

Abstract
Background Recent outbreaks of highly pathogenic avian influenza and multiple occurrences of zoonotic infection and deaths in humans have sparked a dramatic increase in influenza research. In order to rapidly identify and help prevent future influenza outbreaks, numerous laboratories around the world are working to develop new nucleotide‐based diagnostics for identifying and subtyping influenza viruses. While there are several databases that have been developed for manipulating the vast amount of influenza genetic data that have been produced, significant progress can still be made in developing tools for translating the genetic data into effective diagnostics.
Description The Influenza Primer Design Resource (IPDR) is the combination of a comprehensive database of influenza nucleotide sequences and a web interface that provides several important tools that aid in the development of oligonucleotides that may be used to develop better diagnostics. IPDR’s database can be searched using a variety of criteria, allowing the user to align the subset of influenza sequences that they are interested in. In addition, IPDR reports a consensus sequence for the alignment along with sequence polymorphism information, a summary of most published primers and probes that match the consensus sequence, and a Primer3 analysis of potential primers and probes that could be used for amplifying the sequence subset.
Conclusions The IPDR is a unique combination of bioinformatics tools that will greatly aid researchers in translating influenza genetic data into diagnostics, which can effectively identify and subtype influenza strains. The website is freely available at http://www.ipdr.mcw.edu.
Keywords: Bioinfomatics, database, diagnostic, influenza, primer
Introduction
Since the 1997 Hong Kong avian influenza epizootic, the attention of the world and its scientific community has intensified on all aspects of influenza. 1 This has increased even more in the last few years because of the return of H5N1 in 2003. 2 , 3 Significant resources and investment by the United States (NIH, CDC, USDA, etc.) and a large number of countries around the world have been made in all areas of influenza biology, especially in the areas of vaccines, therapeutics, and diagnostics. A result of these efforts has been a significant increase in the amount of genomic data for influenza, which is now greater than 60 000 nucleotide sequences and growing by hundreds weekly. In order to store the ever increasing amount of influenza sequence data and make it accessible to researchers several databases/websites have been formed including NCBI’s Influenza Virus Resource (IVR), LANL’s Influenza Sequence Database (ISD), the NIH‐funded BioHealthBase, and the Influenza Virus Database (IVDB) at the Beijing Institute of Genomics (BIG). 4 , 5 , 6 , 7 All of these databases provide access to varying amounts of influenza sequence data and have an assortment of methods for analyzing the data.
Developing diagnostic assays for influenza can be difficult because of its high mutation rate and the large variety of subtypes for influenza A. An influenza A virus’s subtype is characterized by the hemagglutinin and neuraminidase proteins present on the surface of the virus. 8 , 9 Each influenza A strain’s subtype is determined by the combination of 1 of 16 possible hemagglutinin genes and one of nine possible neuraminidase genes that it contains. Pathogenicity and host range are determined in part by host sialic acid receptors, the HA and NA viral proteins and may be independent of subtype. However, the subtype of the virus is important because it can be directly related to currently circulating strains – both epidemic or pandemic – and suggest pathogenicity (e.g., detecting an H5N1 in a human versus an H5N2 might suggest that the one human has a significantly greater risk of illness and death than the other, also many subtypes have never been identified in humans or many animals).
As it is important to identify the subtype of the virus and not just its presence, there are a variety of different diagnostic techniques being used and developed for both identifying influenza and determining its subtype. Due to their relatively high sensitivity and speed, a large percentage of influenza diagnostic techniques being developed are nucleotide based, including real‐time polymerase chain reaction assays and oligonucleotide microarrays. 10 , 11 , 12 , 13 , 14 The one thing that all nucleotide‐based assays have in common is the need for primers or probes to amplify or detect the sequence of interest. Influenza’s high mutation rate has led to a lot of genetic variability which makes developing oligonucelotides (oligos) for influenza a balancing act between finding regions that are specific yet still well enough conserved to identify the majority of sequences of interest. As influenza viruses mutate to evade the host immune response diagnostic assays must also be adapted to keep up with these changes. Therefore, there will always be a need to develop new influenza diagnostics. In order to aid in primer and probe design there have been a variety of programs developed that vary in functionality and scope. Many of these programs only allow the user to analyze one sequence at a time and those that do analyze multiple sequence alignments have difficulty with the large amount of variability between influenza sequences.
Even though there are multiple databases that provide access to influenza sequence information and there are multiple programs that have been designed to aid in primer design, never have they been combined in a way that significantly aids in the development of influenza diagnostics. To solve this problem we have created the Influenza Primer Design Resource (IPDR), which is an automatically updating database, containing all accessible nucleotide sequences of influenza A, B, and C, and has been integrated into a website that allows all 60 000+ sequences to be searched with a variety of criteria including: gene segment, host, year, species, geographic location, and subtype. The website quickly aligns the sequences using the multiple sequence alignment program (MAFFT) and displays the consensus sequence with the percent conservation at each position and the percentage of sequences contributing to every base pair decision. 15 Additionally, the consensus sequences are blasted against a database of published primers and probes and displayed with the output aligned below the sequence. 16 Each consensus sequence is also analyzed by Primer3 to identify potential primers and probes. 17 We believe this website will significantly advance the development of useful diagnostic assays for human and animal influenza, decrease enormous expenditures in resources around the world, and allow for rapid response as new influenza strains emerge to cause both yearly epidemics or the next pandemic.
Construction and usage
Construction of the database
All of IPDR’s nucleotide sequence information is stored in an Oracle database. The database is populated using a combination of PERL and SHELL scripts to extract the sequence information from LANL (http://www.flu.lanl.gov/) and NCBI (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html) and then insert the information into the database. A UNIX program that executes tasks at scheduled times – called a ‘cron job’– on the server is set to automatically repeat this process weekly. Each record in the sequence information table has the following fields – accession, host, gene segment, subtype, year, country, length, strain, and sequence. In addition to the nucleotide sequence information table there is a separate table of published influenza primers and probes. The information for the primer/probe table comes from several sources including VirOligo, PubMed literature searches, other published resources, and several primers used in our laboratory. 18 , 19 For each oligo the following information is recorded – name, target organism, target gene, strand, location, type, sequence, length, what it was tested against, and the reference it was found in. The primer and probe database will be updated periodically as more are published and a feedback form is provided on the website encouraging researchers to submit primers and probes that have been validated with experimental data.
Searching the database
An interactive web interface for the database has been constructed using Grails, which is a web application framework that uses the Groovy scripting language on the Java platform to help standardize the development of web interfaces. The interface allows the user to search the database using any combination of the following parameters – virus species, host, region/country, segment, H/N type, subtype, years, length, and name (Figure 1). For any parameter that is in a list format multiple values may be selected or deselected by holding down CTRL. Leaving any parameter blank causes all values for that parameter to be selected. The H/N Type and Subtypes parameters both search the subtype field in the database, but they do it in slightly different ways. The H/N Type parameter is setup as two lists of all possible H and N types. If only an H type is selected, then all of the sequences for that H type will be retrieved irrespective of the N type. The same is true if only an N type is selected. If both an H type and an N type are selected, only the sequences for that subtype will be retrieved (e.g. H type = H3, N Type = N2, Subtype = H3N2). If multiple H types and multiple N types are selected every possible combination of the two types will be returned (e.g. H type = H3, H5, N Type = N1, N2, Subtype = H3N1, H3N2, H5N1, H5N2). If the user wants to select only a few specific subtypes, then it is better to type them into the Subtypes box. When using the Subtypes box the specific subtypes that have been entered will be returned. Both the Years and Length parameters allow a beginning and ending value to be entered. If both values are entered they act as a range otherwise if only one of the values is entered it acts as a minimum or maximum. The Name parameter is designed to search the name of the virus strain. A user must be careful when searching by name because abbreviations are commonly used for the host and city names that are usually found in the virus strain name (e.g. New York = NY).
Figure 1.
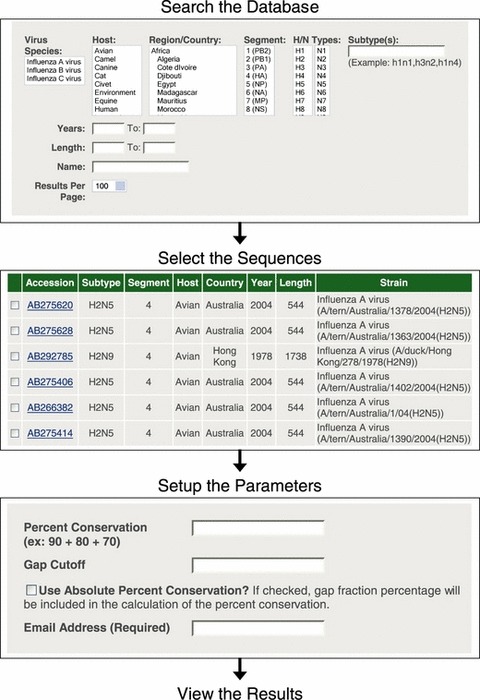
Diagram of the process for using the Influenza Primer Design Resource website. First, the user searches the database for the sequences they are interested in. They then select the sequences that they are interested in. Finally, they set up the parameters for determining the consensus sequence and the Primer3 analysis. The results are returned to the user via email.
The search results
After a search has been submitted the user will be taken to the search results page. On this page there will be a list of the search parameters used, the number of results found, and a table that contains the search results with the following information about each sequence found – Accession, Subtype, Segment, Host, Country, Year, Length, and Strain (Figure 1). From the results page either all of the results may be chosen to be analyzed or a selection of the results may be chosen by clicking on the checkbox next to the individual records. By default, if none of the boxes are checked then all of the results will be used in any further analysis. The results may also be downloaded as seen in the table on the Search Results page with the sequence added in a comma separated values formatted file or as just the sequences in FASTA format.
Setting up the analysis
The next step is for the user to set up the parameters to use for determining the consensus sequence(s) for the alignment and the Primer3 analysis (Figure 1). For calculating the consensus sequence from the alignment there are three parameters that can be adjusted.
Percent conservation
Each ‘Percent Conservation’ entered by the user acts as a cutoff for determining which nucleotide is conserved at each position in the alignment. For example, if the Percent Conservation was 90% and 95% of the sequences in the alignment had a ‘G’ at the position being analyzed then there would be a ‘G’ at that position in the consensus sequence. If instead none of the nucleotides were present in greater than 90% of the sequences at that position the consensus sequence would contain an ‘N’ meaning that position is unconserved. The default value is 0% which causes the program to select which ever nucleotide is most conserved at the position and no positions will be considered unconserved, which is often referred to as the ‘Majority’ consensus. As no single percentage will work best for all sets of data or purpose, the IPDR offers maximum flexibility and multiple Percent Conservations may be entered in order to find the optimal value for each user.
Gap cutoff
The Gap Cutoff is used to determine whether a position in the alignment should be considered a gap in the alignment, which is represented by a ‘‐‘. Frequently when performing alignments, there will be a small percentage of sequences (usually 1–5%) that contain insertions (real or artificial) relative to the majority of sequences in the alignment, which causes a gap in the alignment. The Gap Cutoff is used to prevent these insertions from becoming part of the consensus sequences. Additionally, the cutoff can be used to eliminate other regions of the alignment that have low amounts of sequence information from the consensus sequence, like the extreme ends of the gene segments which are often incompletely sequenced. The program uses the Gap Cutoff value as the minimum percentage of sequences required when determining whether a position in the alignment should be a gap in the consensus sequence. For example, if the Gap Cutoff was 20% (the default) and at the position being analyzed only 15% of the sequences had a nucleotide then that position would be considered a gap in the consensus sequence. In this example with the default Gap Cutoff, if 25% of the aligned sequences had a nucleotide at any position then no gap would be reported in the consensus sequence.
Use absolute percent conservation
In order to understand the last consensus sequence parameter, it is necessary to first look at how the percent conservation is calculated at each position in the alignment when determining the consensus sequence. The following formula is used by default:
The default formula eliminates the sequences without nucleotide information at the position in the alignment being analyzed (# of Sequences with Gap). This is important because the gaps artificially reduce the percent conservation of the nucleotides. For example, if the user was looking for a 90% consensus sequence and at a position only 80% of the sequences have a nucleotide and all 80% are a ‘G’, the formula listed above would calculate the percent conservation at that position to be 100%‘G’ and there would be a ‘G’ in the consensus sequence. In most cases this would be the desired result.
If the user selects to use the absolute percent conservation the sequences with a gap are included in the percent conservation calculation and the following formula is used:
So, in the above example, 100% of the sequences are included in the calculation, which would make the percent conservation 80%‘G’ and therefore an ‘N’ would be added to the consensus sequence.
Primer3 parameters
For each consensus sequence that is generated a Primer3 analysis is also performed. Primer3 is a program that is commonly used to find primers and probes and is useful because it allows users to specify a variety of parameters that will allow the program to select primers that are specific to their interests. Information about Primer3’s parameters can be found at the program’s website. 17
Analysis and results
The analysis that is performed involves several steps. First, the sequences are aligned using MAFFT under the default conditions. The MAFFT alignment program was selected because of its ability to align a large number of sequences in a very short period of time while remaining as accurate as the more commonly used ClustalW program. 20 , 21 After the alignment is completed it is passed to a PERL script that performs three additional analyses.
Sequence conservation analysis
The first analysis performed by the PERL script uses the consensus sequence parameters entered by the user to calculate the consensus sequence from the alignment along with other sequence conservation information. In the results table the user is provided with a variety of information (Figure 2). First, there is a consensus sequence for each of the percents that were entered. With each consensus sequence there is also a conserved sequence. The program determines the conserved sequence by removing regions of the consensus sequence with many unconserved residues in close proximity and replacing them with a ‘‐‘. It is designed to make it easier to find conserved regions in the consensus sequence. Next is the most conserved sequence, which displays the most conserved residue at each position in the alignment. This is followed by a bar graph of the absolute percent conservation of the most conserved nucleotide at each position. Underneath the graph is the sequence polymorphism information with the absolute percent conservation for each nucleotide. The final information in the table is the gap fraction of each position, which shows the percentage of sequences that do not have a nucleotide at each position in the alignment. Users may download either the alignment in ClustalW or FASTA format or the consensus sequences in FASTA format by clicking on the appropriate link.
Figure 2.
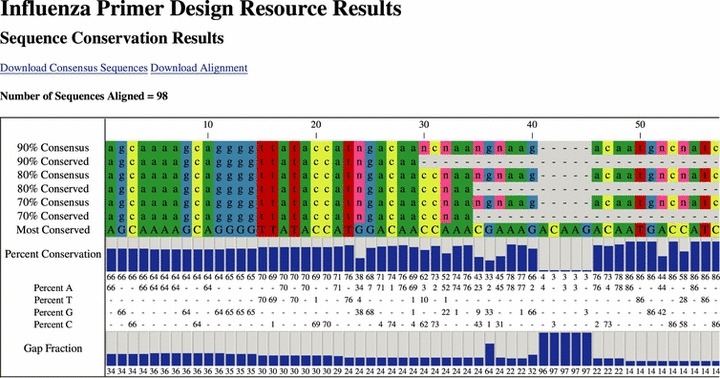
An example of the results returned by Influenza Primer Design Resource. The sequence conservation results display the consensus sequence, conserved regions, percent conservation and sequence polymorphism information.
Primer/probe database blast analysis
For the second analysis, the consensus sequences are blasted against the database of published influenza primers and probes. The PERL script parses the blast results and displays them in both a graphical and table form. In the graphical form the consensus sequence is displayed with the matching oligos aligned with the sequence (Figure 3). Each primer is represented by its name, a colored bar, and the sequence of the matching portion of the primer. The color of the bar represents the score of the blast hit with the darker the blue representing the higher the score.
Figure 3.

The primer/probe database blast results show the published oligos that matched the consensus sequence in a graphical view aligned with the sequence (A) along with a table of published information about the oligos and information from the blast results (B).
The summary table provides both the published information about what the primer was originally used for and how it was designed and a summary of the blast information. In the summary table, in some cases part of the oligo sequence is in capital letters and the rest is in lower case. As it is possible for only part of the sequence to match in blast, the upper case part of the sequence corresponds to the blast hit. It is also important to understand that the values for the rest of the fields that fall under the ‘Published Primer Information’ only represent information from the publication in which the primer was originally described and in many instances will not match with the specific set of sequences being analyzed. For example, a primer originally designed for the NS gene segment may match a set of HA sequences.
Primer3 analysis
For the final analysis, the consensus sequences are analyzed by Primer3 with the parameters that were entered by the user. The Primer3 results are displayed in a manner similar to the blast results with both a graphical view and a summary table for each consensus sequence (Figure 4). In the graphical view each set of oligos is displayed aligned with the consensus sequence. In the summary table each oligo is listed with its start position, length, melting temperature, GC percent, self complementarity, 3′ self‐complementarity, and sequence. Also, for each set, the product size, pair complementarity, and pair 3′ complementarity are listed.
Figure 4.

The Primer3 results display the oligos selected by Primer3 for the consensus sequence based on the user‐entered parameters. The resulting oligos are displayed aligned with the consensus sequence with a summary of the oligo information in a table below.
Discussion
Researchers all over the world are working to develop diagnostic assays for identifying and subtyping influenza virus strains based on their genome sequences. Unfortunately, due to the large amount of sequence variation it can be very challenging for researchers to develop assays that are universally effective at correctly identifying a significant percentage of influenza strains. Also, as the influenza virus is constantly mutating, assays will have to be continuously modified in order to remain effective in the future. These problems highlight the need for better resources for developing the oligos that are essential for these assays to function. There are currently four main resources for influenza researchers to access the vast amount of influenza sequence information and each of these resources provides a different set of tools for data analysis.
The primary source of sequence information for all of the influenza specific databases is NCBI as it houses the primary database for all genetic information, GenBank. 4 NCBI has also developed their own website for analyzing influenza data called the IVR. IVR allows users to search their database for influenza sequences using a variety of parameters. Users may also perform several analyses on the influenza sequences including blast, multiple sequence alignment, and a phylogenetic tree analysis. The other websites are all similar to IVR in design and include most of the same sequences, but vary in their interface, sources of additional sequences, and specific analyses available.
The second resource for influenza sequence information is LANL’s ISD. 5 ISD contains all of the sequence information from NCBI plus several thousand sequences that have not yet been submitted to GenBank. Sequences in ISD undergo a curation process that increases their accuracy and completeness. ISD also features a database search feature similar to, but slightly less functional than, IVR’s. There are multiple data analyses available at ISD, but to use any of them the user must have a paid subscription.
The IVDB from the Beijing Institute of Genomics and the Chinese Academy of Sciences is an integrated information resource and analysis platform for influenza sequence information. 6 Most of the sequence information in IVDB comes from the NCBI and LANL databases but they also include a small number of sequences that have been sequenced at BIG but have not yet been submitted to GenBank. They also perform a manual curation process on all of the sequences to fill out information not included in some of the original sequence records, such as host or subtype. Perhaps as a result of the manual curation process, they only update the database a few times a year and are currently missing ∼5000 nucleotide sequences that have been added to the other databases since their last update. IVDB does provide a variety of analyses for the sequences in their database including blast, multiple sequence alignment, phylogenetic tree builder, a geographic distribution map, and several others.
The Biodefense Public Health Database (BioHealthBase) is the last resource for finding influenza information and analyzing influenza sequence data. 7 BioHealthBase was designed to provide information on multiple infectious agents with influenza only being one of them. Even though users are able to access all of the sequence information for influenza from GenBank, the search and analysis features provided at the website are more limited than those of the other influenza databases. Due to the limits in these features BioHealthBase is more useful for the variety of information it has about influenza than it is for sequence analysis.
The four influenza resources listed above provide access to all of the influenza sequence information and provide multiple tools for analyzing sequence information, but their broad nature and the limits they impose on their analyses prevent them from being optimal tools for influenza oligo design. As IPDR is designed specifically for aiding researchers in designing oligos for influenza diagnostic assays, it provides only the resources needed for oligo design and eliminates the restrictions that hinder analyzing sufficient amounts of data necessary to properly design effective diagnostic assays. IPDR’s sequence database contains all of the publicly available nucleotide sequences from LANL and NCBI and is updated weekly so that users have the most current information available when designing oligos. As both ISD and IPDR collect their sequence information from the same sources and are both updated on a regular basis they are essentially equivalent in the information in their databases. IVDB would be on par with these two databases if not for its manual curation process which causes its time between updates to be several months and several thousand sequences behind. The remaining two databases rely solely on retrieving their sequences from GenBank so they will always have thousands less sequences.
A database is only useful if users are able to extract the information that they want from it. Because IVR’s search interface is well designed and representative of the other websites, we designed IPDR’s search interface to be similar with some minor changes to the subtype search so users would be able to retrieve the exact subset of sequences that they are interested in. IVDB and BioHealthBase also use a similar search interface, but ISD uses a slimmed down search interface that can unfortunately make it more difficult to retrieve the sequences a user is interested in.
Generally, the first step to designing oligos for influenza diagnostics is to perform a multiple sequence alignment on the sequences of interest. For IPDR we decided to use the MAFFT program, which has been shown to be both fast and accurate. We also decided to not set any limits on how many sequences a user can align, and MAFFT can handle alignments of up to ∼10 000 influenza sequences. Both IVR and BioHealthBase use the MUSCLE program for their alignments, which is similar to MAFFT in speed and accuracy, but they impose a limit of 1000 sequences and 50 000 nucleotides (<50 sequences), respectively. IVDB uses the much slower ClustalW program for its alignments and sets a limit of 400 sequences. It is important for users to understand that even though MAFFT has good accuracy no alignment algorithm is 100% accurate. Therefore, users should always download and visualize the alignment to verify its accuracy so that misaligned sequences do not significantly skew subsequent analyses. By utilizing a fast alignment program and removing sequence restrictions IPDR provides users with the best option for aligning influenza sequence data.
The analyses performed on the sequence alignment and its consensus sequences are where IPDR really stands out from the rest of the websites available. After the alignment the other four websites either only provide the user with the alignment or provide the user with just the consensus sequence without the option to set any parameters for calculating the consensus. In addition to providing the user with the alignment and the user‐defined consensus sequences, IPDR also provides users with a sequence highlighting the more conserved regions, sequence polymorphism data, and the gap fraction. These features make it easier for users to find regions to design oligos from and help them decide whether they can use ambiguous nucleotides in the oligo design. While some of the other websites do provide sequence polymorphism data, they only provide it for precomputed alignments, which may not be the exact alignments most users are interested in. Additionally, IPDR provides two other analyses that are not available at the other websites. First, the consensus sequences are compared to a database of published primers and probes using blast. This can eliminate the need for a user to design oligos if they can find some that have already been designed for the sequences that they are interested in or can help them find conserved regions that have been used successfully by other researchers when designing oligos. The final analysis is Primer3, which can design primers for the user so that they do not have to design them, potentially saving the user a significant amount of time.
While there are several other websites currently available that provide access to the vast amounts of influenza sequence data, none of them provide the correct tools to properly facilitate primer and probe design. Through IPDR we provide a combination of analyses that will significantly decrease the time and money that researchers must utilize in order to develop highly effective primers and probes for influenza diagnostics. By utilizing a fast alignment program the website is able to align and analyze thousands of sequences in a matter of minutes. The results of the analyses are then displayed in a user‐friendly format and result files are provided in common formats that can easily be imported into other programs if the user desires. We believe this website will advance the development of useful diagnostic assays for influenza, decrease expenditures around the world, and allow for rapid response to newly emerging influenza strains.
In addition to the functions already available through the website we also have several advancements planned for the future that will further aid researchers in their oligo design. First, we will add a function that allows users to input sequences and then blast against their database search results or add these user‐derived sequences to the IPDR search results prior to alignment. The expanded blast function would be particularly useful for checking oligos for cross‐hybridization with strains or subtypes other than the ones they are intended for. Also we plan to add a function that will geographically map and statistically analyze their search results. The final addition planned is a feature that will allow researchers to check oligos they have designed against the database or subsets of the database to estimate the percentage of sequences that they would be able to amplify/detect (genetic coverage). In order to meet the needs of the influenza research community, we also hope to make additions based on user feedback that we may receive through our comments page. Even though we believe our website already significantly aids in the design of oligos for influenza diagnostics, these additional features will help shape the future of influenza diagnostics.
Note added in proof
During the course of the review process ISD announced that they were planning to merge all of their resources with BioHealthBase by early 2008. Once the merger is complete it is stated that the ISD public website will be shut down. These changes will affect comments made about both websites throughout the manuscript. Additionally, as a part of this process the majority of the sequences unique to ISD have been deposited in Genbank. Therefore, IPDR will no longer be extracting sequence information from ISD, but will still be retrieving all of the sequences from Genbank.
Acknowledgements
We thank Stacy Zacher for Oracle support and Kent Brodie for server support. We also thank Meredith Van Dyke and Lupita Ontiveros for helping retrieve publications for the primer/probe database. This work is supported by grant UO1‐AI070428‐01 from the National Institutes of Health.
References
- 1. Snacken R, Kendal AP, Haaheim LR, Wood JM. The next influenza pandemic: lessons from Hong Kong, 1997. Emerg Infect Dis 1999; 5:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Webster RG, Govorkova EA. H5N1 influenza – continuing evolution and spread. N Engl J Med 2006; 355:2174–2177. [DOI] [PubMed] [Google Scholar]
- 3. Hampson AW, Mackenzie JS. The influenza viruses. Med J Aust 2006; 10(Suppl.):S39–S43. [DOI] [PubMed] [Google Scholar]
- 4. Bao Y, Bolotov B, Dernovoy D, et al. The Influenza Virus Resource at the National Center for Biotechnology Information. J Virol 2008; 82:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Macken C, Lu H, Goodman J, Boykin L. The value of a database in surveillance and vaccine selection; in Osterhaus ADME, Cox N, Hampson AW. (ed): Options for the Control of Influenza IV. Amsterdam: Elsevier Science, 2001. [Google Scholar]
- 6. Squires B, Macken C, Garcia‐Sastre A. et al BioHealthBase: informatics support in the elucidation of influenza virus host–pathogen interactions and virulence. Nucleic Acids Res 2008; 36: D497–D503. Epub [2007 Oct 26]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang S, Zhang J, Liao X et al. Influenza Virus Database (IVDB): an integrated information resource and analysis platform for influenza virus research. Nucleic Acids Res 2007; 35:D376–D380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu G, Yan SM. Mutation trend of hemagglutinin of influenza A virus: a review from a computational mutation viewpoint. Acta Pharmacol Sin 2006; 27:513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stephenson I, Zambon M. The epidemiology of influenza. Occup Med (Lond) 2002; 52:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aguero M, San Miguel E, Sanchez A, Gomez‐Tejedor C, Jimenez‐Clavero MA. A fully automated procedure for the high‐throughput detection of avian influenza virus by real‐time reverse transcription‐polymerase chain reaction. Avian Dis 2007; 51(1 Suppl.):235–241. [DOI] [PubMed] [Google Scholar]
- 11. Townsend MB, Dawson ED, Mehlmann M et al. Experimental evaluation of the FluChip diagnostic microarray for influenza virus surveillance. J Clin Microbiol 2006; 44:2863–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valle L, Amicizia D, Bacilieri S et al. Performance testing of two new one‐step real time PCR assays for detection of human influenza and avian influenza viruses isolated in humans and respiratory syncytial virus. J Prev Med Hyg 2006; 47:127–133. [PubMed] [Google Scholar]
- 13. Henrickson KJ, Kraft A, Shaw J, Canter D. Comparison of electronic microarray (NGEN RVA) to enzyme hybridization assay (Hexaplex) for multiplex RT‐PCR detection of common respiratory viruses in children. Clin Microbiol Newsl 2007; 29: 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Y, Tang H, Duffy SF, et al . A Multiplex Assay for Simultaneously Typing and Subtyping Influenza Viruses Using the Electronic Microarray. 23rd Annual Clinical Virology Symposium, Clearwater, FL, April 29–May 2, 2007. [Google Scholar]
- 15. Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 2005; 33:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Altschul SF, Madden TL, Schaffer AA et al. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers; in Krawetz S, Misener S. (ed): Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ, Humana Press, 2000. [DOI] [PubMed] [Google Scholar]
- 18. Onodera K, Melcher U. VirOligo: a database of virus‐specific oligonucleotides. Nucleic Acids Res 2002; 30:203–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. World Health Organization (WHO), Dept. of Communicable Disease Surveillance and Response . WHO Manual on Animal Influenza Diagnosis and Surveillance. Geneva: World Health Organization, 2002. [Google Scholar]
- 20. Nuin PA, Wang Z, Tiller ER. The accuracy of several multiple sequence alignment programs for proteins. BMC Bioinformatics 2006; 7:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahola V, Aittokallio T, Vihinen M, Uusipaikka E. A statistical score for assessing the quality of multiple sequence alignments. BMC Bioinformatics 2006; 7:484. [DOI] [PMC free article] [PubMed] [Google Scholar]