherapeutic sensitivity of breast cancer in the neo-adjuvant chemotherapy and (3) histological grade, lymph node metastasis, triple-negative breast cancer, and TNM stage were detected as the independent prognostic factors using Cox regression test. Testing the efficacy of bortezomib in one of the most aggressive subtype of BC, TNBC Tseng and group showed that bortezomib induced apoptosis in association with down-regulation of CIP2A and pAKT in a dose- and time-dependent manner in HCC1937, MDA-MB468 and MDA-MB231 but not in hormone positive MCF-7 cells. Their data indicate that inhibition of CIP2A determined the sensitivity of bortezomib to these TNBC cells. Bortezomib showed in vivo efficacy in HCC1937 xenografted tumor and targeting the CIP2A-PP2A-pAKT signaling pathway is a novel approach for the treatment of TNBC [12,13].
Functions and interactions of CIP2A with its major interacting partners like transcription factors including c-MYC, NFkBP65, p53, FOXO1, components of the PI3K-pathway including AKT and mTORC1 and proto-oncogene MDM2 of the “oncogenic nexus” are depicted in Figure 1. Functions of CIP2A in the context of mTOR functions have been demonstrated in tumor samples from breast cancers and breast cancer cell lines. Tissue microarray (TMA) analysis of 210 cancer samples revealed a highly significant, positive correlation between CIP2A expression and phosphorylation of mTORC1 substrate S6K1 [14]. CIP2A expression correlates with mTORC1 activity in primary human breast tumors [11,14] and ectopic expression of CIP2A is sufficient to increase mTORC1 activity in nonmalignant cells [14]. Overexpression of CIP2A has been shown to increase the proliferation of MDA-MB231 cells [15]. CIP2A is associated with clinical aggressiveness in human breast cancers and promotes the malignant growth of breast cancer cells and CIP2A was shown to support MDA-MB231 xenograft growth in nude mice [10].
Figure 1.
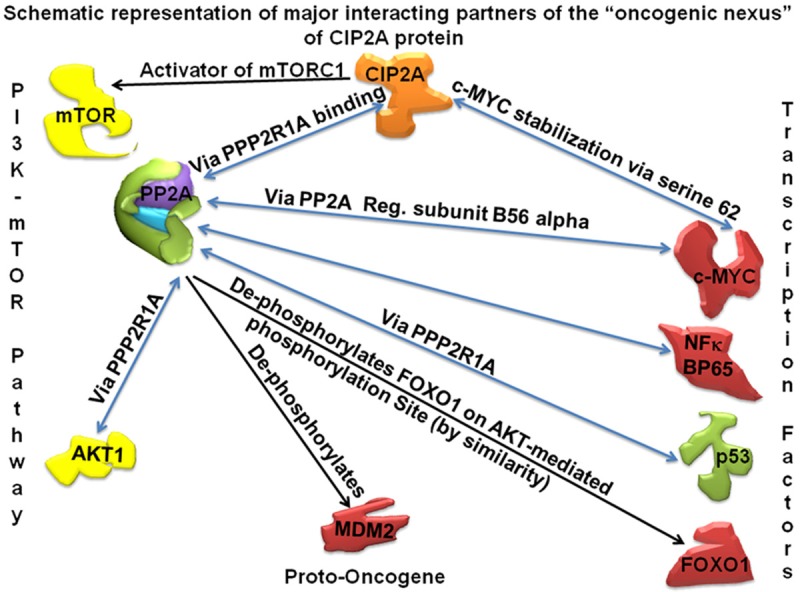
Functions and interactions of CIP2A and the “oncogenic nexus” of CIP2A are presented in a schematic representation of the major interacting partners of “oncogenic nexus” of CIP2A protein. Oncogene CIP2A is depicted in orange. Other proto-oncogenes, oncogenes and oncogenic Transcription Factors (TF) are depicted in red. Tumor suppressors genes (PP2A and p53) are depicted in green. Subunits of PP2A are schematically presented in different colors. Signaling components of the PI3K-mTOR pathway (AKT and mTOR) are depicted in yellow. Functional involvement (post-translational modifications etc) via binding of two proteins is shown in blue bidirectional arrows. Remaining functions are presented in black unidirectional arrows.
CIP2A functions via an “oncogenic nexus” [1]. Being a non-enzymatic integral protein, almost all of its functions are mediated through protein-protein interactions. Thus CIP2A functions via protein binding. Binary interactions of CIP2A occur with (1) PP2A, a tumor suppressor, (2) MYC, a pleiotropic transcription factor (MYC proto-oncogene protein, a Class E basic helix-loop-helix protein 39; Transcription factor p64), (3) polo like kinase (PLK1), (4) NIMA (Never In Mitosis Gene A)-related kinase 2 (NEK2) protein. Table 1 shows reports (PMIDs) on the protein molecules that are known to interact with PP2A and CIP2A. Considering direct interactions between PP2A and CIP2A, it is not surprising that CIP2A forms intricate connections with many proteins pertaining to oncogenesis which are affected by tumor suppressor PP2A phosphatase [7,16] in order to complete its functions. Figure 1 shows a schematic representation of the various functions of CIP2A in a tumor cell. The “oncogenic nexus” of CIP2A consists of (1) an inhibitory effect of CIP2A on the tumor suppressor PP2A phosphatase, (2) a direct interaction of CIP2A with c-MYC protein, (3) PP2A independent functions of CIP2A, (4) regulatory type interactions of different proteins with CIP2A and (5) effect of CIP2A on mTOR functions.
Table 1.
Table showing reports (PMIDs) on the molecules that are known to interact with PP2A and CIP2A proteins
| PP2A interacts with… | References PMID | CIP2A interacts with… | References PMID |
|---|---|---|---|
| CIP2A | 17632056 | PP2A | 17632056 |
| B-Catenin | 23639323 | C-MYC | 17632056 |
| AKT | 16299534 | CHEK1 | 24072747 |
| 18042541 | |||
| 24052256 | |||
| Bcl2 | 18845789 | PLK1 | 23983103 |
| 9852076 | |||
| JAK | 11440634 | DNA-PK | 24072747 |
| 12921784 | |||
| EGFR-MEK-ETS1 | 21445343 | ||
| p53 | 23306062 | ||
| E2F1 binds to CIP2A promoter | 23306062 | ||
| ETS1 | 21445343 | ||
| 23117818 | |||
| ATF2 | 21706057 | ||
| AKT | 20729919 | ||
| BCR/ABL | 21490338 | ||
| APC | 23983103 |
Table 1 shows reports (PMIDs) on the molecules that are known to interact with PP2A and CIP2A. Considering the direct interactions between PP2A and CIP2A, it is not surprising that CIP2A forms intricate connections with many proteins pertaining to oncogenesis.
CIP2A is a positive regulator of mTORC1 activity and inhibitor of autophagy. There is a dynamic interaction of the CIP2A, PP2A, and mTORC1 complex in the regulation of mTORC1 activity. Recently, Puustinen and group identified a novel role of CIP2A as a key modulator of mTORC1 and autophagy in BC. This oncogenic function of CIP2A is via direct regulation of the mTORC1 pathway and autophagy [14]. Using ribonucleic acid interference screens for autophagy-regulating phosphatases in human BC cells, they identified that CIP2A associates with mTORC1 (a PI3K pathway downstream effector which acts as a sensor of nutrients and energy in cells in order to coordinate protein synthesis and autophagy). Their study demonstrated for the first time that the CIP2A-PP2A complex associates with mTORC1 wherein CIP2A functions as a context-specific allosteric inhibitor of PP2A activity in mTORC1 complex and thereby enhances mTORC1-dependent growth signals. They identified CIP2A as a potent inhibitor of autophagic flux in a mTORC1-dependent manner and this regulation of autophagy and mTORC1 activity by CIP2A is independent of CIP2A’s effect on c-MYC and AKT. These results reiterated the existence of the “oncogenic nexus” of CIP2A in breast cancer. CIP2A affects mTORC1-mediated cellular processes, like cell size, proliferation, and morphology. On the other hand, mTORC1-dependent control of CIP2A degradation is a mechanism that links mTORC1 activity with c-MYC stability to coordinate cellular metabolism, growth, and proliferation. Thus CIP2A induces a positive feedback loop that enhances tumorigenesis in breast. Consistent with CIP2A’s reported ability to protect c-MYC against proteasome-mediated degradation, autophagic degradation of CIP2A upon mTORC1 inhibition leads to destabilization of c-MYC.
Data analyses using c-BioPortal: OncoPrint
We studied the alterations in Gene KIAA1524 (GAIN, AMP) in Breast Invasive Carcinoma (TCGA, Provisional) case set and in Breast Invasive Carcinoma (TCGA, Nature 2012 ) case set using c-BioPortal (Figure 2). Genomic study selected were (1) mutations, (2) putative copy-number alteration from GISTIC, (3) mRNA expression Z-scores (microarray) with Z-score thresholds ±2.0, and (4) protein/phosphoprotein level (RPPA) with Z-score thresholds ±2.0. Patient selected were, (1) PAM50 Basal, (2) PAM50 Claudin low, (3) PAM50 HER2 enriched, (4) PAM50 Luminal A, (5) PAM50 Luminal B and (6) PAM50 Luminal (A/B). Advanced cancer genomic data visualization is obtained with the help of “The Onco Query Language (OQL)”. We used the Onco Query Language (OQL) to select and define genetic alterations for all the OncoPrint outputs on the c-BioPortal for Cancer Genomics. OncoPrints are compact means of visualizing distinctive genomic alterations, including somatic mutations, copy number variations (CNV), and mRNA expression changes occurring across a set of cases. In c-BioPortal, OncoPrints are used for visualizing gene set as well as pathway alterations across a set of cases, and for visually identifying trends, such as trends in mutual exclusivity or co-occurrence between gene pairs within a gene set. Individual genes are represented as rows, and individual cases or patients are represented as columns.
Figure 2.
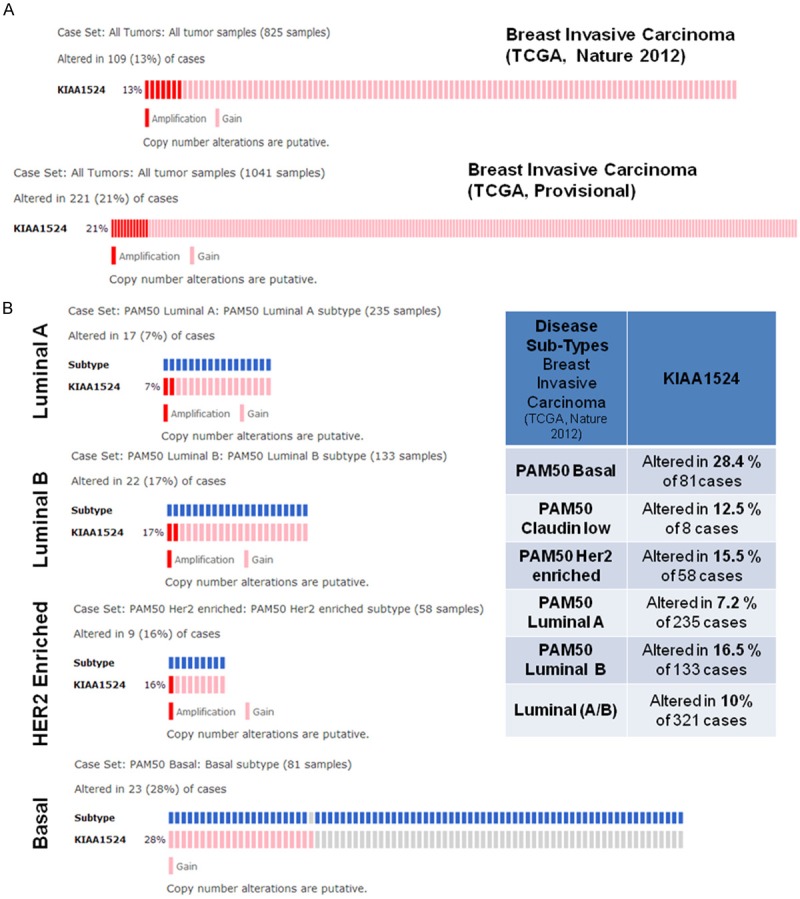
A. Alterations in Gene KIAA1524 (GAIN AMP) in Breast Invasive Carcinoma (TCGA, Provisional) case set (all 1041 tumor samples) as shown in the lower panel and in Breast Invasive Carcinoma (TCGA, Nature 2012) case set (all 825 tumor samples) as shown in the upper panel. Cancer Study Selected was Breast Invasive Carcinoma (TCGA, Nature 2012). Genomic study selected were (1) mutations, (2) putative copy-number alteration from GISTIC, (3) mRNA expression Z-scores (microarray) with Z-score thresholds ±2.0, and (4) protein/phosphoprotein level (RPPA) with Z-score thresholds ±2.0. Patient selected were, (1) PAM50 Basal, (2) PAM50 Claudin low, (3) PAM50 HER2 enriched, (4) PAM50 Luminal A, (5) PAM50 Luminal B and (6) PAM50 Luminal (A/B). Advanced cancer genomic data visualization is obtained with the help of “The Onco Query Language (OQL)”. We used the Onco Query Language (OQL) to select and define genetic alterations for all the OncoPrint outputs on the c-BioPortal for Cancer Genomics. Oncoprints (different levels of zoom) have been generated using c-BioPortal. Unaltered cases were removed. B. Alterations in Gene KIAA1524 (GAIN AMP) in Breast Invasive Carcinoma (TCGA, Nature 2012) from four major case sets: (1) PAM50 luminal A subtype (all 235 tumor samples), (2) PAM50 luminal B subtype (all 133 tumor samples), (3) PAM50 HER2 enriched subtype (all 58 tumor samples) and (4) PAM50 Basal subtype (all 81 tumor samples). Oncoprints (different levels of zoom) have been generated using c-BioPortal (left Panel). Unaltered cases were removed. Table shows cross breast cancer alteration summary for KIAA1524 (GAIN AMP): The table was generated using c-BioPortal. Tumor types (tumor data sets) are chosen in accordance with the publication guidelines (last updated on January 17th, 2014) of TCGA (tcga@mail.nih.gov). Cancer study selected was Breast Invasive Carcinoma (TCGA, Nature 2012). Genomic study selected were (1) mutations, (2) putative copy-number alteration (CNA) from GISTIC, (3) mRNA expression Z-scores (microarray) with Z-score thresholds ±2.0, and (4) protein/phospho-protein level (RPPA) with Z-score thresholds ±2.0. Patient selected were, (1) PAM50 Basal, (2) PAM50 Claudin low, (3) PAM50 HER2 enriched, (4) PAM50 Luminal A, (5) PAM50 Luminal B and (6) PAM50 Luminal (A/B). Advanced cancer genomic data visualization is obtained with the help of “The Onco Query Language (OQL)”. We used the Onco Query Language (OQL) to select and define genetic alterations for all the OncoPrint outputs on the c-BioPortal for Cancer Genomics. The table (right panel) shows cross breast cancer subtypes alteration summary for KIAA1524 (GAIN AMP). The table was generated using c-BioPortal. Tumor types (tumor data sets) are chosen in accordance with the publication guidelines (last updated on January 17th, 2014) of TCGA (tcga@mail.nih.gov). Cancer study selected was Breast Invasive Carcinoma (TCGA, Nature 2012). Genomic study selected were (1) mutations, (2) putative copy-number alteration (CNA) from GISTIC, (3) mRNA expression Z-scores (microarray) with Z-score thresholds ± 2.0, and (4) protein/phospho-protein level (RPPA) with Z-score thresholds ±2.0. Patient selected were, (1) PAM50 Basal, (2) PAM50 Claudin low, (3) PAM50 HER2 enriched, (4) PAM50 Luminal A, (5) PAM50 Luminal B and (6) PAM50 Luminal (A/B). c-BioPortal data is subjected to scheduled updates.
We found alterations (percentage) of the PPP2R1A, PPP2R1B and KIAA1524 genes in (a) breast invasive carcinoma (TCGA, Nature 2012 and TCGA, Provisional) (Figure 3A) and (b) in different subtypes (PAM50 Luminal A, PAM50 Luminal B, PAM50 HER2 enriched, and PAM50 Basal) of breast invasive carcinoma (TCGA, Nature 2012) (Figure 3B) using c-BioPortal. We acknowledge the c-BioPortal for Cancer Genomics site (http://cbioportal.org) which provides a Web resource for exploring, visualizing, and analyzing multidimensional cancer genomics data. The portal reduces molecular profiling data from cancer tissues and cell lines into readily understandable genetic, epigenetic, gene expression and proteomic events (Gao and group 2013, Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the c-BioPortal, Sci. Signal., 2 April, Vol. 6, Issue 269, p. pl1. We acknowledge works of Cerami and group (The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. May 2012 2; 401) and works of Gao and group (Integrative analysis of complex cancer genomics and clinical profiles using the c-BioPortal. Sci. Signal. 6, pl1, 2013). We acknowledge the TCGA Research Network for generating TCGA datasets.
Figure 3.
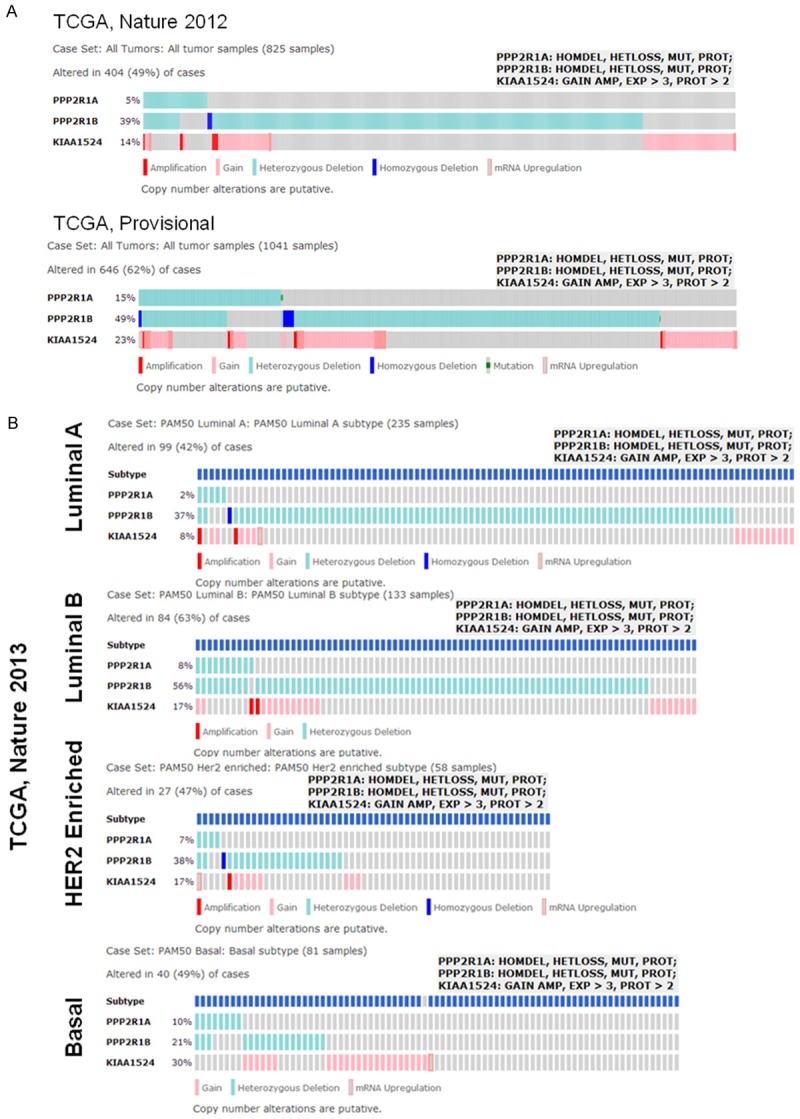
OncoPrints showing alterations (percentage) of the PPP2R1A, PPP2R1B and KIAA1524 genes in breast invasive carcinoma (TCGA, Nature 2012 and TCGA, Provisional) (A) and in different subtypes (PAM50 Luminal A, PAM50 Luminal B, PAM50 HER2 enriched, and PAM50 Basal) of breast invasive carcinoma (TCGA, Nature 2012) (B). OncoPrints (different levels of zoom) are obtained from c-BioPortal. Advanced cancer genomic data visualization is obtained with the help of “The Onco Query Language (OQL)”. We used the Onco Query Language (OQL) to select and define genetic alterations for all the OncoPrint outputs on the c-BioPortal for Cancer Genomics. Onco Query Language (OQL) used for the PPP2R1A, PPP2R1B and KIAA1524 genes in the search are PPP2R1A: HOMDEL, HETLOSS, MUT, PROT; PPP2R1B: HOMDEL, HETLOSS, MUT, PROT; KIAA1524: GAIN AMP, EXP>3, PROT>2 as shown in the figure. Grey bars represent unaltered cases. c-BioPortal data is subjected to scheduled updates. We acknowledge the c-BioPortal for Cancer Genomics site (http://cbioportal.org) which provides a web resource for exploring, visualizing, and analyzing multidimensional cancer genomics data.
Results (Figure 2A) show that there is a comparable pattern of changes in the relative percentage of alterations in KIAA1524 (Gain and Amplification) between two data sets. Considering the intricate relationship of CIP2A with PP2A within the “oncogenic nexus” in breast cancer, we also used c-BioPortal to find the alterations (percentage) of the PPP2R1A, PPP2R1B and KIAA1524 genes in breast invasive carcinoma (TCGA, Nature 2013 and TCGA, Provisional) (Figure 3A). Results also show that there is a comparable pattern of changes in the relative percentage of alterations in PPP2R1A (HOMDEL, HETLOSS, MUT, PROT), PPP2R1B (HOMDEL, HETLOSS, MUT, PROT) and KIAA1524 (GAIN AMP, EXP>3, PROT>2) genes between two data sets. Within a data set, the highest percentages of alterations are observed in PPP2R1B followed by the change in the KIAA1524 and in the PPP2R1A. TCGA provisional data set shows that almost 50% alteration was observed in PPP2R1B (HOMDEL, HETLOSS, MUT, PROT).
Subtype specific changes in CIP2A gene in breast cancers
In BC, CIP2A signature reveals the MYC dependency of CIP2A-regulated phenotypes. Niemelä and group in studying the clinical relevance of the CIP2A-regulated transcriptome in breast cancer subtypes have reported a high-confidence transcriptional signature that is regulated by CIP2A [17]. Bio-informatics pathway analysis of the CIP2A signature revealed that CIP2A regulates several MYC-dependent as well as MYC-independent gene programs [17]. With regard to MYC-independent signaling, JNK2 expression and transwell migration were inhibited by CIP2A depletion. CIP2A depletion was shown to regulate the expression of several established MYC target genes, out of which most were MYC-repressed genes. The CIP2A signature was shown to cluster with basal-type and human epidermal growth factor receptor HER2+ breast cancer signatures. Accordingly, CIP2A protein expression was significantly associated with basal-like (P=0.0014) and HER2+ (P<0.0001) breast cancers. CIP2A expression also associated with MYC gene amplification (P<0.001). With regard to MYC, these results both validate CIP2A’s role in regulating MYC-mediated gene expression and provide a plausible novel explanation for the high MYC activity in basal-like and HER2+ breast cancers.
Using the c-BioPortal for Cancer Genomics, Baldacchino and group reported that PP2A, a phosphatase which is inhibited by CIP2A and which inactivates mTOR effectors is deregulated in 59.6% of basal breast tumours [18]. They tested the viability assays to determine the sensitivity of a panel of breast cancer cell lines (MDA-MB468, MDA-MB436, Hs578T and BT20) to FTY720, a PP2A activator, to indicate that cell lines associated with ER loss are sensitive to lower doses of FTY720. It can be argued that the subset of patients with suppressed the activity of PP2A phosphatase would be potentially eligible for treatment using therapies which target the PI3K/AKT/mTOR pathway, such as phosphatase activators. Inactivation of PP2A by phosphorylation at tyrosine 307 has been demonstrated to be significantly correlated with HER2+ tumor progression [19]. Figure 3B shows that in 38% of the PAM50 HER2-enriched cases the PPP2R1B gene has been altered. The alterations in the PPP2R1B gene were 37% and 56% in the PAM50 luminal A and luminal B subtypes respectively. The PPP2R1B gene is altered in 21% of the PAM50 basal subtype cases. Loss of functions of the PTEN phosphatase [20] and inositol polyphosphate 4-phosphatase type II (INPP4B) [21] has been also associated with aggressive basal-like BC. Since PTEN, INPP4B and PP2A phosphatases are all known antagonists of AKT phosphorylation, a functional loss of phosphatase function would eventually lead to increased AKT activation. Interestingly enough BRCA1 is known to activate PP2A, which dephosphorylates AKT at both threonine 308 and serine 473 [22,23]. Furthermore BRCA1 is known to bind phosphorylated AKT leading to its ubiquitination [24] and subsequent degradation. Thus an enhanced stability and higher expression of pAKT characterizes a mutation in BRCA1 in which the mutant BRCA1 fails to bind to pAKT [24]. This report is substantiated by the findings that BRCA1 loss leads to an increase in AKT activity [24] and a reduction of PP2A activity [22]. Overexpression of the biomarkers of PP2A function including pS6K and pAKT has been reported in breast and ovarian tumors reflecting an attenuation of PP2A activity [25-28]. Thus a contextual downregulation of the tumor suppressor components of the “oncogenic nexus” of CIP2A potentiates the effect of upregulation of the oncogenic components of the “oncogenic nexus” in a concerted way towards the ensuing oncogenesis in the different subtypes of breast cancers.
Studies by Choi and group demonstrated a differential expression of CIP2A in ER-positive tissues as compared to ER-negative tissues which in their study can be explained by the result that CIP2A is a key factor in E2 (17β-estradiol)-enhanced proliferation and that estrogen regulates CIP2A expression by non-genomic action through EGFR [29]. Mechanistically at the translational level E2 (17β-estradiol) increased CIP2A expression in a c-MYC-independent manner in MCF-7 cells. Conversely E2-enhanced proliferation was found impaired without CIP2A expression. E2 (17β-estradiol)-mediated EGFR activation leads to the upregulation of the MAPK and PI3K pathways resulting in the phosphorylation of p70S6 kinase (S424/T421, T229, and T389) which ultimately culminates into an eIF4B-mediated increase in CIP2A translation. Our data derived from the c-BioPortal showed that alterations of CIP2A genes (predominantly copy number gain and amplifications) is highest in the PAM50 basal subtype of BC (Figure 2B) which matched a higher percentage of alterations of gene corresponding to protein subunit (predominantly homozygous and heterozygous deletions) of PP2A gene (Figure 3B). In the context of this result studies by Choi and group had indicated that CIP2A can also be a key factor in E2 (17β-estradiol)-enhanced proliferation wherein estrogen regulates CIP2A expression by non-genomic action through EGFR. However in the same light of this result it will be interesting to study CIP2A expression in an EGFR overexpressed TNBC subset of breast cancers [30] since ETS1 has been proposed to mediate high CIP2A expression in human cancers with increased the EGFR-MEK1/2-ERK pathway activity [31].
CIP2A has been recommended to be a potential therapeutic target in TNBC and CIP2A is a major determinant responsible for bortezomib-induced apoptosis in TNBC cells [13]. Come and group [10] demonstrated that siRNA mediated CIP2A depletion inhibited tumor growth of MDA-MB-231 xenograft tumors. Similarly Tseng and group showed that bortezomib downregulated CIP2A in HCC1937 xenograft tumors and inhibited their tumor growth [13]. We computed the alterations in Gene KIAA1524 (GAIN, AMP) in Breast Invasive Carcinoma (TCGA, Nature 2012) from four major subtypes, PAM50 luminal A, PAM50 luminal B, PAM50 HER2 enriched and PAM50 Basal (Figure 2B). The data showed that the more aggressive subtype of luminal B exhibits more than double percent higher alteration (17%) than the less aggressive luminal A (7%). Interestingly, luminal B subtype has a similar percentage of alterations to the other aggressive HER2 enriched subtype (16%). The most aggressive PAM50 basal subtype exhibited the highest percentage of alterations (28%). OncoPrints (Figure 3B) showed alterations (percentage) of the PPP2R1A, PPP2R1B and KIAA1524 genes in different subtypes (PAM50 Luminal A, PAM50 Luminal B, PAM50 HER2 enriched, and PAM50 Basal) of breast invasive carcinoma (TCGA, Nature 2013). Data showed that PAM50 luminal A, PAM50 luminal B and PAM50 HER2 enriched subtypes exhibit the same pattern of relative changes between PPP2R1A, PPP2R1B and KIAA1524 genes (PPP2R1B > KIAA1524 > PPP2R1A). Interestingly PAM50 basal on the other hand exhibited a distinct pattern of alterations (KIAA1524 > PPP2R1B > PPP2R1A). The significance of this characteristic pattern is not known yet. It is interesting that the ratio of percentage of alterations of PPP2R1B to KIAA1524 of each subtype was negatively correlated to the overall aggressive nature of the subtype. The ratio of alterations in PAM50 luminal A subtype (4.6) was greater than PAM50 luminal B subtype (3.2) which was greater than the ratio in PAM50 HER2 enriched subtype (2.2). The lowest ratio of alterations of PPP2R1B to KIAA1524 was observed in PAM50 basal subtype (0.7). The significance of this observation at the subtype level of BC remains to be identified.
Understanding the interaction between different proteins within the “oncogenic nexus of CIP2A”: network summary using STRING10
We used the network image of the “Action View” of the network summary of proteins pertaining to the components of the “oncogenic nexus of CIP2A” using STRING10 (Figure 4). These proteins are termed as “inputs”. Network is displayed as nodes which are either colored (as they are directly linked to the input of protein corresponding to genes). Edges, i.e. predicted functional links, consist of eight lines: one color for each type of evidence (as shown in the picture) (Figure 4A). We used STRING10 to predict the interactions and associations between different components (“input”) of the “oncogenic nexus of CIP2A”. The “input” proteins are as follows:
Figure 4.
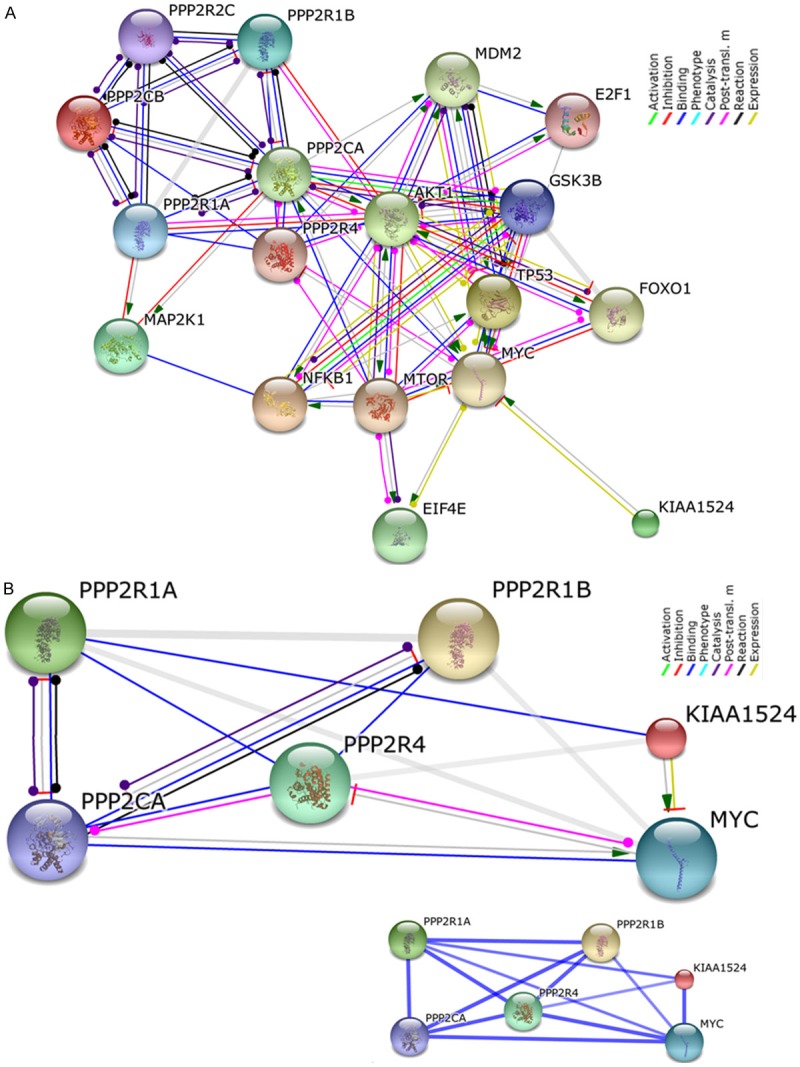
Network display of the interaction between major components of the “oncogenic nexus” of CIP2A (as represented in the Figure 1 and [1]): A. The interaction included different subunits of PP2A tumor suppressor protein, different transcription factors including MYC oncogene and the interacting components of major oncogenic pathways like PI3K-mTOR pathway as well as MAPK-ERK pathway (the details about the method of analyses are described in the text). B. Network display of the interaction between CIP2A, MYC and genes corresponding to subunits of PP2A protein, the PPP2R1A, and PPP2R1B which are mostly altered in BC. “Action View” is presented with “Confidence View” as the inset. The “input” proteins (KIAA1524, PPP2R1B, PPP2R1A, PPP2R4, MYC, and PPP2CA) were entered into STRING10 in order to find the association at the medium confidence view, 0.400. Stronger associations are represented by thicker lines. The details regarding the active prediction methods are included in the text. We acknowledge the STRING10 (Search Tool for the Retrieval of Interacting Genes/Proteins).
PPP2CB: protein phosphatase 2, catalytic subunit, beta isozyme; NFKB1: nuclear factor of kappa light polypeptide gene enhancer in B-cells 1; NF-kappa-B is a pleiotropic transcription factor. TP53: tumor protein p53; Acts as a tumor suppressor in tumor types; induces growth arrest/apoptosis in a context dependent manner. AKT1: v-akt murine thymoma viral oncogene homolog 1; AKT1 is serine/threonine-protein kinase which regulates metabolism, proliferation, cell survival, growth and angiogenesis. KIAA1524: Oncoprotein which inhibits PP2A and stabilizes MYC in many human malignancies. MAP2K1: mitogen-activated protein kinase kinase 1; Dual specificity protein kinase which is an essential component of the MAP kinase signal transduction pathway. PPP2R1B protein phosphatase 2, regulatory subunit A, beta; The PR65 subunit of protein phosphatase 2A acts as a scaffolding molecule to assemble of the catalytic subunit and a variable regulatory B subunit. PPP2R1A: protein phosphatase 2, regulatory subunit A, alpha; The PR65 subunit of protein phosphatase 2A acts as a scaffolding molecule to manage the assembly of the catalytic subunit and a variable regulatory B subunit. GSK3B: glycogen synthase kinase 3 beta; Components of PI3K pathway and Wnt-beta catenin pathway. PPP2R2C: protein phosphatase 2, regulatory subunit B, gamma; The B regulatory subunit modulates substrate selectivity and catalytic activity, and also might direct the localization of the catalytic enzyme to a particular subcellular compartment. E2F1, E2F transcription factor 1; Transcription activator which binds DNA cooperatively with DP proteins through the E2 recognition site, 5’-TTTC[CG]CGC-3’. It is found in the promoter region of a number of genes involved in cell cycle regulation/DNA replication. E2F1 binds preferentially RB1 in a cell-cycle dependent manner. PPP2R4: protein phosphatase 2A activator, regulatory subunit 4. mTOR: mechanistic target of rapamycin (serine/threonine kinase); serine/threonine protein kinase which regulates cellular metabolism, mRNA translation and ribosome synthesis, growth and survival in response to hormones, growth factors, nutrients, energy and stress signals. mTOR functions as mTORC1 and mTORC2 complexes. MYC: v-MYC myelocytomatosis viral oncogene homolog (avian); Participates in the regulation of gene transcription associated to oncogenesis. FOXO1: forkhead box O1; Transcription factor and the main target of insulin signaling (via binding to the insulin response element) and regulate metabolic homeostasis in response to oxidative stress. MDM2: Mdm2, p53 E3 ubiquitin protein ligase homolog (mouse). PPP2CA: protein phosphatase 2, catalytic subunit, alpha isozyme; PP2A is the major phosphatase for microtubule-associated proteins (MAPs). EIF4E: eukaryotic translation initiation factor 4E; It stimulates translation. These proteins were used as “input” to STRING10 in order to find the association at the confidence view, 0.900 (highest confidence). Action view is presented with the modes of interaction in different color as the inset. The active prediction methods included “Neighborhood”, “gene Fusion”, “Co-occurrence”, “Co-expression”, “Experiments” “Databases” and “Textmining”. Custom limit was chosen for the interactors (as shown in Figure 4A). The subunits of PP2A gene, the PPP2R1A, PPP2R1B are altered (percentage) genes in breast invasive carcinoma (TCGA, Nature 2013 and TCGA, Provisional) and in different subtypes (PAM50 Luminal A, PAM50 Luminal B, PAM50 HER2 enriched, and PAM50 Basal) of breast invasive carcinoma (TCGA, Nature 2013) presented OncoPrints (different levels of zoom) as obtained from c-BioPortal in Figure 3. The interaction between CIP2A, MYC and two protein products of PP2A gene, the PPP2R1A, and PPP2R1B are studied using STRING10 (as shown in the Network display; Figure 4B). “Action View” is presented with “Confidence View” as the inset. The active prediction methods included “Neighborhood”, “gene Fusion”, “Co-occurrence”, “Co-expression”, “Experiments” “Databases” and “Textmining”. Custom limit was chosen for the interactors. The “input” proteins (KIAA1524, PPP2R1B, PPP2R1A, PPP2R4, MYC, and PPP2CA) were entered into STRING10 in order to find the association at the medium confidence view, 0.400. STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) is being developed at CPR (NNF Center for Protein Research), EMBL (European Molecular Biology Laboratory), SIB (Swiss Institute of Bioinformatics), KU (SUND-KU, University of Copenhagen), TUD (Technical University Dresden, Biotec) and UZH (University of Zurich). The version 10 of STRING covers more than 2000 organisms, and is equipped with improved prediction algorithms. We acknowledge the version 10 of STRING, and authorities/institutions/organizations/Universities/resources which institutionally and financially support the STRING.
Computation of scores using STRING10: The selected reasonable score cut-off value for the analyses was carried out by using STRING10. The setting of the cutoff level of confidence at the lower level increased coverage for interactions. We used the score cut-off to limit the number of interactions to those that have higher confidence and are more likely to be true positives. In order to eliminate the fraction of false positives we have chosen a high confidence level. The combined score in the STRING10 are computed by combining the probabilities from the different evidence channels, correcting for the probability of randomly observing an interaction. For a more detailed description please see Jensen et al. Nucleic Acids Res. 2009, 37 (Database issue): D412-6.
The interaction of different subunits of PP2A tumor suppressor protein, different transcription factors including MYC oncogene and the interacting components of major oncogenic pathways like the PI3K-mTOR pathway as well as the MAPK-ERK pathway are presented in Figure 4A. The interaction(s) amoung these “input” proteins were generated using STRING10 at the confidence view, 0.900 (highest confidence). Action view is presented with the modes of interaction in different color as the inset (Figure 4A). The data show that the major point of interaction between the CIP2A and the nexus is gated through MYC protein which is well connected to different units of PP2A, PI3K pathway (AKT1, GSK3Beta, mTOR), MAP2K1, transcriptional factors (NFkB1, EIF4E, FOXO1, E2F1, TP53) and MDM2 via different categories of actions (activation, inhibition, binding, catalyses, post-translational modification etc). The confidence score presented is at the highest confidence level (0.900). The confidence score is the approximate probability that a predicted link exists between two enzymes in the same metabolic map in the KEGG database. We observed that there are two genes corresponding to subunits of PP2A protein those are frequently altered in breast cancers as obtained from c-BioPortal; they are PPP2R1A and PPP2R1B genes (Figure 3). Figure 4B shows the Action view of the interaction of PPP2R1A and PPP2R1B with MYC and CIP2A proteins at medium confidence level (0.400). “Action View” is presented with “Confidence View”. “Confidence View” of the association is presented as an inset. Stronger associations are represented by thicker lines. Active prediction methods included Databases and Text mining. “Confidence View” of the association is presented as an inset.
CIP2A controls oncogenic signals: antagonizes phosphatase functions of tumor suppressor PP2A
CIP2A functions as an oncogenic protein by suppressing functions of tumor suppressor PP2A [2,3,32] which constitutes one of the major tenets of the “oncogenic nexus” of CIP2A. Hence understanding of the molecular structure, function and the regulation of PP2A functions is crucial to envisage the “oncogenic nexus” of CIP2A. Ser/Thr phosphatase PP2A is conserved throughout eukaryotic tree [33]. PP2A protein is also one amongst the very few serine/threonine-specific phosphatases in the cell. The complex structure and regulation of PP2A insinuate its many functions. PP2A forms a heterotrimeric PP2A complex. The crystal structure of an AB’C (subunits of PP2A protein) heterotrimeric PP2A holoenzyme has been reported [34]. The PP2A holoenzyme consists of a heterodimeric core enzyme, which comprises a scaffolding subunit and a catalytic subunit, and a variable regulatory subunit [35]. PP2A enzymes typically exist as heterotrimers comprising catalytic C-, structural A- and regulatory B-type subunits. The B-type subunits function as targeting and substrate-specificity factors; hence, holoenzyme assembly with the appropriate B-type subunit is crucial for PP2A specificity and regulation [33]. A common heterodimeric core of tumor suppressor enzyme PP2A is composed of (1) PPP2CA a 36 kDa catalytic subunit (subunit C) and (2) PPP2R1A protein a 65 kDa constant regulatory subunit (PR65 or subunit A) forming the core dimer. Proteins that associate with the core dimer include three families of regulatory subunits B, the 48 kDa variable regulatory subunit, viral proteins, and cell signaling molecules. PP2A holoenzyme composition allows an amazingly diverse enzyme with a vast array of substrate specificities [3]. The structural details of PP2A form a crucial foundation for understanding PP2A assembly, its substrate recruitment, its function, and regulation of its function [35,36]. Thus a de-regulation of PP2A culminates into specific pathologies [37].
CIP2A binds to tumor suppressor PP2A and inhibits phosphatase functions resulting in tumorigenic transformations. The binary interaction of CIP2A occurs with serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform, PPP2R1A. Since the PR65 subunit of protein phosphatase 2A serves as a scaffolding molecule to coordinate the assembly of the catalytic subunit and a variable regulatory B subunit, the interaction of PP2A with CIP2A inhibits tumor suppressor function of PP2A. A number of studies have confirmed the role of PP2A in the oncogenic transformation [16,38-41]. Studies have demonstrated a number of ways in which PP2A is known to exert its tumor suppressor actions. Chen and group have reported that cancer-associated alpha mutations contribute to cancer development by inducing functional haplo-insufficiency, disturbing PP2A holoenzyme composition, and altering the enzymatic activity of PP2A [42]. One of the mechanisms by which PP2A functions as a tumor suppressor occurs via a critical interconnection with the a potent oncoprotein, c-MYC. PP2A regulatory subunit B56alpha has been reported to be associated with c-MYC and negatively regulates c-MYC accumulation. A specific PP2A regulatory subunit, B56alpha selectively associates with the N terminus of c-MYC. B56alpha directs intact PP2A holoenzymes to c-MYC, resulting in a dramatic reduction in c-MYC protein levels. Inhibition of PP2A-B56alpha holoenzymes results in c-MYC overexpression, elevated levels of c-MYC serine 62 phosphorylation, and increased c-MYC function [43]. PP2A interacts with a substantial number of other cellular and viral proteins, which are PP2A substrates, in order to target PP2A to different sub-cellular compartments or affect its enzymatic activity [37]. Suppression of PR61/B’gamma, a specific third regulatory subunit of PP2A, is reported to substitute for the viral SV40 protein small t antigen in causing tumorigenic transformation of several human cell lines-provided that telomerase, SV40 large T antigen and oncogenic RAS are also present. Accumulation of c-MYC protein seems to be the common denominator of cellular transformation [38]. SV40 (simian virus 40) ST (the early region of SV40 encodes two oncoproteins: the large T, LT and small t, ST antigens) inhibits the activity of PP2A and this interaction is required for SV40-mediated transformation of human cells. Mutations in PP2A subunits occur at low frequency in human tumors, suggesting that alterations of PP2A signaling play a role in both experimentally induced and spontaneously arising cancers [44]. Alterations in different subunits of PP2A have been reported to be associated with human malignancies. The gene encoding for the beta isoform of its subunit A, PPP2R1B is found to be altered in human lung and colorectal carcinomas [45]. Calin and group reported a low frequency of alterations of PPP2R1A and PPP2R1B isoforms of PP2A in human neoplasms. They have detected mutations in breast, lung carcinomas and melanomas in the genes of both alpha (PPP2R1A) and beta isoforms. Recently Zhou and group reported that in tumors, knockdown of PPP2R2D, a regulatory subunit of PP2A, inhibited T-cell apoptosis and enhanced T-cell proliferation as well as cytokine production indicting its key role as regulator of immune function in relevant microenvironment [46]. This observation becomes more relevant in the light of recent reports regarding the facilitating role of tumor infiltrating lymphocytes on the clinical efficacy of several anti-cancer drugs [47,48]. Mutations affecting PPP2R1B gene (exons deletions, suggesting abnormal splicing) were found in four breast carcinomas, while mutations in PPP2R1A (nucleotide substitutions changing highly conserved amino acids and one frame-shift) were found in carcinomas of the breast and of the lung and in one melanoma patients [45]. As mentioned earlier we also studied the percentage alterations in genes corresponding to subunits of PP2A gene using c-BioPortal (Figure 3A, 3B). The data showed alterations (percentage) of the PPP2R1A, PPP2R1B genes along with alterations of KIAA1524 genes in breast invasive carcinoma (TCGA, Nature 2013 and TCGA, Provisional) (Figure 3A). Within a particular data set, the highest percentages of alterations are observed in PPP2R1B gene followed by the change in the KIAA1524 gene and in the PPP2R1A gene. The highest change in percentage was observed in TCGA provisional data set showing that almost 50% alteration was observed in PPP2R1B (HOMDEL, HETLOSS, MUT, PROT). We extended our study regarding the similar percentage of changes in the PP2A subunits in different subtypes of BC using c-BioPortal. OncoPrints of Figure 3B shows alterations (percentage) of the PPP2R1A, PPP2R1B as well as KIAA1524 genes in PAM50 Luminal A, PAM50 Luminal B, PAM50 HER2 enriched, and PAM50 Basal subtype of breast invasive carcinoma (TCGA, Nature 2013). The appearance of a distinct pattern of percentage of alterations of these genes in a subtype specifc manner warrants further in-depth study in this direction.
CIP2A-PP2A signal actively regulates cell cycle and apoptosis via its control over p53-MDM2 axis. Moule and group studied the role of PP2A in ARF signaling to p53 [49]. They demonstrated that polyoma virus (Py) small T-antigen (PyST) blocks ARF-mediated activation of p53. This inhibition requires the small T-antigen PP2A-interacting domain. Their results reveal a previously unrecognized role of PP2A in the modulation of the ARF-p53 tumor suppressor pathway. Studies by Neviani and group showed that the tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein and functional inactivation of PP2A is essential for BCR/ABL leukemogenesis and, perhaps, required for blastic transformation [50]. Another mechanism by which PP2A has shown to protect cells from tumorigenic transformation is via the MDM2-p53 pathway. Okamoto and group demonstrated that cyclin G (cyclin G, a commonly induced p53 target) forms a quaternary complex in vivo and in vitro with enzymatically active phosphatase 2A (PP2A) holoenzymes containing B’ subunits. Cyclin G also binds in vivo and in vitro to MDM2 and markedly stimulates the ability of PP2A to dephosphorylate MDM2 at T216 suggesting that cyclin G recruits PP2A in order to dephosphorylate MDM2 and thereby to regulate both MDM2 and p53 [51] indicating the cross-talk between two tumor suppressors. PP2A has been identified as a crucial regulator of the NF-κB feedback loop [52]. Figure 4A represents the network image of the “Action view” of the network summary of proteins inputs pertaining to the components of the “oncogenic nexus of CIP2A” as presented in the Figure 1 and our previous article [1] using STRING10 at the highest confidence level (0.900). Interestingly the above studies by several independent groups can be substantiated by the “Action view” of the Figure 4A thus demonstrating the functional relevancy of the “oncogenic nexus of CIP2A”.
Recently, it was demonstrated that the inhibition of PP2A is a key determinant in acquiring TRAIL-sensitivity during tumorigenesis, with c-Fos/AP-1 as an essential mediator [53].The inhibition of PP2A activity confers a TRAIL-sensitive phenotype during malignant transformation and the down-regulation of PP2A activity is a critical step for normal cells to acquire a TRAIL-sensitive phenotype during tumorigenesis. The level of PP2A activity foretold cellular sensitivity to TRAIL-induced apoptosis. Biological function of CIP2A in breast cancer was explored by Liu and group using shRNA knockdown in MDA-MB231 and LM2-4 cell lines. CIP2A depletion inhibited proliferation and increased paclitaxel-induced apoptosis, and down-regulated the expression of c-MYC and p-ERK1/2 [54]. Interestingly both of their observed effects of CIP2A depletion in cells can be explained by the network display of the interaction (“action view”) between major components of the “oncogenic nexus” of CIP2A [as represented in the Figure 1 and also in our previous article [1] as well as (Figure 4A) which presented an interaction between different subunits of PP2A tumor suppressor protein, protein product of MYC oncogene and the interacting components of major oncogenic pathways like the MAPK-ERK pathway. These results substantiate the fact that the “oncogenic nexus” of CIP2A oncoprotein is functional in breast carcinigenesis.
Functions of “oncogenic nexus” of CIP2A in cancers: stabilization of MYC protein
The “oncogenic nexus” of CIP2A regulates transcription of MYC targeted genes. As an integral protein, CIP2A achieves oncogenic credential via inhibition of PP2A capacity to destabilize MYC. CIP2A interacts directly with c-MYC protein, inhibits PP2A activity toward c-MYC serine 62, and thereby prevents c-MYC proteolytic degradation. Detailed mechanism of this action has been reviewed in past. In studies by Junttila and group co-immunoprecipitation of endogenous PR65 from cytosolic HeLa extracts revealed an interaction between endogenous CIP2A, PR65 and PP2Ac proteins [3]. As a pleiotropic oncogenic transcription factor, MYC activates the transcription of growth/transformation-related genes. Thus aberrant MYC signaling observed in human cancers have been demonstrated to promote cell transformation and tumor progression [5]. Accumulation of c-MYC (stability) is one of the common denominator of cellular transformation [38], especially in MYC-mediated malignancies. The “oncogenic nexus” of CIP2A protein in human malignancies is executed through the stabilization MYC protein. PP2A has been identified as a new protein involved in regulating c-MYC expression [43]. It has been established that the amplification of MYC gene is one of the major causes of the development of the drug-induced resistance inhibitors of the PI3K-mTOR pathway [55]. PP2A-mediated c-MYC dephosphorylation can be adequate for SV40 small-t antigen in human transformation assays [56]. This data is in line with the report that CIP2A has been overexpressed in a number of common human malignancies. Studies by Junttila and group identified CIP2A as an endogenous PP2A-associated protein that promotes c-MYC protein stability by inhibiting c-MYC-associated PP2A-mediated de-phosphorylation which culminates into cellular transformation and tumor growth strongly indicating that CIP2A is a human oncoprotein that inhibits PP2A in human malignancies [3]. Thus CIP2A is required for the malignant cellular transformation. Stabilization of MYC protein represents PP2A phosphatase dependent functions of CIP2A in cancer cells.
Regulation of the “oncogenic nexus” of CIP2A in breast cancers: hormonal control, autophagy & intrinsic senescence
The expression of CIP2A has been regulated both via several transcription factors as well as through the downstream effectors of classical growth promoting steroid hormone-dependent mitogenic pathways in breast cancers. CIP2A expression has been reported to be enhanced by estradiol (E2) in ER-positive breast cancer cells via the activation of p70 S6 kinase as well as ETS1 transcription factor following E2 mediated activation of MEK1/2 and PI3K pathways via EGFR signaling. Choi and group also showed that E2 increased CIP2A expression at the translational level in a c-MYC-independent manner in MCF-7 cells. E2-enhanced proliferation was impaired without CIP2A expression. E2-stimulated EGFR activated the MAPK and PI3K pathways, which converged to activate p70 S6 kinase (S6K) (phosphorylation sites S424/T421, T229, and T389) that was required for the phosphorylation of eukaryotic initiation factor 4B (eIF4B), leading to the increase in CIP2A translation [29]. Studies have also shown that ETS1 mediated MEK1/2-dependent overexpression of CIP2A occurs in human cancer cells [31].
CIP2A itself is degraded by autophagy. CIP2A is rapidly degraded upon proteasome inhibition, a known activator of autophagy [58]. p62/SQSTM1 guides CIP2A to autophagic degradation. Notably, this model is highly meaningful to recently reported p62/SQSTM1-dependent autophagic degradation of the Dishevelled, which leads to the attenuation of oncogenic Wnt signaling [59]. This effect on the autophagic degradation of the Dishevelled protein is thought provoking in the context of BC since the involvement of Wnt-beta-catenin signals has been closely associated to the outcome of the triple negative subtype of BC. We have demonstrated a differential upregulation of Wnt-beta-catenin pathway in TNBC and its association with metastasis [60]. Our study also identified that the differential activation of Wnt-beta-catenin pathway in triple negative breast cancer increases MMP7 in a PTEN dependent manner [61]. This observation of ours has been further put into the perspective of promotion of invasion and metastasis of breast cancer cells by the works of Ma and group [62] who reported that Wnt-beta-catenin signaling was hyperactivated in metastatic breast cancer cells that express miR-301a, and mediated miR-301a-induced invasion and metastasis while miR-301a directly targeted and suppressed PTEN, one of the negative regulators of the Wnt/β-catenin signaling cascade. Furthermore, mTORC1- and autophagy-regulated stability of CIP2A provides a molecular explanation for the coordinated activation of mTORC1 and c-MYC, which together alter cellular metabolism to achieve excessive cell growth and proliferation [14].
CIP2A has been associated to the phenomenon of senescence in BC cells. Senescence in addition to apoptosis is cellular failsafe program which counteracts the disproportionate mitogenic signals arising from the upregulation of oncogenes. Thus either an effective bypass or a robust inhibition of senescence and apoptosis is required for a cell destined to a tumorigenic transformation [63]. In BC cells, a positive feedback loop between CIP2A and E2F1 had been shown to define the cell-intrinsic senescence sensitivity [64]. Laine and group identified that the E2F1-CIP2A positive feedback loop is a key determinant of BC cell sensitivity to senescence and growth arrest induction, the results of which may also facilitate novel stratification strategies for selection of patients to receive senescence-inducing cancer therapies. In their study Laine and group also reported that mammary tumorigenesis is impaired in a CIP2A-deficient mouse model and CIP2A-deficient tumors display markers of senescence induction which is in line with the fact that high CIP2A expression predicts for poor prognosis in a subgroup of patients with breast cancer treated with senescence-inducing chemotherapy [64]. The findings from their study that (1) wild type p53 negatively regulates CIP2A expression, (2) E2F1 upregulates CIP2A expression downstream of inactivated p53, (3) inhibition of CIP2A expression is a prerequisite for p53-mediated senescence induction, (4) positive feedback loop between CIP2A and E2F1 functions as a barrier for p21-mediated senescence induction, and (5) inhibition of CIP2A inhibits growth and induces senescence in mouse embryonic fibroblasts demonstrates that an increased activity of E2F1-CIP2A feedback renders breast cancer cells resistant to senescence induction. It is remarkable to note here that this feedback loop involving the interplay between these two frequently overexpressed oncoproteins may also be considered as a promising pro-senescence target for therapy of triple negative or basal-type breast cancers with an inactivated p53-p21 pathway. It will be interesting and important to find the role of CIP2A “oncogenic nexus” in mediating drug induced senescence that generates chemotherapy resistance in cancers with different genetic backgrounds. Tumor stem cells contribute to tumor recurrence after chemotherapy. Interestingly, drug-induced senescence has been found to generate chemo-resistant stem-like cells in breast cancer [65]. Thus it will provide a rationale to target this CIP2A “oncogenic nexus” for chemotherapy resistance or drug-induced senescence. Studies in gastric cancer demonstrated that CIP2A depletion leads to senescence of tumor cells [66]. Gastric cancer-derived AGS cells underwent senescence following the inhibition of CIP2A expression.
CIP2A as prognostic biomarker in breast cancers
The relevance of a tumor marker in the early immuno-diagnosis depends on its high sensitivity and specificity. Auto-antibodies directed against tumor-associated antigens (TAAs) have been shown to be relevant tumor markers fulfilling these criteria. Antibodies against KIAA1524 are present in sera from many patients with gastric or prostate cancer, suggesting that it may act as a marker for such cancers. Clinical implications of CIP2A protein expression in breast cancer have been reported. CIP2A expression may be considered as a potential biomarker for chemotherapeutic sensitivity and prognosis in breast cancer [11]. Liu and group reported that auto-antibodies against p90/CIP2A may be useful serum biomarker for early stage breast cancer screening and immuno-diagnosis. They evaluated whether or not auto-antibodies to a tumor-associated antigen p90/CIP2A can be used as immuno-diagnostic markers in breast cancer to demonstrate that auto-antibodies against p90/CIP2A may be a useful serum biomarker for early stage breast cancer screening and immuno-diagnosis [67]. The frequency of p90/CIP2A expression in breast cancer tissues was significantly higher than that in adjacent normal tissues (P<0.01) and p90/CIP2A induced a relatively higher frequency of auto-antibody response in breast cancer (19.1%) compared to the sera of normal individuals (2.3%). CIP2A expression may be a potential biomarker for chemotherapeutic sensitivity and prognosis of breast cancer [11]. High expression of CIP2A has also been proposed by Khanna and Pimanda as a useful biomarker that predicts therapeutic response to chemotherapeutics such as Bortezomib, Erlotinib, Checkpoint Kinase 1 (CHK1) inhibitors and pro-senescence based therapies [68]. In their review they also highlighted the ambiguity in CIP2A’s prognostic role in different human cancers and its role in modulating response and resistance to chemotherapeutics. A report by Ventelä and group demonstrating that CIP2A is a Oct4 target gene in stem cells and in human Head & Neck cancer cell lines has provided CIP2A with an interesting perspective in solid tumors [69] since the role of stem cell has been emerging more convincingly than ever in the mechanism governing metastatic dormancy in the newly homed microenvironment of the tumor cells and reactivation in the solid tumors [70]. The emerging role of the “oncogenic nexus” of CIP2A in molecular underpinning of the metastatic dormancy and the reactivation is worth perusing in the future.
Acknowledgements
We acknowledge Genomic Oncology Institute, Department of Molecular and Experimental Medicine, Avera Cancer Institute, Sioux Falls, SD, USA. We acknowledge c-BioPortal and STRING10. Cancer Discovery. May 2012, 2; 401. [71] and Gao and group Integrative analysis of complex cancer genomics and clinical profiles using the c-BioPortal. Sci. Signal. 6, pl1 [72]. We acknowledge the TCGA Research Network for generating TCGA datasets. We acknowledge the version 10 of STRING, and authorities/institutions/organizations/Universities/resources which institutionally and financially support the STRING10. The authors also acknowledge the Department of Molecular & Experimental Medicine, Avera Research Institute, Sioux Falls, SD and the Department of Internal Medicine, USD, SD, USA.
Disclosure of conflict of interest
None.
References
- 1.De P, Carlson J, Leyland-Jones B, Dey N. Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A (CIP2A): an oncoprotein with many hands. Oncotarget. 2014;5:4581–4602. doi: 10.18632/oncotarget.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khanna A, Pimanda JE, Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res. 2013;73:6548–6553. doi: 10.1158/0008-5472.CAN-13-1994. [DOI] [PubMed] [Google Scholar]
- 3.Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, Lu SL, Lin S, Chan EK, Wang XJ, Grenman R, Kast J, Kallunki T, Sears R, Kahari VM, Westermarck J. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 4.Cristobal I, Gonzalez-Alonso P, Daoud L, Solano E, Torrejon B, Manso R, Madoz-Gurpide J, Rojo F, Garcia-Foncillas J. Activation of the Tumor Suppressor PP2A Emerges as a Potential Therapeutic Strategy for Treating Prostate Cancer. Mar Drugs. 2015;13:3276–3286. doi: 10.3390/md13063276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Junttila MR, Westermarck J. Mechanisms of MYC stabilization in human malignancies. Cell Cycle. 2008;7:592–596. doi: 10.4161/cc.7.5.5492. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 7.Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007;130:21–24. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 8.Soo Hoo L, Zhang JY, Chan EK. Cloning and characterization of a novel 90 kDa ‘companion’ auto-antigen of p62 overexpressed in cancer. Oncogene. 2002;21:5006–5015. doi: 10.1038/sj.onc.1205625. [DOI] [PubMed] [Google Scholar]
- 9.Peng B, Samuel , Liu WQ, Edward C, Eng MT, Jianying Z. Immune response to a fetal oncoprotein p90/CIP2A in lung cancer. Cancer Res. 2010;70:4792. [Google Scholar]
- 10.Come C, Laine A, Chanrion M, Edgren H, Mattila E, Liu X, Jonkers J, Ivaska J, Isola J, Darbon JM, Kallioniemi O, Thezenas S, Westermarck J. CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res. 2009;15:5092–5100. doi: 10.1158/1078-0432.CCR-08-3283. [DOI] [PubMed] [Google Scholar]
- 11.Yu G, Liu G, Dong J, Jin Y. Clinical implications of CIP2A protein expression in breast cancer. Med Oncol. 2013;30:524. doi: 10.1007/s12032-013-0524-9. [DOI] [PubMed] [Google Scholar]
- 12.Tseng LM, Liu CY, Chang KC, Chu PY, Shiau CW, Chen KF. CIP2A mediates the apoptotic effect of bortezomib on triple negative breast cancer cells. Cancer Res. 2011;71:2622. doi: 10.1186/bcr3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tseng LM, Liu CY, Chang KC, Chu PY, Shiau CW, Chen KF. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012;14:R68. doi: 10.1186/bcr3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puustinen P, Rytter A, Mortensen M, Kohonen P, Moreira JM, Jaattela M. CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J Cell Biol. 2014;204:713–727. doi: 10.1083/jcb.201304012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi YA, Park JS, Park MY, Oh KS, Lee MS, Lim JS, Kim KI, Kim KY, Kwon J, Yoon do Y, Moon EY, Yang Y. Increase in CIP2A expression is associated with doxorubicin resistance. FEBS Lett. 2011;585:755–760. doi: 10.1016/j.febslet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 16.Van Hoof C, Goris J. PP2A fulfills its promises as tumor suppressor: which subunits are important? Cancer Cell. 2004;5:105–106. doi: 10.1016/s1535-6108(04)00027-3. [DOI] [PubMed] [Google Scholar]
- 17.Niemela M, Kauko O, Sihto H, Mpindi JP, Nicorici D, Pernila P, Kallioniemi OP, Joensuu H, Hautaniemi S, Westermarck J. CIP2A signature reveals the MYC dependency of CIP2A-regulated phenotypes and its clinical association with breast cancer subtypes. Oncogene. 2012;31:4266–4278. doi: 10.1038/onc.2011.599. [DOI] [PubMed] [Google Scholar]
- 18.Baldacchino S, Saliba C, Petroni V, Fenech AG, Borg N, Grech G. Deregulation of the phosphatase, PP2A is a common event in breast cancer, predicting sensitivity to FTY720. EPMA J. 2014;5:3. doi: 10.1186/1878-5085-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong LL, Zhang D, Chang CF, Koay ES. Silencing of the PP2A catalytic subunit causes HER-2/neu positive breast cancer cells to undergo apoptosis. Exp Cell Res. 2010;316:3387–3396. doi: 10.1016/j.yexcr.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M, Lebigot I, Djelti F, Tourdes A, Gestraud P, Hupe P, Barillot E, Cruzalegui F, Tucker GC, Stern MH, Thiery JP, Hickman JA, Dubois T. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008;10:R101. doi: 10.1186/bcr2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, Pandolfi PP, Cantley LC. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, Hu C, Riegel AT, Fan S, Rosen EM. Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-alpha activity. Mol Endocrinol. 2007;21:1905–1923. doi: 10.1210/me.2006-0397. [DOI] [PubMed] [Google Scholar]
- 23.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y, Kashiwagi A, Olefsky JM. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol. 2004;24:8778–8789. doi: 10.1128/MCB.24.19.8778-8789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiang T, Ohashi A, Huang Y, Pandita TK, Ludwig T, Powell SN, Yang Q. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008;68:10040–10044. doi: 10.1158/0008-5472.CAN-08-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filonenko VV, Tytarenko R, Azatjan SK, Savinska LO, Gaydar YA, Gout IT, Usenko VS, Lyzogubov VV. Immunohistochemical analysis of S6K1 and S6K2 localization in human breast tumors. Exp Oncol. 2004;26:294–299. [PubMed] [Google Scholar]
- 26.Zhou X, Tan M, Stone Hawthorne V, Klos KS, Lan KH, Yang Y, Yang W, Smith TL, Shi D, Yu D. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin Cancer Res. 2004;10:6779–6788. doi: 10.1158/1078-0432.CCR-04-0112. [DOI] [PubMed] [Google Scholar]
- 27.Stal O, Perez-Tenorio G, Akerberg L, Olsson B, Nordenskjold B, Skoog L, Rutqvist LE. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 2003;5:R37–44. doi: 10.1186/bcr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi YA, Koo JS, Park JS, Park MY, Jeong AL, Oh KS, Yang Y. Estradiol enhances CIP2A expression by the activation of p70 S6 kinase. Endocr Relat Cancer. 2014;21:189–202. doi: 10.1530/ERC-13-0453. [DOI] [PubMed] [Google Scholar]
- 30.Dey N, Smith BR, Leyland-Jones B. Targeting basal-like breast cancers. Curr Drug Targets. 2012;13:1510–1524. doi: 10.2174/138945012803530116. [DOI] [PubMed] [Google Scholar]
- 31.Khanna A, Okkeri J, Bilgen T, Tiirikka T, Vihinen M, Visakorpi T, Westermarck J. ETS1 mediates MEK1/2-dependent overexpression of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancer cells. PLoS One. 2011;6:e17979. doi: 10.1371/journal.pone.0017979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sablina AA, Hahn WC. The role of PP2A A subunits in tumor suppression. Cell Adh Migr. 2007;1:140–141. doi: 10.4161/cam.1.3.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail) Trends Biochem Sci. 2008;33:113–121. doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445:53–57. doi: 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- 35.Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Yu JW, Strack S, Jeffrey PD, Shi Y. Structure of the protein phosphatase 2A holoenzyme. Cell. 2006;127:1239–1251. doi: 10.1016/j.cell.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 36.Kalev P, Sablina AA. Protein phosphatase 2A as a potential target for anticancer therapy. Anticancer Agents Med Chem. 2011;11:38–46. doi: 10.2174/187152011794941172. [DOI] [PubMed] [Google Scholar]
- 37.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. Curr Opin Genet Dev. 2005;15:34–41. doi: 10.1016/j.gde.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 39.Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- 40.Sablina AA, Hector M, Colpaert N, Hahn WC. Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res. 2010;70:10474–10484. doi: 10.1158/0008-5472.CAN-10-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14:152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005;65:8183–8192. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 43.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol. 2006;26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- 45.Calin GA, di Iasio MG, Caprini E, Vorechovsky I, Natali PG, Sozzi G, Croce CM, Barbanti-Brodano G, Russo G, Negrini M. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene. 2000;19:1191–1195. doi: 10.1038/sj.onc.1203389. [DOI] [PubMed] [Google Scholar]
- 46.Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS, Elpek K, Brogdon J, Lamb J, Turley SJ, Ploegh HL, Root DE, Love JC, Dranoff G, Hacohen N, Cantor H, Wucherpfennig KW. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014;506:52–57. doi: 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soria JC, Marabelle A, Brahmer JR, Gettinger S. Immune checkpoint modulation for non-small cell lung cancer. Clin Cancer Res. 2015;21:2256–2262. doi: 10.1158/1078-0432.CCR-14-2959. [DOI] [PubMed] [Google Scholar]
- 49.Moule MG, Collins CH, McCormick F, Fried M. Role for PP2A in ARF signaling to p53. Proc Natl Acad Sci U S A. 2004;101:14063–14066. doi: 10.1073/pnas.0405533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, Roy DC, Valtieri M, Bruner-Klisovic R, Caligiuri MA, Bloomfield CD, Marcucci G, Perrotti D. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–368. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 51.Okamoto K, Li H, Jensen MR, Zhang T, Taya Y, Thorgeirsson SS, Prives C. Cyclin G recruits PP2A to dephosphorylate Mdm2. Mol Cell. 2002;9:761–771. doi: 10.1016/s1097-2765(02)00504-x. [DOI] [PubMed] [Google Scholar]
- 52.Barisic S, Strozyk E, Peters N, Walczak H, Kulms D. Identification of PP2A as a crucial regulator of the NF-kappaB feedback loop: its inhibition by UVB turns NF-kappaB into a proapoptotic factor. Cell Death Differ. 2008;15:1681–1690. doi: 10.1038/cdd.2008.98. [DOI] [PubMed] [Google Scholar]
- 53.Yang H, Chen X, Wang X, Li Y, Chen S, Qian X, Wang R, Chen L, Han W, Ruan A, Du Q, Olumi AF, Zhang X. Inhibition of PP2A activity confers a TRAIL-sensitive phenotype during malignant transformation. Mol Cancer Res. 2014;12:217–227. doi: 10.1158/1541-7786.MCR-13-0441. [DOI] [PubMed] [Google Scholar]
- 54.Liu X, Peng B, Li Y, Lei N, Li W, Zhang JY. p90/CIP2A mediates breast cancer cell proliferation and apoptosis. Mol Biol Rep. 2014;41:7471–7478. doi: 10.1007/s11033-014-3635-2. [DOI] [PubMed] [Google Scholar]
- 55.Dey N, Leyland-Jones B, De P. MYC-xing it up with PIK3CA mutation and resistance to PI3K inhibitors: summit of two giants in breast cancers. Am J Cancer Res. 2015;5:1–19. [PMC free article] [PubMed] [Google Scholar]
- 56.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 57.De Geest AF, Schoolmeesters I, Willems JL, De Geest H. Dental health, prophylactic antibiotic measures and infective endocarditis: an analysis of the knowledge of susceptible patients. Acta Cardiol. 1990;45:441–453. [PubMed] [Google Scholar]
- 58.Lamark T, Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol. 2010;22:192–198. doi: 10.1016/j.ceb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 59.Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, Fu W, Zhang J, Wu W, Zhang X, Chen YG. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol. 2010;12:781–790. doi: 10.1038/ncb2082. [DOI] [PubMed] [Google Scholar]
- 60.Dey N, Barwick BG, Moreno CS, Ordanic-Kodani M, Chen Z, Oprea-Ilies G, Tang W, Catzavelos C, Kerstann KF, Sledge GW Jr, Abramovitz M, Bouzyk M, De P, Leyland-Jones BR. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer. 2013;13:537. doi: 10.1186/1471-2407-13-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dey N, Young B, Abramovitz M, Bouzyk M, Barwick B, De P, Leyland-Jones B. Differential activation of Wnt-beta-catenin pathway in triple negative breast cancer increases MMP7 in a PTEN dependent manner. PLoS One. 2013;8:e77425. doi: 10.1371/journal.pone.0077425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma F, Zhang J, Zhong L, Wang L, Liu Y, Wang Y, Peng L, Guo B. Upregulated microRNA-301a in breast cancer promotes tumor metastasis by targeting PTEN and activating Wnt/beta-catenin signaling. Gene. 2014;535:191–197. doi: 10.1016/j.gene.2013.11.035. [DOI] [PubMed] [Google Scholar]
- 63.Schmitt CA. Senescence, apoptosis and therapy--cutting the lifelines of cancer. Nat Rev Cancer. 2003;3:286–295. doi: 10.1038/nrc1044. [DOI] [PubMed] [Google Scholar]
- 64.Laine A, Sihto H, Come C, Rosenfeldt MT, Zwolinska A, Niemela M, Khanna A, Chan EK, Kahari VM, Kellokumpu-Lehtinen PL, Sansom OJ, Evan GI, Junttila MR, Ryan KM, Marine JC, Joensuu H, Westermarck J. Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov. 2013;3:182–197. doi: 10.1158/2159-8290.CD-12-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Achuthan S, Santhoshkumar TR, Prabhakar J, Nair SA, Pillai MR. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem. 2011;286:37813–37829. doi: 10.1074/jbc.M110.200675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li W, Ge Z, Liu C, Liu Z, Bjorkholm M, Jia J, Xu D. CIP2A is overexpressed in gastric cancer and its depletion leads to impaired clonogenicity, senescence, or differentiation of tumor cells. Clin Cancer Res. 2008;14:3722–3728. doi: 10.1158/1078-0432.CCR-07-4137. [DOI] [PubMed] [Google Scholar]
- 67.Liu X, Chai Y, Li J, Ren P, Liu M, Dai L, Qian W, Li W, Zhang JY. Autoantibody response to a novel tumor-associated antigen p90/CIP2A in breast cancer immunodiagnosis. Tumour Biol. 2014;35:2661–2667. doi: 10.1007/s13277-013-1350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khanna A, Pimanda JE. Clinical significance of Cancerous Inhibitor of Protein Phosphatase 2A (CIP2A) in human cancers. Int J Cancer. 2015 doi: 10.1002/ijc.29431. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 69.Ventela S, Sittig E, Mannermaa L, Makela JA, Kulmala J, Loyttyniemi E, Strauss L, Carpen O, Toppari J, Grenman R, Westermarck J. CIP2A is an Oct4 target gene involved in head and neck squamous cell cancer oncogenicity and radioresistance. Oncotarget. 2015;6:144–158. doi: 10.18632/oncotarget.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell. 2013;155:750–764. doi: 10.1016/j.cell.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the c-BioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]