

Abstract
Background
H3N2 influenza viruses circulating in humans and European pigs originate from the pandemic A/Hong Kong/68 virus. Because of slower antigenic drift in swine, the antigenic divergence between swine and human viruses has been increasing. It remains unknown to what extent this results in a reduced cross‐protection between recent human and swine H3N2 influenza viruses.
Objectives
We examined whether prior infection of pigs with an old [A/Victoria/3/75 (A/Vic/75)] or a more recent [A/Wisconsin/67/05 (A/Wis/05)] human H3N2 virus protected against a European swine H3N2 virus [sw/Gent/172/08 (sw/Gent/08)]. Genetic and antigenic relationships between sw/Gent/08 and a selection of human H3N2 viruses were also assessed.
Results
After challenge with sw/Gent/08, all challenge controls had high virus titers in the entire respiratory tract at 3 days post‐challenge and nasal virus excretion for 5–6 days. Prior infection with sw/Gent/08 or A/Vic/75 offered complete virological protection against challenge. Pigs previously inoculated with A/Wis/05 showed similar virus titers in the respiratory tract as challenge controls, but the mean duration of nasal shedding was 1·3 days shorter. Unlike sw/Gent/08‐ and A/Vic/75‐inoculated pigs, A/Wis/05‐inoculated pigs lacked cross‐reactive neutralizing antibodies against sw/Gent/08 before challenge, but they showed a more rapid antibody response to sw/Gent/08 than challenge controls after challenge. Cross‐protection and serological responses correlated with genetic and antigenic differences.
Conclusions
Infection immunity to a recent human H3N2 virus confers minimal cross‐protection against a European swine H3N2 virus. We discuss our findings with regard to the recent zoonotic infections of humans in the United States with a swine‐origin H3N2 variant virus.
Keywords: Cross‐protection, Europe, H3N2 swine influenza virus, human H3N2 virus, zoonosis
Introduction
The antigenic and genetic characteristics of swine influenza viruses (SIVs) vary in different continents and regions of the world, but most if not all H3N2 SIVs contain hemagglutinin (HA) and neuraminidase (NA) genes from early or more recent human H3N2 viruses.1 In Europe, H3N2 SIVs are derived from descendants of the 1968 “Hong Kong” pandemic virus, but they have evolved further through genetic reassortment with the endemic avian‐like H1N1 SIV in the mid‐1980s. This has resulted in H3N2 SIVs with human‐like HA and NA genes and avian‐like internal genes.2, 3 In North America, H3N2 viruses did not become established in the pig population until 1998. These viruses are “triple” reassortants with the HA, NA, and PB1 genes of human H3N2 viruses from the mid‐1990s and the remaining genes of classical swine and avian origin.4 Multiple H3N2 SIV lineages have been found in Asia, and all have HA and NA genes derived from human H3N2 viruses from various time periods.5, 6, 7 Because of slower antigenic drift in the H3 HA in swine than in humans,5, 8 human‐lineage H3N2 viruses in swine over time become increasingly divergent from contemporary human H3N2 viruses. As an example, European H3N2 SIVs from 1983 to 1999 showed a six times slower rate of antigenic drift than their human counterparts.8 Consequently, European H3N2 SIVs from recent years still show some degree of serological cross‐reactivity with human viruses from the 1970s to 1980s, but not with human viruses isolated after 1990.9, 10
Swine influenza viruses usually do not infect humans, but sporadic dead‐end zoonotic infections have been reported with most SIV subtypes or lineages.11, 12, 13 Such infections are generally mild and clinically indistinguishable from infections with human influenza viruses. Most cases of swine influenza occur in people with direct or indirect exposure to swine, frequently children, or young adults. The European H3N2 SIV has been isolated from a 1‐year‐old girl and a 2‐year‐old boy in the Netherlands in 1993.14 A closely related virus was also isolated from a 10‐month‐old girl in Hong Kong in 1999.15 In North America, nine cases of human infection with triple reassortant H3N2 SIV have been reported from 2005 to 2010.12, 16, 17 A variant of the original triple reassortant H3N2 SIVs, which contains the matrix gene of the pandemic 2009 H1N1 virus, has been detected in 13 humans in the United States from July 2011 to April 2012,18 and in 306 cases in 10 US states from July to September 2012, mainly in children visiting county or state fairs.19 The increasing numbers of human cases may be in part due to heightened awareness and surveillance. No H3N2 SIV has so far shown the capacity to spread efficiently between humans.
Already in 1977, Shortridge et al.20 suggested that pigs could serve as a reservoir for old, A/Hong Kong/68‐like human H3N2 viruses and that such viruses might be re‐introduced into humans when their immunity has waned. However, it remains unknown to what extent immunity to human H3N2 viruses may offer protection against H3N2 SIVs. We have therefore examined the effect of prior infection with an old (1975) or a more recent (2005) human H3N2 influenza virus on challenge with a recent European H3N2 SIV in the pig model.
Materials and methods
Genetic and antigenic characterization of viruses
Sw/Gent/172/08 (sw/Gent/08) is representative of H3N2 SIVs that are enzootic in Western Europe. The human H3N2 influenza viruses A/Victoria/3/75 (A/Vic/75) and A/Wisconsin/67/05 (A/Wis/05) have been WHO vaccine reference strains in 1976–1978 and 2006–2008, respectively. All viruses were propagated in the allantoic cavity of 10‐day‐old embryonated chicken eggs for less than four passages.
The HA1, NA, matrix (M), and nucleoprotein (NP) sequences of A/Vic/75, A/Wis/05, and sw/Gent/08 were compared at the nucleotide and amino acid (aa) level using megalign program within dnastar 5.01 software (DNASTAR, Inc., Madison, WI, USA). In addition, the HA1 and NA of a selection of human H3N2 viruses circulating from 1973 to 2009 (Table 2) were compared with sw/Gent/08. Amino acid differences at putative antigenic sites of the HA and NA, as defined by others,21, 22, 23 were identified by alignment using mega 5.05 software (http://www.megasoftware.net/)24. N‐linked glycosylation sites were predicted by the NetNGlyc 1.0 web server (http://www.cbs.dtu.dk/services/NetNGlyc) as described elsewhere.25 The HA1, NA, M, and NP of sw/Gent/08 and the HA1 of A/Vic/75 and A/Wis/05 were (re‐)sequenced for this study (GenBank accession numbers KC142126–32). All other sequences used for comparison were downloaded from GenBank.
A/Vic/75, A/Wis/05, and sw/Gent/08 were characterized in hemagglutination inhibition (HI) and neuraminidase inhibition (NI) tests using post‐infection ferret or hyperimmune swine sera, or both.
Experimental design
Thirty‐two 5‐week‐old pigs were obtained from an influenza virus‐seronegative farm. Pigs were randomly assigned to four groups of eight pigs. Each group was housed in a separate biosafety level‐2 HEPA‐filtered isolation unit. Before the start of the experiment, all pigs were seronegative to any of the European endemic H1N1, H1N2, and H3N2 SIVs, as determined by HI test, immunoperoxidase monolayer assay, and a competitive anti‐influenza A NP enzyme‐linked immunosorbent assay (Idexx Laboratories, Hoofddorp, the Netherlands). After acclimation for 1 week, three groups were inoculated with A/Vic/75, A/Wis/05, or sw/Gent/08, respectively. One group was left uninoculated and served as the challenge control group. Six weeks later, all groups were challenged with sw/Gent/08. All inoculations were performed intranasally with 7·0 log10 50% egg infective doses (EID50) influenza virus in 3 ml (1·5 ml per nostril) as described elsewhere.26 Pigs were observed daily for clinical signs from 4 days before until 7 days after each inoculation, or until euthanasia. To determine virus excretion, nasal swabs were collected daily from all pigs from 0–7 days post‐primary inoculation (dpi) and from 0–7 days post‐challenge (dpc), or until euthanasia. Four pigs per group were euthanized at 3 dpc and gross lung lesions were assessed as described elsewhere.27 To determine the extent of replication of the sw/Gent/08 challenge virus in the respiratory tract, tissue samples of the upper (nasal mucosa respiratory part and olfactory part, tonsil, and trachea) and lower (apical, cardiac, and diaphragmatic lobes of the left and right lung) respiratory tract were collected and titrated separately. Blood samples for serological examinations were collected at the start of the experiment, 14 and 42 dpi, that is, at the time of challenge with sw/Gent/08. The remaining pigs were also bled at 5, 7, 10, and 14 dpc.
Virus titration
Cotton swabs were weighed before and after collection to determine virus titers per 100 mg nasal secretions. Swabs from both nostrils were suspended in 1 ml of phosphate‐buffered saline supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin and mixed vigorously at 4°C for 1 hour. Tissue samples were weighed and ground in PBS containing 10 IU/ml penicillin and 10 μg/ml streptomycin to obtain 20% (w/v) tissue homogenates. Nasal swab samples and tissue homogenates were clarified by centrifugation (16 000 g for 3 minutes) and stored at −70°C until titration. All samples were titrated on Madin–Darby canine kidney (MDCK) cells in serum‐free medium with trypsin. Briefly, confluent monolayers of cells were inoculated with 10‐fold serial dilutions of samples. Cells were washed 2 hours after inoculation and subsequently observed for development of cytopathic effect over 7 days. Virus titers were expressed as log10 50% tissue culture infective doses (TCID50) per 100 mg (nasal swabs) or per gram (tissues).
Serological assays
Serum antibody responses were examined by HI, virus neutralization (VN), and NI tests. All sera collected at 0, 14 dpi, and 0 and 14 dpc were examined against all three viruses in HI and VN tests, while only the sera collected at 0 and 14 dpc were tested in NI tests. Additional VN tests against the challenge virus sw/Gent/08 were performed on the sera collected at 5, 7, and 10 dpc. All sera were heat inactivated (56°C, 30 minutes) before use. The HI test was performed according to standard procedures with 0·5% turkey erythrocytes and 4 hemagglutinating units of virus.28 The VN test was performed in MDCK cells in microplates with 100 TCID50 of virus per well as previously described.29 The NI test was based on the colorimetric analysis of sialic acid release from fetuin substrate and conducted in 96‐well PCR plates as described elsewhere.30 Starting dilutions were 1:2 in the VN test and 1:10 in the HI and NI tests.
Statistics
Nasal virus shedding in each group was quantified by calculation of the area under the curve (AUC), which is obtained by plotting viral titers versus each time point of sample collection. Samples that tested negative for virus were given a numeric value of 1·6 log10 TCID50 per 100 mg or gram. Samples that tested negative in the serological assays were assigned a value corresponding to half of the minimum detectable titer. Mann–Whitney tests were used to compare virus titers and antibody levels between any two experimental groups. P < 0·05 was considered statistically significant. graphpad prism 5.0 software (GraphPad Software, Inc., San Diego, CA, USA) was used for all analyses.
Results
Genetic and antigenic relationships between human and European swine H3N2 viruses
Percentages of nucleotide and aa identity between the HA1, NA, M, and NP of A/Vic/75, A/Wis/05, and sw/Gent/08 are summarized in Table 1. The HA1 and NA of sw/Gent/08 were more similar to A/Vic/75 than to A/Wis/05 at both levels. The M and NP genes of both human viruses were equally similar to those of the swine virus.
Table 1.
Percent identity of the nucleotide and amino acid sequences of the hemagglutinin (HA1), neuraminidase (NA), matrix (M), and nucleoprotein (NP) genes of sw/Gent/08 with those of the human H3N2 viruses A/Vic/75 and A/Wis/05
| % Identify compared with sw/Gent/08 | ||||||||
|---|---|---|---|---|---|---|---|---|
| HA1 | NA | M | NP | |||||
| N | aa | N | aa | N | aa | N | aa | |
| A/Vic/75 | 88 | 87 | 89 | 91 | 87 | 91 | 84 | 91 |
| A/Wis/05 | 80 | 80 | 85 | 87 | 86 | 90 | 82 | 91 |
N, nucleotide; aa, amino acid.
Amino acid differences at presumed antigenic sites of the HA are shown in Figure 1. The HA1 segment of A/Vic/75 contained 42 aa differences compared with sw/Gent/08, with 9 of these occurring in each of the 5 recognized antigenic sites: 4 in antigenic site A, 1 in B, 2 in C, 1 in D, and 1 in E. The HA1 of A/Wis/05 contained as much as 64 aa differences compared with sw/Gent/08. Fourteen differences were located in antigenic sites: 2 in antigenic site A, 7 in B, 3 in C, 1 in D, and 1 in E. Three additional N‐glycosylation sites in the globular head of HA1, at positions 133, 144, and 246, were found in A/Wis/05 but absent in A/Vic/75 and sw/Gent/08. Positions 133 and 144 were located in antigenic site A and position 133 was in the receptor binding site. The NA gene of A/Vic/75 was also more closely related to sw/Gent/08 (44 aa differences, with 10 in putative antigenic sites) than that of A/Wis/05 (62 aa differences, 15 in antigenic sites). Compared with sw/Gent/08, one additional N‐glycosylation site was predicted for A/Vic/75 (position 234) and two for A/Wis/05 (positions 93 and 234).
Figure 1.

Alignment of deduced amino acid sequences in the HA1 of sw/Gent/08, A/Vic/75, and A/Wis/05. Residues in the open boxes represent previously identified antigenic sites (A, B, C, D, and E) of H3. Underlined residues represent potential N‐glycosylation sites in the globular head of the HA1. Only amino acids different from those in the sw/Gent/08 sequence are shown, conserved residues are shown as dots.
Table 2 compares the HA1 and NA protein sequences and presumed antigenic sites of sw/Gent/08 with those of additional human H3N2 viruses collected over time. The HA1 of sw/Gent/08 was most similar to that of A/Vic/75. Starting in 1989, all human strains showed less than 85% aa homology with the swine virus, and 14–17 aa differences in antigenic sites of the HA. N‐glycosylation site 246 and 133 were consistently present in the globular head of the HA1 of all human H3N2 viruses examined since 1987 and 1997, respectively. The NA of sw/Gent/08 was most similar to that of A/Port Chalmers/1/73. Human viruses from 1989 or later showed <89% aa homology with the swine virus and 13–15 aa differences in antigenic sites of the NA.
Table 2.
Comparison of hemagglutinin (HA1) and neuraminidase (NA) protein sequences of epidemic human H3N2 influenza viruses from 1973 to 2009 and their antigenic sites with those of sw/Gent/08
| Virus strain | HA1 | NA | ||||
|---|---|---|---|---|---|---|
| GenBank accession no. | % aa identity | No. of aa differences in antigenic sites | GenBank accession no. | % aa identity | No. of aa differences in antigenic sites | |
| A/Port Chalmers/1/73 | ABE12532 | 86·9 | 11 | ABE12548 | 91·6 | 6 |
| A/Victoria/3/75 | AFY08275 | 87·2 | 9 | AAB03361 | 90·5 | 10 |
| A/Texas/1/77 | ABQ58940 | 86·9 | 11 | AFM68968 | 90·7 | 9 |
| A/Bangkok/01/79 | ABF21268 | 85·4 | 13 | ABF21324 | 90·3 | 9 |
| A/Philippines/2/82 | ADJ41805 | 85·4 | 11 | ADJ41808 | 89·7 | 10 |
| A/Leningrad/360/86 | AAB69845 | 85·1 | 13 | AFN11845 | 89·7 | 11 |
| A/Sichuan/02/87 | D10161 | 85·1 | 15 | – | – | – |
| A/Beijing/353/89 | AAB58297 | 83·9 | 16 | AAB06969 | 88·6 | 14 |
| A/Shangdong/9/93 | ACL12129 | 82·4 | 16 | ACL12132 | 88·6 | 13 |
| A/Wuhan/359/95 | AFR42694 | 83·0 | 16 | AAB06998 | 87·7 | 13 |
| A/Sydney/5/97 | ACO95259 | 81·8 | 17 | ACO95262 | 88·1 | 13 |
| A/Moscow/10/99 | ABE73115 | 82·7 | 16 | ABE73101 | 86·4 | 15 |
| A/California/7/04 | ABO37490 | 81·2 | 14 | ABV24038 | 86·9 | 15 |
| A/Wisconsin/67/05 | AFY08274 | 80·2 | 14 | ABW80983 | 86·6 | 15 |
| A/Brisbane/10/07 | ABW23353 | 81·2 | 15 | ACO95273 | 86·2 | 14 |
| A/Perth/16/09 | ACS71642 | 80·9 | 16 | ADW80519 | 86·0 | 14 |
Aa, amino acid; –, not available in GenBank.
Table 3 shows the antigenic relationship in the HA and NA of the three viruses used in infection‐challenge studies. Low to moderate cross‐reactivity between A/Vic/75 and sw/Gent/08 was observed in both HI and NI tests, whereas antisera against the two viruses lacked cross‐reactivity with A/Wis/05 in both tests. For an unknown reason, ferret serum against A/Wis/05 showed similar NI antibody titers to all three viruses, while the swine serum showed minimal reaction with sw/Gent/08 or A/Vic/75.
Table 3.
Cross‐reactivity between the human H3N2 viruses A/Vic/75 and A/Wis/05 and the swine H3N2 virus sw/Gent/08 in hemagglutination inhibition (HI) and neuraminidase inhibition (NI) assays
| Antibody titers with serum to··· | ||||
|---|---|---|---|---|
| A/Vic/75 (F) | A/Wis/05 (F) | A/Wis/05 (S) | sw/Gent/08 (S) | |
| A/Vic/75 | ||||
| HI | 160 | <10 | <10 | 40 |
| NI | 2560 | 40 | <10 | 320 |
| A/Wis/05 | ||||
| HI | <10 | 2560 | 2560 | <10 |
| NI | <10 | 80 | 640 | <10 |
| sw/Gent/08 | ||||
| HI | 40 | <10 | <10 | 1280 |
| NI | 320 | 40 | 20 | 5120 |
F, post‐infection ferret serum; S, hyperimmune swine serum.
Virus excretion and serological response after primary inoculation
As expected after intranasal inoculation of pigs with influenza virus,1 clinical symptoms were absent in most pigs except for two sw/Gent/08‐inoculated pigs with dyspnea at 3 dpi. All pigs excreted virus in nasal swabs. Virus shedding was detected for 5–6 consecutive dpi with sw/Gent/08 or A/Vic/75 and for 4–5 dpi with A/Wis/05. Mean virus titers in nasal swabs are shown in Figure 2. The average AUC value was highest for sw/Gent/08 (21·8), followed by A/Vic/75 (16·6) and A/Wis/05 (12·1).
Figure 2.
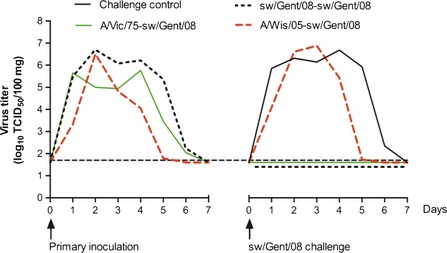
Nasal virus excretion after primary inoculation with human or swine H3N2 influenza virus and after challenge with sw/Gent/08. Mean virus titers in nasal swabs of each group are given. The horizontal broken line represents the detection limit (<1·7 log10 TCID 50/100 mg).
All pigs were seronegative against the three viruses prior to the start of the experiment. Antibody titers at 2 and 6 weeks after the primary inoculation are shown in Table 4. All challenge controls remained seronegative until the time of challenge, whereas the other pigs developed HI, VN, and NI antibodies to the influenza virus used for inoculation. These homologous antibody titers were significantly higher in sw/Gent/08‐inoculated pigs than in the pigs inoculated with human H3N2 viruses (P < 0·05), except for the HI titers at 14 dpi (P > 0·05). Inoculation with A/Vic/75 induced cross‐reactive VN antibodies against sw/Gent/08 in all pigs. Cross‐reactive HI antibodies were detected in only 3 of 8 pigs, at 14 dpi only, and cross‐reactive NI antibodies in 6 pigs. In A/Wis/05‐inoculated pigs, serological cross‐reaction with sw/Gent/08 was negligible in the VN test and undetectable in the HI or NI test.
Table 4.
Antibody response before and after challenge with sw/Gent/08 in challenge controls and pigs first infected with human or swine H3N2 influenza virus
| Geometric mean antibody titers of positive pigs (no. of positive pigs/total no.) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 14 days post first inoculation | Time of challenge | 14 days post challenge | ||||||||
| Group | A/Vic/75 | A/Wis/05 | sw/Gent/08 | A/Vic/75 | A/Wis/05 | sw/Gent/08 | A/Vic/75 | A/Wis/05 | sw/Gent/08 | |
| Challenge control | ||||||||||
| HI | <a | < | < | < | < | < | 12 (4/4) | < | 160 (4/4) | |
| VN | < | < | < | < | < | < | 20 (4/4) | < | 347 (4/4) | |
| NI | n.d. | n.d. | n.d. | < | < | < | 34 (4/4) | < | 381 (4/4) | |
| sw/Gent/08‐sw/Gent/08 | ||||||||||
| HI | 13 (5/8) | < | 104 (8/8) | 10 (4/8) | < | 52 (8/8) | 10 (1/4) | < | 57 (4/4) | |
| VN | 9 (8/8) | 2 (1/8) | 268 (8/8) | 5 (8/8) | 2 (4/8) | 206 (8/8) | 12 (4/4) | 3 (3/4) | 292 (4/4) | |
| NI | n.d. | n.d. | n.d. | 17 (5/8) | < | 320 (8/8) | 25 (3/4) | < | 269 (4/4) | |
| A/Vic/75‐sw/Gent/08 | ||||||||||
| HI | 52 (8/8) | < | 13 (3/8) | 13 (8/8) | < | < | 67 (4/4) | < | 80 (4/4) | |
| VN | 83 (8/8) | 2 (3/8) | 15 (8/8) | 58 (8/8) | 3 (3/8) | 14 (8/8) | 491 (4/4) | 5 (2/4) | 457 (4/4) | |
| NI | n.d. | n.d. | n.d. | 20 (8/8) | < | 13 (6/8) | 381 (4/4) | < | 269 (4/4) | |
| A/Wis/05‐sw/Gent/08 | ||||||||||
| HI | < | 80 (8/8) | < | < | 24 (8/8) | < | 14 (4/4) | 453 (4/4) | 226 (4/4) | |
| VN | 2 (3/8) | 41 (8/8) | 4 (1/8) | 2 (1/8) | 27 (8/8) | 3 (2/8) | 17 (4/4) | 431 (4/4) | 264 (4/4) | |
| NI | n.d. | n.d. | n.d. | < | 10 (8/8) | < | 135 (4/4) | 381 (4/4) | 1076 (4/4) | |
HI, hemagglutination inhibition; VN, virus neutralization; NI, neuraminidase inhibition; n.d., not determined.
Antibody titer below the detection limit, that is, <10 in HI and NI tests and <2 in VN test.
Virological protection after challenge with sw/Gent/08
No clinical signs were observed in any pigs after challenge infection. Mean sw/Gent/08 titers in nasal swabs are shown in Figure 2. Virus shedding was detectable in all challenge controls during 5–6 consecutive days or until euthanasia. All pigs of the sw/Gent/08‐sw/Gent/08 and A/Vic/75‐sw/Gent/08 groups tested negative for virus excretion. Pre‐infection with A/Wis/05 failed to prevent challenge infection or to reduce peak virus titers in nasal swabs. However, the mean duration of nasal shedding was 1·3 days shorter than in the challenge control group.
Sw/Gent/08 virus titers in the respiratory tract of the pigs euthanized at 3 dpc are shown in Table 5. Sw/Gent/08 was isolated from the nasal mucosa, tonsil, trachea, left lung, and right lung of all challenge control pigs. In contrast, all pigs of the sw/Gent/08‐sw/Gent/08 and A/Vic/75‐sw/Gent/08 groups were completely virus negative. In the A/Wis/05‐sw/Gent/08 group, virus isolation rates and virus titers were similar as in the challenge control group (P > 0·05). Lung lesions characterized by dark‐red consolidated areas, involving only 1–2% of the lung surface, were present in two challenge control pigs and two A/Wis/05‐sw/Gent/08 pigs, but absent in both other groups.
Table 5.
Virus titers in the respiratory tract 3 days after challenge with sw/Gent/08 in challenge controls and pigs first infected with human or swine H3N2 influenza virus
| Group | Mean virus titers (log10 TCID50/g) ± SEMa | |||||
|---|---|---|---|---|---|---|
| Nasal mucosa respiratory part | Nasal mucosa olfactory part | Tonsil | Trachea | Left lung | Right lung | |
| Challenge control | 6·4 ± 0·1 | 5·4 ± 0·5 | 4·1 ± 0·7 | 7·1 ± 0·1 | 6·2 ± 0·1 | 6·7 ± 0·3 |
| sw/Gent/08‐sw/Gent/08 | <b | < | < | < | < | < |
| A/Vic/75‐sw/Gent/08 | < | < | < | < | < | < |
| A/Wis/05‐sw/Gent/08 | 7·6 ± 0·4 | 4·6 ± 1·1 | 4·6 ± 0·7 | 7·3 ± 0·2 | 6·2 ± 0·3 | 6·1 ± 0·3 |
Standard error of the mean.
Virus titers below the detection limit (<1·7 log10 TCID50/g).
Serological profile after challenge with sw/Gent/08
All pigs of the challenge control group had developed HI, VN, and NI antibodies to sw/Gent/08 at 14 dpc (Table 4), whereas anti‐sw/Gent/08 antibody titers remained at pre‐challenge levels in the sw/Gent/08‐sw/Gent/08 group (P > 0·05). Pigs of the A/Vic/75‐sw/Gent/08 and A/Wis/05‐sw/Gent/08 groups developed antibodies to sw/Gent/08 or showed a considerable increase in pre‐existing antibody titers. As shown in Figure 3, VN antibodies to the challenge virus developed more rapidly in the A/Wis/05‐sw/Gent/08 group than in the challenge control group, but all 4 groups had similar antibody titers at 10 dpc. Homologous antibody titers to the human H3N2 viruses also increased after challenge with sw/Gent/08. Many pigs showed low cross‐reactive antibody titers to A/Vic/75 without being exposed to it, but cross‐reaction with A/Wis/05 was rare.
Figure 3.
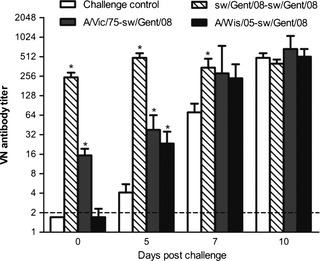
Evolution of virus‐neutralizing (VN) antibody titers against the challenge virus sw/Gent/08 during the first 10 days post challenge. Bars represent group geometric mean VN antibody titers with standard error of the mean (SEM); the asterisk denotes a statistically significant difference (P < 0·05) with the challenge control group. The horizontal broken line represents the detection limit of the assay (<2).
Discussion
The increasing antigenic divergence between H3N2 viruses from swine and humans may have important implications for zoonotic transmission of H3N2 SIVs. This is the first study to evaluate cross‐protection between human and swine H3N2 viruses in pigs. It shows a complete virological protection against infection with a contemporary European H3N2 SIV in pigs infected with A/Vic/75 6 weeks earlier, but only a minimal protection in pigs pre‐infected with the more recent human H3N2 virus A/Wis/05. We did not identify the immune mechanisms underlying the observed cross‐protection, but several findings point toward a role for cross‐reactive antibodies against the HA and/or NA of the swine virus. Indeed, the difference in protective immune response induced by the two human viruses was in line with the genetic and antigenic differences in their HA and NA. Antigenic sites A and B are located on the tip of the HA molecule, and they are supposed to be the primary targets for neutralizing antibodies.21, 22 A/Wis/05 differed from sw/Gent/08 in as much as seven of the total nine amino acids of site B, and it had two additional N‐glycosylations in site A, which make it more distinct from sw/Gent/08 than A/Vic/75. In addition, low‐titered cross‐reactive VN antibodies against sw/Gent/08 were found in all pigs previously infected with A/Vic/75, but they were rare after infection with A/Wis/05. Post‐challenge, however, such antibodies developed more rapidly in A/Wis/05 pre‐infected pigs than in challenge controls, and this coincided with the enhanced clearance of the challenge virus. The HI test, as well as the NI test, is less sensitive than the VN test.26 This can explain the lack of cross‐reactive HI and NI antibodies in most A/Vic/75‐inoculated pigs. Given the low cross‐reactive serum antibody titers in A/Vic/75‐inoculated pigs, it is likely that local antibodies in the respiratory tract and cell‐mediated immunity, which are more cross‐reactive,31 also contribute to the cross‐protection.
Previous studies by Kyriakis et al.9 have shown that H3N2 SIVs from 2007 to 2008 cross‐react in the HI test with hyperimmune sera against the human viruses A/Vic/75 and A/Philippines/2/82, but not with sera against A/Sydney/5/97 or A/Wis/05. The present comparison of sw/Gent/08 with human H3N2 viruses showed increased genetic differences since 1987–1989, as reflected by an increased number of aa differences in the HA (≥14) and NA (≥13), and additional N‐glycosylations in the HA1 globular head. This suggests that especially people born after the mid‐1980s are at a higher risk for infection with European H3N2 SIVs. Additional in vivo cross‐protection studies with more, antigenically distinct human viruses would be required for a better understanding of the nature of the human viruses that could offer cross‐protection against swine viruses. Still, genetic and antigenic analyses should be interpreted with caution. Our cross‐NI test results with A/Wis/05 serum from ferrets versus swine, for example, illustrate that sera from different animal species may yield discrepant results. As for the definition of antigenic sites, this is based on studies of the oldest human viruses and their reaction with mouse monoclonal antibodies. It is questionable whether the antigenic sites of such historical viruses will overlap exactly with those of recent human or swine H3N2 viruses. Furthermore, different animal species will likely mount antibodies recognizing different epitopes. Finally, some amino acids will be more important for antigenic drift than others. According to recent studies, only 7 aa positions in the HA were largely responsible for the antigenic evolution of human H3N2 viruses over a period of 35 years.32 It is of great interest to identify the immunodominant amino acids in the HA of SIVs and those that are cross‐reactive with human H3N2 viruses.
Virus titers in nasal swabs and antibody titers in serum were lower in pigs inoculated with human H3N2 virus than in pigs inoculated with sw/Gent/08. A/Wis/05 also seemed to replicate less efficiently in pigs than A/Vic/75, which is more closely related to swine‐adapted H3N2 viruses. Similar findings were made in a comparative pig infection study with a triple reassortant H3N2 SIV from North America and a non‐reassortant wholly human H3N2 virus.33 Like the authors of that study, we suspect that the lower antibody responses to human influenza viruses in pigs are in part due to their lower replication efficiencies. Virus titers in the ferret model also differ for different H3N2 virus strains.34, 35 Comparative studies of the pathogenesis of human and swine H3N2 viruses in pigs and ferrets would help to select the best animal models and experimental conditions for cross‐protection studies with such viruses. On the other hand, a separate experiment in our laboratory has demonstrated complete protection against A/Wis/05 in pigs that had been previously infected with the homologous virus under the same conditions as in the present study, and with similar antibody titers. Thus, human H3N2 viruses can elicit an efficient protective immune response in pigs.
The recent infections of over 300 humans in the United States, mainly children in a fair setting, with a variant H3N2 virus (A(H3N2)v) of swine origin have raised concerns about the extent of pre‐existing immunity against this virus in the human population. Serological studies have shown cross‐reactive HI antibodies in ≥50% of late adolescents and young adults, whereas such antibodies were generally lacking in children under 10.18, 36, 37 Overall, these serological findings and the present study support the idea that various H3N2 SIV lineages could cause epi‐ or pandemics in the younger population, should they acquire the capacity to spread efficiently between people. The H3 of A(H3N2)v is phylogenetically most closely related to human H3N2 viruses from the 1995 to 1997 era and slightly more related to human H3N2 viruses from the 21st century than European H3N2 SIVs.38 We therefore assume that the immune status of young people may be even lower for the European H3N2 SIV than for A(H3N2)v. Both serological investigation of humans and experimental cross‐protection studies, as well as studies of the host barriers, are needed to further explore the susceptibility of humans to various H3N2 SIVs. Experimental cross‐protection studies are performed under artificial conditions, but they will assess all arms of the immune response and true protection. Many serological studies only consider subjects with HI titers ≥40 as protected, and they may therefore underestimate protection against SIVs in the human population.
Acknowledgements
We thank Dr. John McCauley for providing the human H3N2 influenza viruses, Prof. Dr. Guus Rimmelzwaan for providing the ferret sera, and Lieve Sys, Melanie Bauwens, Philippe Elskens and Zeger van den Abeele for excellent technical assistance. This study was supported by the Belgian Federal Public Service Health, Food Chain Safety, and Environment (RT 09/6227, FLUCROSS) and the European Commission (FP7‐GA 258084, FLUPIG).
Qiu et al (2013). Prior infection of pigs with a recent human H3N2 influenza virus confers minimal cross‐protection against a European swine H3N2 virus. Influenza and Other Respiratory Viruses 7(6), 1260–1268.
References
- 1. Van Reeth K, Brown IH, Olsen CW. Swine influenza; in Zimmerman JJ, Karriker LA, Ramirez A, Schwartz KJ, Stevenson GW. (eds): Diseases of Swine, 10th edn Hoboken, NJ: John Wiley & Sons, Inc., 2012; 557–571. [Google Scholar]
- 2. Castrucci MR, Donatelli I, Sidoli L, Barigazzi G, Kawaoka Y, Webster RG. Genetic reassortment between avian and human influenza A viruses in Italian pigs. Virology 1993; 193:503–506. [DOI] [PubMed] [Google Scholar]
- 3. Campitelli L, Donatelli I, Foni E et al Continued evolution of H1N1 and H3N2 influenza viruses in pigs in Italy. Virology 1997; 232:310–318. [DOI] [PubMed] [Google Scholar]
- 4. Webby RJ, Swenson SL, Krauss SL, Gerrish PJ, Goyal SM, Webster RG. Evolution of swine H3N2 influenza viruses in the United States. J Virol 2000; 74:8243–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nerome K, Kanegae Y, Shortridge KF, Sugita S, Ishida M. Genetic analysis of porcine H3N2 viruses originating in southern China. J Gen Virol 1995; 76(Pt 3):613–624. [DOI] [PubMed] [Google Scholar]
- 6. Yu H, Hua RH, Zhang Q et al Genetic evolution of swine influenza A (H3N2) viruses in China from 1970 to 2006. J Clin Microbiol 2008; 46:1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ngo LT, Hiromoto Y, Pham VP et al Isolation of novel triple‐reassortant swine H3N2 influenza viruses possessing the hemagglutinin and neuraminidase genes of a seasonal influenza virus in Vietnam in 2010. Influenza Other Respi Viruses 2012; 6:6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Jong JC, Smith DJ, Lapedes AS et al Antigenic and genetic evolution of swine influenza A (H3N2) viruses in Europe. J Virol 2007; 81:4315–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kyriakis CS, Brown IH, Foni E et al Virological surveillance and preliminary antigenic characterization of influenza viruses in pigs in five European countries from 2006 to 2008. Zoonoses Public Health 2011; 58:93–101. [DOI] [PubMed] [Google Scholar]
- 10. de Jong JC, van Nieuwstadt AP, Kimman TG et al Antigenic drift in swine influenza H3 haemagglutinins with implications for vaccination policy. Vaccine 1999; 17:1321–1328. [DOI] [PubMed] [Google Scholar]
- 11. Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans: a review of the literature. Clin Infect Dis 2007; 44:1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shu B, Garten R, Emery S et al Genetic analysis and antigenic characterization of swine origin influenza viruses isolated from humans in the United States, 1990–2010. Virology 2012; 422:151–160. [DOI] [PubMed] [Google Scholar]
- 13. Van Reeth K. Avian and swine influenza viruses: our current understanding of the zoonotic risk. Vet Res 2007; 38:243–260. [DOI] [PubMed] [Google Scholar]
- 14. Claas EC, Kawaoka Y, de Jong JC, Masurel N, Webster RG. Infection of children with avian‐human reassortant influenza virus from pigs in Europe. Virology 1994; 204:453–457. [DOI] [PubMed] [Google Scholar]
- 15. Gregory V, Lim W, Cameron K et al Infection of a child in Hong Kong by an influenza A H3N2 virus closely related to viruses circulating in European pigs. J Gen Virol 2001; 82(Pt 6):1397–1406. [DOI] [PubMed] [Google Scholar]
- 16. Olsen CW, Karasin AI, Carman S et al Triple reassortant H3N2 influenza A viruses, Canada, 2005. Emerg Infect Dis 2006; 12:1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robinson JL, Lee BE, Patel J et al Swine influenza (H3N2) infection in a child and possible community transmission, Canada. Emerg Infect Dis 2007; 13:1865–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skowronski DM, Janjua NZ, De Serres G et al Cross‐reactive and vaccine‐induced antibody to emerging swine influenza A(H3N2)v. J Infect Dis 2012; 206:1852–1861. [DOI] [PubMed] [Google Scholar]
- 19. CDC : Update: influenza activity – United States and worldwide, May 20‐September 22, 2012. MMWR Morb Mortal Wkly Rep 2012; 61:785–789. [PubMed] [Google Scholar]
- 20. Shortridge KF, Webster RG, Butterfield WK, Campbell CH. Persistence of Hong Kong influenza virus variants in pigs. Science 1977; 196:1454–1455. [DOI] [PubMed] [Google Scholar]
- 21. Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody‐binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981; 289:373–378. [DOI] [PubMed] [Google Scholar]
- 22. Underwood PA. Mapping of antigenic changes in the haemagglutinin of Hong Kong influenza (H3N2) strains using a large panel of monoclonal antibodies. J Gen Virol 1982; 62(Pt 1):153–169. [DOI] [PubMed] [Google Scholar]
- 23. Colman PM, Varghese JN, Laver WG. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983; 303:41–44. [DOI] [PubMed] [Google Scholar]
- 24. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Das SR, Puigbo P, Hensley SE, Hurt DE, Bennink JR, Yewdell JW. Glycosylation focuses sequence variation in the influenza A virus H1 hemagglutinin globular domain. PLoS Pathog 2010; 6:e1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Vleeschauwer AR, Van Poucke SG, Karasin AI, Olsen CW, Van Reeth K. Cross‐protection between antigenically distinct H1N1 swine influenza viruses from Europe and North America. Influenza Other Respi Viruses 2011; 5:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Atanasova K, Van Gucht S, Barbe F, Duchateau L, Van Reeth K. Lipoteichoic acid from Staphylococcus aureus exacerbates respiratory disease in porcine respiratory coronavirus‐infected pigs. Vet J 2011; 188:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leuwerke B, Kitikoon P, Evans R, Thacker E. Comparison of three serological assays to determine the cross‐reactivity of antibodies from eight genetically diverse U.S. swine influenza viruses. J Vet Diagn Invest 2008; 20:426–432. [DOI] [PubMed] [Google Scholar]
- 29. Van Reeth K, Gregory V, Hay A, Pensaert M. Protection against a European H1N2 swine influenza virus in pigs previously infected with H1N1 and/or H3N2 subtypes. Vaccine 2003; 21:1375–1381. [DOI] [PubMed] [Google Scholar]
- 30. Sandbulte MR, Gao J, Straight TM, Eichelberger MC. A miniaturized assay for influenza neuraminidase‐inhibiting antibodies utilizing reverse genetics‐derived antigens. Influenza Other Respi Viruses 2009; 3:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tamura S, Kurata T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn J Infect Dis 2004; 57:236–247. [PubMed] [Google Scholar]
- 32. Koel BF, Burke DF, Bestebroer TM et al 35 years of antigenic evolution of influenza A/H3N2 virus is dictated by 7 amino acid positions flanking the hemagglutin receptor binding site. Proceedings of the 4th ESWI Influenza Conference. Malta, 2011:45.
- 33. Landolt GA, Karasin AI, Phillips L, Olsen CW. Comparison of the pathogenesis of two genetically different H3N2 influenza A viruses in pigs. J Clin Microbiol 2003; 41:1936–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van den Brand JM, Stittelaar KJ, van Amerongen G et al Comparison of temporal and spatial dynamics of seasonal H3N2, pandemic H1N1 and highly pathogenic avian influenza H5N1 virus infections in ferrets. PLoS ONE 2012; 7:e42343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Svitek N, Rudd PA, Obojes K, Pillet S, von Messling V. Severe seasonal influenza in ferrets correlates with reduced interferon and increased IL‐6 induction. Virology 2008; 376:53–59. [DOI] [PubMed] [Google Scholar]
- 36. CDC : Antibodies cross‐reactive to influenza A (H3N2) variant virus and impact of 2010‐11 seasonal influenza vaccine on cross‐reactive antibodies – United States. MMWR Morb Mortal Wkly Rep 2012; 61:237–241. [PubMed] [Google Scholar]
- 37. Waalen K, Kilander A, Dudman SG, Ramos‐Ocao R, Hungnes O. Age‐dependent prevalence of antibodies cross‐reactive to the influenza A(H3N2) variant virus in sera collected in Norway in 2011. Euro Surveill 2012; 17:pii:20170. [PubMed] [Google Scholar]
- 38. Lina B, Bouscambert M, Enouf V, Rousset D, Valette M, van der Werf S. S‐OtrH3N2 viruses: use of sequence data for description of the molecular characteristics of the viruses and their relatedness to previously circulating H3N2 human viruses. Euro Surveill 2011; 16:pii:20039. [PubMed] [Google Scholar]