

Abstract
Autosomal recessively inherited glucocerebrosidase 1 (GBA1) mutations cause the lysosomal storage disorder Gaucher's disease (GD). Heterozygous GBA1 mutations (GBA1+/−) are the most common risk factor for Parkinson's disease (PD). Previous studies typically focused on the interaction between the reduction of glucocerebrosidase (enzymatic) activity in GBA1+/− carriers and alpha-synuclein-mediated neurotoxicity. However, it is unclear whether other mechanisms also contribute to the increased risk of PD in GBA1+/− carriers. The zebrafish genome does not contain alpha-synuclein (SNCA), thus providing a unique opportunity to study pathogenic mechanisms unrelated to alpha-synuclein toxicity. Here we describe a mutant zebrafish line created by TALEN genome editing carrying a 23 bp deletion in gba1 (gba1c.1276_1298del), the zebrafish orthologue of human GBA1. Marked sphingolipid accumulation was already detected at 5 days post-fertilization with accompanying microglial activation and early, sustained up-regulation of miR-155, a master regulator of inflammation. gba1c.1276_1298del mutant zebrafish developed a rapidly worsening phenotype from 8 weeks onwards with striking reduction in motor activity by 12 weeks. Histopathologically, we observed marked Gaucher cell invasion of the brain and other organs. Dopaminergic neuronal cell count was normal through development but reduced by >30% at 12 weeks in the presence of ubiquitin-positive, intra-neuronal inclusions. This gba1c.1276_1298del zebrafish line is the first viable vertebrate model sharing key pathological features of GD in both neuronal and non-neuronal tissue. Our study also provides evidence for early microglial activation prior to alpha-synuclein-independent neuronal cell death in GBA1 deficiency and suggests upregulation of miR-155 as a common denominator across different neurodegenerative disorders.
Introduction
Gaucher's disease (GD) is the most common lysosomal storage disorder with a prevalence of 1:40 000 (1). It is caused by autosomal recessively inherited homozygous or compound heterozygous mutations in glucocerebrosidase 1 (GBA1). GBA1 is a lysosomal enzyme required for the breakdown of glucosylceramide to ceramide and glucose and forms part of the sphingolipid pathway. The pathological hallmark of GD is the accumulation of characteristic macrophages engorged with glycolipids also known as Gaucher cells. Clinically, GD can present heterogeneously with three different subtypes, categorized by severity and distribution of symptoms. Patients with type I can be virtually asymptomatic, type II presents with rapid neurological decline and subsequent death within the first 3 years of life, whereas type III presents with neurological decline during adolescence (2). Current treatment options largely focus on enzyme replacement therapy, which is effective for the treatment of non-neurological complications of GD but ineffective for the treatment or prevention of neurological complications due to its inability to cross the blood–brain barrier (3).
Heterozygous GBA1 mutations (GBA1+/−) are the most common risk factor for Parkinson's disease (PD) with an odds ratio of >5 (4–6). PD patients carrying such a heterozygous GBA1 mutation have an earlier age of onset and are more likely to develop impaired cognitive function (7,8).
Both toxic gain of function and loss of function mechanisms have been proposed to explain the link between heterozygous GBA1 mutations and PD with particular focus on an interaction between glucocerebrosidase 1 (GCase) enzymatic activity and alpha-synuclein (6,9).
GBA1 knock out (KO) mouse die shortly after birth due to skin defects leading to a loss of hydration. Conditional GBA1 KO mice with isolated neuronal GCase deficiency have an initial, symptom-free period of 10 days, followed by rapid neurological decline and subsequent death due to excessive seizures. Conditional KO mice in the hematopoietic and mesenchymal cell lineages model the major visceral symptoms of GD, but otherwise have a normal life span and fail to model the neuropathic forms (10).
Zebrafish have become a versatile disease model for studying neurodegeneration (11). As vertebrates, they are more closely related to humans than Drosophila or Caenorhabditis elegans, develop externally and are transparent. We and others have previously demonstrated their usefulness to identify novel drug targets in zebrafish models of PD and other neurodegenerative disorders (12,13).
We have used the TALEN (transcription activator-like effector nucleases) approach to create a gba1 mutant zebrafish. Homozygous gba1 mutant zebrafish (gba1−/−) develop normally but already display sphingolipid dysregulation and accumulation as early as 5 days post-fertilization (dpf) with marked alterations of the GD biomarkers β-hexosaminidase and chitotriosidase in juvenile brain tissue. We further demonstrate early microglial activation with marked upregulation of miRNA-155 (miR-155) which precedes subsequent organ infiltration with Gaucher cells in juvenile gba1−/−. These gba1−/− zebrafish also develop progressive neurodegeneration, mitochondrial dysfunction and loss of dopaminergic neurons with ubiquitin-positive inclusions in the absence of alpha-synuclein. This new vertebrate model of GCase deficiency is likely to have utility for future gene–gene interaction studies and in vivo drug screens. The identification of distinct and potentially ‘druggable’ molecular targets such as miR-155 will facilitate these in vivo drug screens.
Results
Zebrafish possess a single GBA1 orthologue
A BLAST search identified a single zebrafish orthologue of human GBA1 on chromosome 16 (ENSDARG00000076058) of the zebrafish genome. The zebrafish gene (gba1) encodes a single protein of 518 amino acids and 57% identity with the human orthologue. The genetic loci of both (human) GBA1 and (Danio rerio) gba1 shared conserved synteny, both containing the genes RUSC1, FDPS and DAP3 within 500 kb of each orthologue. gba1 was expressed at constant levels through 1–5 dpf with more marked expression in the brain. Expression was also detected in adult brain and liver tissue, organs specifically affected by GD pathology (Fig. 1A–D).
Figure 1.

gba1 expression in wild-type (WT) zebrafish and loss of function studies. gba1 expression through early development and in adult organs particularly affected by GD (namely brain and liver) was confirmed by RT–PCR (A); ef1a was used as a loading control. WISH confirmed early expression of gba1 in brain tissue at 1 dpf (B), 2 dpf (C) and 3 dpf (D). Using TALENs, a 23 bp deletion in exon 7 of gba1 (gba1c.1276_129del) was generated which could be genotyped by PCR. A representative genotyping gel (E) shows WT (lane 1), gba1+/− (lane 2) and gba1−/− (lane 3). The gba1c.1276_129del mutation resulted in a >50% decrease in gba1 transcript levels in gba1−/− brain tissue (P < 0.01, F) and a decrease in enzymatic GCase activity (P < 0.05, G). *P < 0.05; **P < 0.01.
gba1 TALEN-generated mutants are loss of function
Using TALEN technology, we generated a gba1 mutant containing a 23 bp deletion in exon 7 (c.1276_1298del, Fig. 1E and Supplementary Material, Fig. S1). The deletion results in a frame-shift at position c.1276 and a subsequent premature stop codon 66 bp downstream, within exon 7 at c.1342 (p.379). The gba1c.1276_1298del (from hereon referred to as gba1−/−) resulted in a reduction of gba1 mRNA by >50% (P < 0.01, Fig. 1F). Similarly, GCase activity was reduced in gba1−/− brains by >50% (P < 0.05) compared with wild-type (Fig. 1G).
Analysis of sphingolipid metabolites
GCase deficiency leads to marked sphingolipid dysregulation and accumulation of GCase substrates in Gba1 KO mice and patients with GD (14–16). We analyzed sphingolipid metabolites by mass spectrometry across all gba1 genotypes and identified marked accumulation of sphingolipid metabolites as early as 5 dpf in gba1−/−, with increases in the C18 molecular weight species of each glycolipid being the most pronounced (Fig. 2). Hexosylsphingosine was virtually undetectable in wild-type samples but increased to 1573% in gba1−/− of the level seen in controls (Fig. 2C; P < 0.0001), glucosylceramide was increased to ∼360% (Fig. 2D; P < 0.0001). Substrates upstream of GCase also accumulated, namely lactosylceramide to nearly 300% (Fig. 2F; P < 0.0001) whereas galactosylceramide was notably decreased by 50% (Fig. 2E; P < 0.01). Mass spectrometric analysis was repeated in juvenile brain tissue at 12 weeks post-fertilization (wpf) across all gba1 genotypes. Again, direct substrates of GCase had the largest increases in gba1−/− brains: hexosylsphingosine was virtually undetectable in wild-type brains but increased in gba1−/− to 2734% of the level seen in controls (Fig. 2I; P < 0.0001), whereas glucosylceramide increased to 14 000% (Fig. 2J; P < 0.0001). Galactosylceramide was now increased as well (Fig. 2K; P < 0.0001) but not as strongly as lactosylceramides which increased above wild-type levels to 2000% of the level seen in controls (Fig. 2L; P < 0.0001). Sphingosine levels were unaltered in 5 dpf larval homogenates but doubled in gba1−/− juvenile brains (Fig. 2A and G, P < 0.0001), sphinganine levels were increased to a similar extent in gba1−/− larvae and juvenile brains (Fig. 2B and H, P < 0.01). In contrast, there were no significant changes for any of the analyzed sphingolipid metabolites in either gba1+/− larvae or gba1+/− juvenile brains compared with wild-type (see also Supplementary Material, Table S1 which lists all metabolites analyzed).
Figure 2.

Sphingolipid metabolites accumulate in zebrafish larvae and brain tissue. Sphingolipid metabolites were analyzed across gba1 genotypes in 5 dpf larvae and 12 wpf brain tissue. Sphingosine levels remained unaltered across all genotypes at 5 dpf (A) but were approximately doubled in gba1−/− brains at 12 wpf (G). In contrast, sphinganine (B), hexosylsphingosine (C), glucosylceramide C18.0 (D) and lactosylceramide C18.0 (F) had already accumulated in gba1−/− larvae up to 1500% of control values, whereas galactosylceramide C18.0 (E) levels were reduced by 50%. In 12 wpf brain tissue, all these sphingolipid metabolites had accumulated in gba1−/− by up to 14 000% (G–L). The concentration of each sphingolipid is given in ng/mg (ns: non-significant; **P < 0.01; ****P < 0.0001).
gba1−/− zebrafish mirror key Gaucher's disease phenotypes
gba1−/− and gba1+/− did not develop an overt morphological phenotype during early development. By 8 wpf, gba1−/− first began to swim more slowly and to generally look less well. By 12 wpf, juvenile gba1−/− developed a curvature of the spine, reminiscent of the gibbus formation seen in conditional mouse KO models (Fig. 3A and B) (17). The oldest gba1−/− fish reached an age of 14 wpf before death during pilot longevity studies. Consequently, all gba1−/− fish were culled at 12 wpf for humane reasons.
Figure 3.
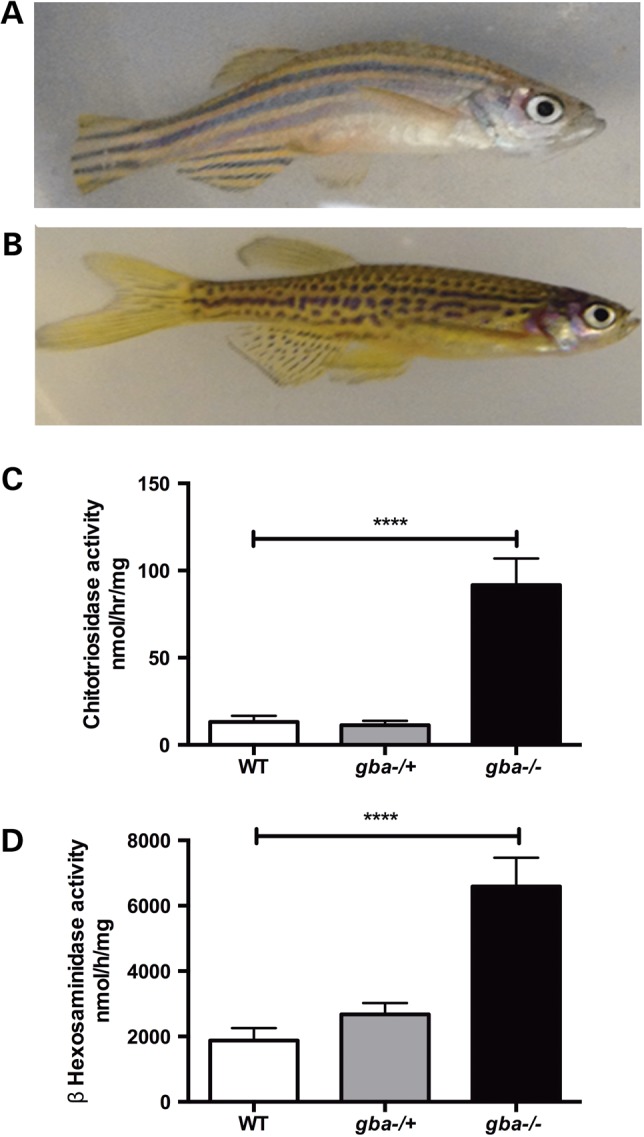
Skeletal and biochemical indices of GCase deficiency. At 12 wpf gba1−/− (A) developed a curve to their spine compared with wild-type (WT) controls (B) in a similar manner to some conditional GBA1 KO mice (17). The difference in the stripe pattern of gba1−/− (A) and wild-type zebrafish (B) is due to the gba1+/− line being of mixed lineage with (wild-type) AB and (wild-type) TL genetic background. Classical Gaucher disease biomarkers were markedly elevated in gba1−/− brains at 12 wpf, with a 10-fold increase in chitotriosidase activity (P < 0.0001) (C) and a 4-fold increase in β-hexosaminidase activity (P < 0.0001) (D). No significant changes were detected in either assay in gba1+/− brains. ****P <0.0001.
Chitotriosidase and β-hexosaminidase activity are markedly increased in the serum of GD patients and used as biomarkers to monitor disease activity (1). In gba1−/− zebrafish brains, chitotriosidase activity was increased ∼10-fold in gba1−/− brains (P < 0.0001; Fig. 3C) without a change in gba1+/−. Similarly, β-hexosaminidase activity was increased to 350% of values observed in controls (P < 0.0001) at 12 wpf but no difference was observed in gba1+/− brains (P > 0.05; Fig. 3D). In contrast, β-galactosidase activity remained unchanged in its activity across all genotypes (data not shown).
At 12 wpf, gba1−/− showed a reduction in total displacement of 50% (P < 0.001), with a reduction by 25% in gba1+/− (P > 0.05) (Fig. 4A). When individual swimming movements were assigned to low, medium and high speeds, wild-type fish spent the majority of their time making fast movements (Fig. 4B and C). The opposite was true of gba1−/− fish, which spent most of their time making slow movements or remaining stationary (P < 0.0001, Fig. 4B and E). gba1+/− fish had an intermediate phenotype for all speeds, but these changes were not significantly different to either wild-type or gba1−/− (Fig. 4B and D). In addition, there were obvious defects of balance, with the gba1−/− animals showing severe variability of vertical body axis orientation (roll) during swimming, resulting in a ‘corkscrew’ pattern of motion. Occasional episodes were observed in which gba1−/− animals showed bursts of high-velocity movements, often violently moving in circles. These abnormalities were frequently interrupted by longer periods of inactivity during which the gba1−/− zebrafish lay on the tank floor (Supplementary Material, Video). These abnormalities were not seen in any of the heterozygous or wild-type sibling controls.
Figure 4.

Marked slowing of spontaneous motor activity in gba1−/−. Video-tracking software was utilized to measure locomotion in gba1 genotypes. All fish were filmed from the side. gba1−/− exhibited a 50% decrease in total displacement (A) (P < 0.001). When speeds were segregated into small medium and high speeds (B), gba1−/− spent more time moving at low speeds (300%, P < 0.0001) and a spent less time moving at high speeds (88% less, P < 0.0001). For representative movements traces of wild-type (WT) (C), gba1+/− (D) and gba1−/− (E), red lines represent high-speed movements, green represents medium-speed movements and black represents low-speed movements. ns, P >0.05; ***P < 0.001; ****P < 0.0001.
gba1−/− exhibit Gaucher cell organ invasion and microglial activation
The primary histopathological hallmark of GD is the formation and accumulation of lipid-engorged macrophages known as Gaucher cells leading to visceral organomegaly. Microglial activation and other immune mechanisms have also been implicated in the pathogenesis of neuronal cell death in both GD and PD (18–20). Hematoxylin and eosin (H&E) staining in 12 wpf gba1−/− revealed marked infiltration with enlarged ‘Gaucher-like’ cells not only in the brain (Fig. 5B), but also in liver (Fig. 5C), thymus (Fig. 5D) and pancreas (data not shown). As expected, no overt pathology could be detected in wild-type control individuals (Fig. 5A). Gaucher cells were periodic acid Schiff (PAS)-positive, indicative of glycolipid accumulation (data not shown). No abnormalities could be detected in the wild-type or gba1+/− fish. There was no overt pathology at all in any of the three genotypes at 4 wpf (data not shown).
Figure 5.

gba1 deficiency leads to Gaucher cell invasion and increased abundance of activated microglia in gba1−/− brain. H&E staining of 12 wpf sections demonstrated Gaucher cell (black arrows) organ invasion of the tectal ventricle within the brain of gba1−/− (B), compared with wild-type (WT) brains (A). Gaucher cell organ invasion was also present in the visceral organs of gba1−/− such as the liver (C) and thymus (D). Fluorescent images show confocal micrographs of gba1−/− and wild-type siblings control brains labeled by indirect immunofluorescence for 4.C4 (macrophages and microglia; green), DAPI (nuclei; blue) and P0 (myelin; red). Low-power images through the tectal ventricle showed accumulation of 4.C4 immunoreactive macrophages in the ventricle and periventricular region of gba1−/− (F) but not wild-type brain (E). Microglia within the brain parenchyma were identified by their immunoreactivity to 4.C4 and typical morphology (G and I). Compared with wild-type brain (G), microglial were more numerous and brightly labeled in gba1−/− brain (I). In addition, compared with the typical quiescent morphology of microglia seen in wild-type brain (H), marked rounding of the cell body and retraction of processes was apparent in gba1−/− brain (J). The white arrows point at a normal microglial cell body in a wild-type control brain (H) and a rounded microglial cell body in a gba1−/− brain (J); the white arrow heads point at a normal, extended microglial process in a wild-type control brain (H) and at a retracted microglial process in a gba1−/− brain (J). These morphological changes are typical of microglial activation.
These Gaucher-like cells around the tectal ventricle labeled strongly with the 4.C4 monoclonal antibody marker for zebrafish monocyte/macrophage lineage cells (Fig. 5E and F). In addition, there was a marked increase in microglial cells in the brain parenchyma of gba1−/− zebrafish compared with controls (Fig. 5G and I). The microglia in gba1−/− brains showed swollen cell bodies and retracted processes typical of microglial activation (Fig. 5H and J).
The transparent nature of zebrafish embryos allows the assessment of microglial activation in vivo in a zebrafish transgenic line in which the membrane-targeted fluorescent reporter (GFP-CAAX) expression is driven by the promoter of macrophage-expressed gene 1 (mpeg1). We crossed this mpeg1:GFP-CAAX transgenic line with gba1+/− zebrafish and then assessed microglial activation in larvae at 4 dpf across the three different genotypes to further determine whether altered immune mechanisms may precede overt neuropathology. gba1+/− and gba1−/− had altered microglial shape, reflecting microglial activation (shape factor in wild-type controls: 0.2077; gba1+/−: 0.2319; gba1−/−: 0.2356; P < 0.001 for both gba1+/− and gba1−/−; Fig. 6A). Microglia vacuole count was also increased in gba1−/− microglia by 40% with average count across genotypes being 3.737 (wild-type), 4.015 (gba1+/−, P > 0.05) and 5.273 (gba1−/−, P < 0.0001) per microglia (Fig. 6B). In contrast, microglia volume and absolute count were unchanged across the three genotypes (data not shown). miR-155 is a key regulator of inflammation (21). We hypothesized that miR-155 up-regulation may be an early feature in gba1−/−. As predicted, miR-155 levels were increase by 88% in gba1−/− larvae at 5 dpf compared with values observed in controls (P < 0.05, Fig. 6C), with an even more marked increase by 470% in juvenile gba1−/− brain tissue (P < 0.01, Fig. 6D).
Figure 6.

Activation of inflammatory/immune mechanisms during larval stages. Microglia in 4 dpf gba1+/− and gba1−/− had an increase in shape factor (sphericity) (P < 0.001 for both) indicative of microglial activation (A). Additionally, the number of vacuoles in gba1−/− was increased by 40% compared with the values observed in wild-type (WT) controls (P < 0.0001) (B). Levels of miR-155, a master regulator of inflammatory/immune mechanisms, were analyzed in 5 dpf larvae (C) and 12 wpf brain tissue (D) across gba1 genotypes. miR-155 was increased 2-fold in gba1−/− larvae (P < 0.05) and 4-fold in 12 wpf gba1−/− brains (P < 0.01). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
gba1−/− undergo alpha-synuclein-independent neurodegeneration
The alpha-synuclein (SNCA) gene is notably absent in the zebrafish genome, but zebrafish possess orthologues of beta- and gamma-synuclein (22). To further investigate the effect of partial or complete GCase deficiency in the absence of alpha-synuclein (protein), dopaminergic neuronal cells were counted both during development and in juvenile zebrafish. At 5 dpf, there was no difference between either gba1+/− or gba1−/− and wild-type controls in the number of ascending dopaminergic neurons within the posterior tuberculum, the anatomical structure in zebrafish analogous to the human substantia nigra pars compacta (Fig. 7A). By 12 wpf, however, there was a marked reduction of dopaminergic neurons in both the caudal hypothalamus by 40% (P < 0.01; Fig. 7B) and the posterior tuberculum by ∼30% (P < 0.01; Fig. 7C). These data show unequivocally that dopaminergic neurons degenerate in gba1−/− zebrafish. Unexpectedly, both beta- and gamma-synuclein protein levels were markedly reduced by 60% in gba1−/− brains (P < 0.0001; Fig. 7D and E). Microscopically, there was an abundance of ubiquitylated neuronal cytoplasmic inclusions as well as occasional ubiquitylated neurites throughout the CNS, but most prominently in the larger hindbrain neurons of gba1−/− fish at 12 wpf (Fig. 7H and I) which bear resemblance to Lewy bodies and Lewy neurites in postmortem PD brain tissue (Fig. 7J).
Figure 7.

Dopaminergic neuronal cell loss and ubiquitinated inclusions in gba1−/− brains. The number of ascending diencephalic dopaminergic neurons (Rink–Wullimann groups 1, 2, 4 and 5) was similar across the three gba1 genotypes at 5 dpf (A). In contrast, there was a 40% loss (P < 0.01) of the dopaminergic neurons in the caudal hypothalamus (B) and a 30% loss (P < 0.01) in the posterior tuberculum at 12 wpf (C). β (D) and γ1 (E) synuclein proteins levels were reduced by 60% in gba1−/− brain tissue suggesting a distinct loss of synapses due to global neurodegeneration. F–J: IHC labels ubiquitin brown by 3,3′-DAB. Glial cell nuclei are highlighted by hematoxylin counterstaining (blue). At 12 weeks of age, there is no significant pathology in wild-type (WT) (F) or gba1+/− fish (G). In contrast, gba1−/− fish (H and I) have granular ubiquitylated neuronal cytoplasmic inclusions (black arrows) and ubiquitylated neuritic pathology (white arrow). These granular neuronal cytoplasmic inclusions and neurites resemble the granular aggregates of a-synuclein (black arrow) and Lewy neurites (white arrow) as seen in sporadic PD (J; substantia nigra) (scale bar = 50 μm throughout). ns, P > 0.05; **P < 0.01; ****P < 0.0001.
gba1−/−-induced mitochondrial dysfunction and impaired autophagy
Mitochondrial dysfunction has been demonstrated in other models of gba1 deficiency (23). We analyzed the activity of the mitochondrial respiratory chain in 12 wpf brain tissue across the gba1 genotypes. Complex III and IV activity was lower by ∼50% in gba1−/− compared with wild-type (P < 0.05). Both complex III and IV activity in gba1+/− fish had intermediate values between those seen in gba1−/− and wild-type, but did not differ significantly from either (Fig. 8A and B). We hypothesized that the observed specific abnormalities in mitochondrial function seen in gba1−/− fish may be due to impaired mitochondrial biogenesis or mitochondrial protein turnover, possibly linked to impaired mitophagy. However, the outer mitochondrial membrane protein TOMM20 and TIMM9 (located in the inter membrane space) levels were similar across the three genotypes (data not shown). In contrast, NDUFA9 (encoding a complex I subunit) and Cox4i1 (encoding a complex IV subunit) were reduced in gba1−/− brains compared with controls (Fig. 8C and D, P < 0.01). The reduction of Cox4i1 may at least in part underlie the observed lowering of complex IV activity. ATP5A (encoding a complex V subunit) was also somewhat lower in gba1−/− fish but this difference was not significant (P > 0.05; data not shown).
Figure 8.

Mitochondrial dysfunction and autophagy in gba1−/− zebrafish. There was a 50% decrease (P < 0.05) in complex III (A) and complex IV (B) activity in gba1−/− brains at 12 wpf. There was also a decrease in the protein levels of the complex I subunit NDUFA9 (C, P < 0.01) and the complex IV subunit Cox4i1 (D, P < 0.01). LC3-II was increased 2-fold in gba1−/− brains at 12 wpf compared with wild-type (WT) (E, P < 0.01). (F) A representative western blot of LC3-II protein levels in wild-type and gba1−/− zebrafish brains. *P < 0.05; **P < 0.01.
GCase deficiency results in lysosomal dysfunction due to the accumulation of its substrate, glucocerebroside and in mice lacking Gba1, decreased autophagosome formation and accumulation of autophagy substrates in the brain as well as decreased mitophagy has been observed (23). Therefore, we investigated whether autophagy was disrupted in the brains of 12 wpf gba1 mutants compared with wild-type siblings. LC3-II is specifically targeted to autophagosomal membranes and strongly correlates with the number of autophagosomes (24). Brains from gba1−/− fish had more than 2-fold increase in LC3-II levels compared with wild-type siblings (Fig. 8E and F; P < 0.01). Whereas this difference in LC3-II levels clearly demonstrates that autophagosome number is altered in gba1−/− brains compared with those of wild-types, it is unclear whether autophagosome formation is increased or whether autophagosome degradation is defective, because both of these scenarios would lead to an increase in LC3-II levels.
Discussion
Modern gene editing techniques such as the TALEN strategy have transformed zebrafish research (25). We have used the TALEN approach to generate a gba1 mutant zebrafish line which faithfully resembles key pathological and biochemical features of human GCase deficiency. We provide data on gba1−/− mRNA stability, reduced GCase enzymatic activity and other biochemical readouts including extensive mass spectrometry-based analysis of sphingolipids which all support the presence of a marked biological effect caused by the TALEN-induced 23 bp deletion in gba1 on GCase function.
This zebrafish model of GCase deficiency is the first vertebrate model to faithfully replicate key GD pathology in both visceral and neural tissue simultaneously. Conventional KO and conditional KO mice model either neuropathic or non-neuropathic Gaucher disease but not both (10). Our extensive glycolipid mass spectrometry analysis suggests that it is mostly lower MW species which accumulate, with high MW species either unchanged or decreased compared with wild-type. The predominant increase of C18 metabolites is in keeping with similar studies in other model systems (16,26,27). Our observation of a marked increase in the accumulation of distinct glycosphingolipids prior to the onset of marked inflammation and neuronal cell loss in GCase deficient zebrafish larvae is in keeping with similar observations in a mouse model of neuronopathic GD (14). miR-155 is a master regulator of pathways involved in the regulation of immune mechanisms (21) that is expressed in both the innate and the adaptive immune system and predominantly acts via moderate mRNA degradation. Of note, miR-155 upregulation has already been implicated in the pathogenesis of different neurodegenerative disorders. Early miR-155 upregulation contributes to neuroinflammation in an Alzheimer's disease transgenic mouse model as well as in Aβ-activated microglial and astrocyte cultures (28). Expression levels of miR-155 are increased in the spinal cord of both familial and sporadic amyotrophic lateral sclerosis and genetic ablation of miR-155 markedly increased survival in SOD1 mice with restoration of abnormal microglia (29). However, miR-155 had not been implicated in the pathogenesis of GD or PD before now. Our study clearly suggests that activation of immune mechanisms precedes neuronal cell loss rather than being a consequence of it. Future work needs to determine whether miR-155 may also be a promising ‘druggable’ target for neuroprotective therapy in both GD and PD. Of note, an association of GBA mutation status with an increase in the plasma levels of different inflammatory mediators such as interleukin 8 has been reported in PD patients (30).
Both loss of GCase function and toxic gain of function have been proposed to explain the increased risk of PD for GBA1+/− carriers (6,31). There is also strong evidence for an interplay between GCase activity and alpha-synuclein levels (9,16,32). The marked loss of dopaminergic neurons in gba1−/− zebrafish in the absence of alpha-synuclein indicates that alpha-synuclein-independent mechanisms can contribute to the neurodegeneration resulting from GCase deficiency. The extensive accumulation of ubiquitin-positive intra-neuronal inclusions in the brains of juvenile gba1−/− zebrafish further suggests that proteins other than alpha-synuclein accumulate in this model. Obvious candidates are the β- and γ1-synucleins expressed in the zebrafish CNS (22). However, western blot analysis showed that these are both markedly reduced in juvenile gba1−/− brains, possibly as a consequence of extensive synaptic loss accompanying neurodegeneration in this model. Indeed, neuronal ubiquitinopathy preceding an increase in alpha-synuclein levels has been described in a GBA1 knock-in mouse model (33) and it is likely that gba1−/− zebrafish represent an example of non-synuclein proteinopathy and synuclein-independent neurodegeneration occurring in the absence of GCase activity.
Mitochondrial dysfunction with impaired quality control has been reported in a mouse model of GD and iPSC-derived GBA1+/− neurons (16,23). However, we observed reduced complex III and IV activity rather than reduced complex I activity as observed in GBA1−/− mice (23). We hypothesize that this may at least in part be due to alpha-synuclein-mediated mitochondrial toxicity in GBA1−/− mice which typically affects complex I activity (34). The reduced complex IV activity in gba1−/− juvenile zebrafish brains may be due to a direct effect of the markedly elevated glucosylsphingosine, a potent inhibitor of the mitochondrial cytochrome c oxidase on the environment of this membrane-bound enzyme (35). Interestingly, magnetic resonance spectroscopic imaging data in human patients also provide circumstantial evidence of altered membrane phospholipid metabolism in GBA1-associated PD (36). Alternatively, the reduced complex IV activity may at least partially be due to the observed reduction in the Cox4i1 protein level the gba1−/− brains (Fig. 8D). Autophagy plays an essential role in the clearance of aggregate-prone proteins and damaged mitochondria, and dysfunctional autophagy has been implicated in the pathogenesis of PD (37). In mouse models of GCase deficiency, autophagosome formation is decreased and ubiquitylated proteins monomeric and oligomeric forms of alpha-synuclein and ubiquitylated proteins accumulate in the brain (23). In juvenile gba1−/− zebrafish brains, we observed a significant increase in LC3-II levels which may result from either an increase in autophagosome formation or a defect in degradation.
Some of our findings are remarkably similar to observations in a GBA1 nonsense medaka (Oryzias latipes) model of Gaucher disease (27). Future studies need to reveal whether the observed early microglial activation and subsequent neuronal cell loss is linked to the recently reported Wnt signaling abnormalities in GCase1 deficient D. rerio zebrafish with reduced CGase activity caused by transient antisense knockdown of gba1 early in development (38).
Conclusion
Zebrafish are an excellent vertebrate model to study human brain diseases and increasingly used for high-throughput drug screens (39,40). The large sphingolipid accumulation and microglial dysfunction during larval stages shows the potential to use the gba1 mutant zebrafish as a tool for phenotypic drug discovery to identify new disease modifying therapies for neuronopathic GD and to aid in the identification of novel PD toxins that may act synergistically in conjunction with gba1+/−. There is growing evidence of lysosomal impairment in PD in general and decreased activity of GCase in particular, even in the absence of GBA1 mutations (32,41–44). Augmenting CNS GCase activity has been proposed as a promising therapeutic strategy for PD and other GD-related synucleinopathies (45). A further promising aim for zebrafish in vivo high-throughput screens could therefore be to identify compounds which would upregulate neuronal GCase activity.
Materials and Methods
Zebrafish husbandry
All larval and adult zebrafish were housed at the University of Sheffield; experimental procedures being in accordance with UK Home Office Animals (Scientific Procedures) Act 1986 (Project license PPL 70/8437, held by Dr Oliver Bandmann). Adult zebrafish were housed at a density of 40 per tank, whereas on a cycle of 14 h of light, 10 h of dark. Adults and embryos were kept at constant temperature of 28°C.
gba1 stable mutant line
A stable loss of function allele was generated with the TALEN genome editing system targeting an mwoI restriction enzyme site located within exon seven of gba1. A pair of TALENs binding 5′TCTGTACCCTGATTACTT (right TALEN) and 5′ATGCGCTGGGTGGAGTCCA (left TALEN) were chosen by the TALEN targeter (https://boglab.plp.iastate.edu/node/add/talen). TALEN mRNA was generated and injected into one cell stage Zebrafish embryos. F0 mosaic founders were identified and outcrossed to wild-type TL adults. A (heterozygous) allele was identified in the F1 generation containing a 23 bp deletion (gba1+/−) and outcrossed again to TL until the F3 generation was reached. Zebrafish homozygous for this 23 bp deletion (gba1−/−) used for all experiments were generated from an incross of F3 gba1+/−. All zebrafish were genotyped using primers F-5′AAAGCAGCACGATATGTCCA and R-5′ATGTCATGGGCGTAGTCCTC. DNA was amplified and analyzed on a 2% agarose gel.
Gene expression analysis
RNA was extracted from 20 zebrafish embryos/zebrafish caudal hypothalamus by 40% (P < 0.01; Fig. 7B) and the posterior tuberculum by ∼30% (P < 0.01; Fig. 7C). brains (two per replicate) at specific time points using TRIzol® (Life Technologies™). A Verso cDNA synthesis kit (Thermo Scientific) was used to generate cDNA. Quantitative real-time PCR (qPCR)-based quantification of gba1 expression was undertaken using primers F-5′GGCACAGGCTCTATCTGCTC and R-5′TCTAGAAACCTGATATAGT. SYBR (Life Technologies™) green was used for all qPCR experiments, with ef1a as a reference gene (ef1a primers: F-5′TGGTACTTCTCAGGCTGACT and R-5′TGACTCCAACGATCAGCTGT). For microRNA expression analysis, RNA was harvested from embryos and brain tissue as previously described. RNA concentration was accurately quantified using the QuantiFluor™ RNA system (Promega) and the Qubit® fluorometer (Life Technologies). 100 ng of total RNA was reverse-transcribed and subsequently qPCR was performed using Taqman miRNA assays (Applied Biosystems). A Taqman probe (sequence: 5′UUAAUGCUAAUCGUGAUAGGGG) was used to quantify miR-155 levels.
Lysosomal enzyme analysis, assessment of mitochondrial respiratory chain function and mass spectrometry
All lysosomal enzyme assays were performed on homogenates of whole zebrafish brain at 12 wpf with a protein concentration of 1 mg/ml and at 28°C unless otherwise stated. All assays were stopped with 1 M glycine NaOH buffer pH 10.4 and used 1 nm 4-methylumbelliferone (Sigma) as a standard to calculate the final result. Chitotriosidase activity was measured using 4-methylumbelliferyl-β-d-N,N′,N″-triacetyl-chitotriose (Sigma) in McIlvaine citrate–phosphate buffer pH 5.2. β-Hexosaminidase activity was measured using 4-methylumbelliferyl-2-acetamido-2-deoxy-β-d-gluco-pyranoside (Sigma) in McIlvaine citrate–phosphate buffer pH 4.5. Beta-galactosidase activity was measured using 1 mm 4-methylumbelliferyl-d-galactopyranoside dissolved in McIlvaine citrate–phosphate buffer pH 4.1. GCase activity was measured with 5 mm 4-methylumbelliferyl-β-d-glucopyransoside (Sigma) in McIlvaine citrate–phosphate buffer pH 5.4 in the presence and absence of conduritol B epoxide at 37°C.
Mitochondrial complex activities I–IV were assessed in whole brain homogenates at 12 wpf as previously described (46).
Mass spectrometry for the detection of sphingolipid metabolites was undertaken at 5 dpf and 12 wpf as previously described (47). Larvae were genotyped as previously described (48). Genotype larvae were then frozen in liquid nitrogen in groups of 20 per genotype (wild-type, gba1+/− and gba1−/−) and stored at −80°C prior to mass spectrometric analysis. Mass spectrometry was then also undertaken in brains of juvenile zebrafish (12 wild-type, 10 gba1+/− and 10 gba1−/−) at 12 wpf.
Assessment of dopaminergic nervous system
Dopaminergic neurons were first counted at 5 dpf using whole mount in situ hybridization (WISH) staining with a probe for tyrosine hydroxylase (TH) (n = 10 embryos per genotype and biological replicate). Dopaminergic neurons were counted by eye using an axioplan compound microscope (Zeiss) at 20× magnification as previously described (46). The counter was blinded to the genotype and condition. The dopaminergic neuron count was assessed by counting the distinct neuronal subgroups one, two, four and five in the diencephalon, defined according to the Rink and Wullimann classification (49,50). The mean neuron count of each control group was normalized to 100% and all other group counts expressed as a percentage of the control group. Juvenile zebrafish were culled and brains fixed in paraformaldehyde (PFA) to enable dopaminergic neuronal cell count at 12 wpf in wild-type, gba1+/− and gba1−/− zebrafish. Dopaminergic neurons were stained using a TH1 antibody (Mouse monoclonal anti-TH, DiaSorin Inc.) and then counted in the posterior tuberculum and caudal hypothalamus as previously described (51).
Movement analysis
Locomotion was quantified using Viewpoint analysis software version 3, 22, 3, 9 (Viewpoint). Fish were filmed individually from the side, for 10 min following 10 min acclimation time. Low-speed movements were defined as <5 cm/s. Medium-speed movements were defined as 5< X <7 cm/s. High-speed movements were defined as movements >7 cm/s.
Microglial activation
gba1+/− were crossed to Tg(mpeg1:GFP-CAAX) (mpeg1,sh425), similar to a previously published protocol (52). Details on the transgenesis methods are available from the authors. All subsequent embryo work was generated from an incross of gba1+/− and mpeg1 and imaged at 4 dpf. High-resolution imaging was performed using an inverted UltraViewVoX spinning-disk confocal microscope (PerkinElmer Life and Analytical Sciences). Imaging was performed to a depth of ∼150 µm from the dorsal surface of the brain using 2 µm z-sections. Volumetric and shape factor analyses were performed using Volocity 6.3 (PerkinElmer Life and Analytical Sciences) software, using intensity of fluorescence to identify individual cells. Measurements of vacuole diameter were performed manually using the line tool. Data for the assessment of microglial shape and vacuole count were pooled form three independent experiments including a total of 177 wild-type, 94 gba1+/− and 82 gba1−/− microglial cells from 15 wild-type, 9 gba1+/− and 7 gba1−/− larvae. All measurements were performed blind to the gba1 genotype. Following microscopy and image analysis, embryos were genotyped for the gba1 mutation (as described earlier).
Histology
For H&E and PAS staining, zebrafish were fixed in Bouins fixative for 2 weeks and embedded in paraffin. Ubiquitylation was assessed in zebrafish fixed in 10% buffered formalin solution for 1–2 weeks with subsequent decalcification for 7 days in ethylenediaminetetraacetic acid. Coronal or sagittal sections were made of ∼4 μm thickness. Each zebrafish was sectioned completely and every 10th and 11th slide was used for subsequently staining with either H&E or PAS. Ubiquitin immunohistochemistry (IHC) was performed with antigen retrieval by pressure cooker at pH 6, using a polyclonal anti-ubiquitin antibody (Dako Z 0458) at a 1:1000 dilution, standard ABC methods and diaminobenzidine (DAB) as chromogen. Prepared microscope slides were viewed by a board-certified pathologist (A.M.), using conventional bright-field microscopy.
Sample preparation for IHC and confocal microscopy to investigate microglial activation in juvenile zebrafish brains was carried out as reported previously (53). Zebrafish were perfused and brains post-fixed in 4% PFA, followed by cryoprotection in PBS-sucrose. Fourteen micrometer thick cryosections were mounted on glass slides, treated with PBS-T (0.3% Triton-X) for 1 h, blocked with 10% goat serum in PBS for 2 h and then incubated overnight at 4°C with primary antibodies diluted 1:20 (7.4.C4, purified from hybridoma clone; #92092321, HPA Culture Collections, UK), 1:500 (P0) in PBS with 1% goat serum (54). Primary antibodies were detected using Alexa-488 (anti-mouse), and Alexa-555 (anti-rabbit) conjugated secondary antibodies (Life Technologies, Grand Island, NY) diluted 1:1000 in carrier buffer and sections counter labeled with 4′, 6-diamidino-2-phenylindole (DAPI). Images were acquired using an Olympus Fluoview confocal microscope and multi-field collages made with Adobe Photoshop.
Western blotting
Mitochondrial primary antibodies: TOMM20 (Santa Cruz), TIMM9 (Abcam), NDUFA9 (Abcam), COX4i1 (Abcam), ATP5A (Abcam), β-ACTIN (Sigma-Aldrich). Horseradish peroxidase (HRP)-linked secondary antibodies were used (Sigma-Aldrich). LC3 primary antibodies: Rabbit anti-LC3 (Novus Biologicals; NB100–2220) used at 1:1000 dilution; mouse anti-actin (Sigma A5316) used at 1:500 dilution. Secondary antibodies: Polyclonal goat anti-rabbit immunoglobulins/HRP (Dako P0448) used at 1:5000; polyclonal goat anti-mouse immunoglobulins/HRP (P0447) used at 1:5000. Beta- and Gamma-synuclein primary antibodies: mAb α/β-Synuclein (Syn205, Cell Signaling; 1:1000) or pAb γ1-Synuclein (1:1000). γ1-Synuclein polyclonal antibody was raised to the peptide DFSHGGMEGGEGGEGY by immunization of rabbits (New England Peptide) and affinity purified as previously described (54). IRDye-800 and IRDye-680 (LI-COR, Lincoln, NE) conjugated secondary antibodies (1:10 000) enabled the blot to be imaged using an Odyssey Infrared Imager (catalog no. 9120; LI-COR) with a wide linear range.
Statistical tests and analysis
Graphpad prism V.5 software (Graphpad) was used for statistical analysis and all errors bars shown denote the mean ± SE of the mean. All experiments were performed in biological triplicate unless otherwise. All data were analyzed with either T test, one-way ANOVA or two-way ANOVA.
Supplementary Material
Funding
This work was supported by BBSRC/Lilly (PhD CASE studentship for M.K.); Parkinson's UK (G-1404 for O.B.); The Academy of Finland and Sigrid Juselius Foundation (for P.P.); FishMed/EU Seventh Framework Programme (no. 316125 for A.S.); Wellcome Trust/MRC Joint call in Neurodegeneration award to the UK Parkinson’s Disease Consortium (WT089698 for M.G. and A.H.V.S.); National Institute for Environmental Health Sciences (ES022644 for E.A.B.); Australian NHMRC C.J. Martin fellowship to F.E. (GNT1054664), MRC Programme Grant (SAR; reference number MR/M004864/1), MRC Center grant (G0700091), University of Sheffield Vice-Chancellor's Fellowship (PME). Microscopy studies were supported by a Wellcome Trust grant to the MBB/BMS Light Microscopy Facility (GR077544AIA). Funding to pay the Open Access publication charges for this article was provided by Parkinson’s UK.
Supplementary Material
Acknowledgements
We thank the aquarium staff at the Bateson Centre and the histopathology technicians at the Sheffield Institute for Translational Neuroscience (SITraN), both University of Sheffield, UK as well as Michal Bazala (International Institute of Molecular and Cell Biology, Warsaw, Poland) for expert assistance. The photomicrograph in Figure 7J is from a microscope slide supplied by the Parkinson's UK Brain Bank, funded by Parkinson's UK, a charity registered in England and Wales (258197) and in Scotland (SC037554).
Conflict of Interest statement. None declared.
References
- 1.Grabowski G.A. (2012) Gaucher disease and other storage disorders. Hematology Am. Soc. Hematol. Educ. Program, 2012, 13–18. [DOI] [PubMed] [Google Scholar]
- 2.Baris H.N., Cohen I.J., Mistry P.K. (2014) Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev., 12(Suppl. 1), 72–81. [PMC free article] [PubMed] [Google Scholar]
- 3.Zimran A., Elstein D. (2014) Management of Gaucher disease: enzyme replacement therapy. Pediatr. Endocrinol. Rev., 12(Suppl. 1), 82–87. [PubMed] [Google Scholar]
- 4.Neumann J., Bras J., Deas E., O'Sullivan S.S., Parkkinen L., Lachmann R.H., Li A., Holton J., Guerreiro R., Paudel R. et al. (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain, 132, 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sidransky E., Nalls M.A., Aasly J.O., Aharon-Peretz J., Annesi G., Barbosa E.R., Bar-Shira A., Berg D., Bras J., Brice A. et al. (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. New Engl. J. Med., 361, 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siebert M., Sidransky E., Westbroek W. (2014) Glucocerebrosidase is shaking up the synucleinopathies. Brain, 137, 1304–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alcalay R.N., Caccappolo E., Mejia-Santana H., Tang M., Rosado L., Orbe Reilly M., Ruiz D., Ross B., Verbitsky M., Kisselev S. et al. (2012) Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology, 78, 1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark L.N., Ross B.M., Wang Y., Mejia-Santana H., Harris J., Louis E.D., Cote L.J., Andrews H., Fahn S., Waters C. et al. (2007) Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology, 69, 1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazzulli J.R., Xu Y.H., Sun Y., Knight A.L., McLean P.J., Caldwell G.A., Sidransky E., Grabowski G.A., Krainc D. (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell, 146, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farfel-Becker T., Vitner E.B., Futerman A.H. (2011) Animal models for Gaucher disease research. Dis. Model. Mech., 4, 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandmann O., Burton E.A. (2010) Genetic zebrafish models of neurodegenerative diseases. Neurobiol. Dis., 40, 58–65. [DOI] [PubMed] [Google Scholar]
- 12.Flinn L.J., Keatinge M., Bretaud S., Mortiboys H., Matsui H., De Felice E., Woodroof H.I., Brown L., McTighe A., Soellner R. et al. (2013) TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann. Neurol., 74, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGown A., McDearmid J.R., Panagiotaki N., Tong H., Al Mashhadi S., Redhead N., Lyon A.N., Beattie C.E., Shaw P.J., Ramesh T.M. (2013) Early interneuron dysfunction in ALS: insights from a mutant sod1 zebrafish model. Ann. Neurol., 73, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farfel-Becker T., Vitner E.B., Kelly S.L., Bame J.R., Duan J., Shinder V., Merrill A.H. Jr, Dobrenis K., Futerman A.H. (2014) Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet., 23, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaye E.M., Ullman M.D., Wilson E.R., Barranger J.A. (1986) Type 2 and type 3 Gaucher disease: a morphological and biochemical study. Ann. Neurol., 20, 223–230. [DOI] [PubMed] [Google Scholar]
- 16.Schondorf D.C., Aureli M., McAllister F.E., Hindley C.J., Mayer F., Schmid B., Sardi S.P., Valsecchi M., Hoffmann S., Schwarz L.K. et al. (2014) iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun., 5, 4028. [DOI] [PubMed] [Google Scholar]
- 17.Mistry P.K., Liu J., Yang M., Nottoli T., McGrath J., Jain D., Zhang K., Keutzer J., Chuang W.L., Mehal W.Z. et al. (2010) Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl Acad. Sci. U.S.A., 107, 19473–19478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirsch E.C., Vyas S., Hunot S. (2012) Neuroinflammation in Parkinson's disease. Parkinsonism Relat. Disord., 18(Suppl. 1), S210–4212. [DOI] [PubMed] [Google Scholar]
- 19.Holmans P., Moskvina V., Jones L., Sharma M., Vedernikov A., Buchel F., Saad M., Bras J.M., Bettella F., Nicolaou N. et al. (2013) A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson's disease. Hum. Mol. Genet., 22, 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vitner E.B., Farfel-Becker T., Eilam R., Biton I., Futerman A.H. (2012) Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher's disease. Brain, 135, 1724–1735. [DOI] [PubMed] [Google Scholar]
- 21.Vigorito E., Kohlhaas S., Lu D., Leyland R. (2013) miR-155: an ancient regulator of the immune system. Immunol. Rev., 253, 146–157. [DOI] [PubMed] [Google Scholar]
- 22.Milanese C., Sager J.J., Bai Q., Farrell T.C., Cannon J.R., Greenamyre J.T., Burton E.A. (2012) Hypokinesia and reduced dopamine levels in zebrafish lacking beta- and gamma1-synucleins. J. Biol. Chem., 287, 2971–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osellame L.D., Rahim A.A., Hargreaves I.P., Gegg M.E., Richard-Londt A., Brandner S., Waddington S.N., Schapira A.H., Duchen M.R. (2013) Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson's disease. Cell Metab., 17, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klionsky D.J., Abeliovich H., Agostinis P., Agrawal D.K., Aliev G., Askew D.S., Baba M., Baehrecke E.H., Bahr B.A., Ballabio A. et al. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy, 4, 151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auer T.O., Del Bene F. (2014) CRISPR/Cas9 and TALEN-mediated knock-in approaches in zebrafish. Methods, 69, 142–150. [DOI] [PubMed] [Google Scholar]
- 26.Enquist I.B., Lo Bianco C., Ooka A., Nilsson E., Mansson J.E., Ehinger M., Richter J., Brady R.O., Kirik D., Karlsson S. (2007) Murine models of acute neuronopathic Gaucher disease. Proc. Natl Acad. Sci. U.S.A., 104, 17483–17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uemura N., Koike M., Ansai S., Kinoshita M., Ishikawa-Fujiwara T., Matsui H., Naruse K., Sakamoto N., Uchiyama Y., Todo T. et al. (2015) Viable neuronopathic Gaucher Disease Model in Medaka (Oryzias latipes) displays axonal accumulation of alpha-synuclein. PLoS Genet., 11, e1005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guedes J.R., Custodia C.M., Silva R.J., de Almeida L.P., Pedroso de Lima M.C., Cardoso A.L. (2014) Early miR-155 upregulation contributes to neuroinflammation in Alzheimer's disease triple transgenic mouse model. Hum. Mol. Genet., 23, 6286–6301. [DOI] [PubMed] [Google Scholar]
- 29.Butovsky O., Jedrychowski M.P., Cialic R., Krasemann S., Murugaiyan G., Fanek Z., Greco D.J., Wu P.M., Doykan C.E., Kiner O. et al. (2015) Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol., 77, 75–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chahine L.M., Qiang J., Ashbridge E., Minger J., Yearout D., Horn S., Colcher A., Hurtig H.I., Lee V.M., Van Deerlin V.M. et al. (2013) Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol, 70, 852–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schapira A.H. (2015) Glucocerebrosidase and Parkinson disease: recent advances. Mol. Cell. Neurosci., 66, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy K.E., Gysbers A.M., Abbott S.K., Tayebi N., Kim W.S., Sidransky E., Cooper A., Garner B., Halliday G.M. (2014) Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson's disease. Brain, 137, 834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cullen V., Sardi S.P., Ng J., Xu Y.H., Sun Y., Tomlinson J.J., Kolodziej P., Kahn I., Saftig P., Woulfe J. et al. (2011) Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol., 69, 940–953. [DOI] [PubMed] [Google Scholar]
- 34.Devi L., Raghavendran V., Prabhu B.M., Avadhani N.G., Anandatheerthavarada H.K. (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem., 283, 9089–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Igisu H., Hamasaki N., Ito A., Ou W. (1988) Inhibition of cytochrome c oxidase and hemolysis caused by lysosphingolipids. Lipids, 23, 345–348. [DOI] [PubMed] [Google Scholar]
- 36.Brockmann K., Hilker R., Pilatus U., Baudrexel S., Srulijes K., Magerkurth J., Hauser A.K., Schulte C., Csoti I., Merten C.D. et al. (2012). Neurodegeneration, altered membrane metabolism, and lack of energy failure. Neurology, 79, 213–220. [DOI] [PubMed] [Google Scholar]
- 37.Frake R.A., Ricketts T., Menzies F.M., Rubinsztein D.C. (2015) Autophagy and neurodegeneration. J. Clin. Invest., 125, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zancan I., Bellesso S., Costa R., Salvalaio M., Stroppiano M., Hammond C., Argenton F., Filocamo M., Moro E. (2015) Glucocerebrosidase deficiency in zebrafish affects primary bone ossification through increased oxidative stress and reduced Wnt/beta-catenin signaling. Hum. Mol. Genet., 24, 1280–1294. [DOI] [PubMed] [Google Scholar]
- 39.Ablain J., Zon L.I. (2013) Of fish and men: using zebrafish to fight human diseases. Trends Cell. Biol., 23, 584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wyatt C., Bartoszek E.M., Yaksi E. (2015) Methods for studying the zebrafish brain: past, present and future. Eur. J. Neurosci., 42, 1746–1763. [DOI] [PubMed] [Google Scholar]
- 41.Dehay B., Martinez-Vicente M., Caldwell G.A., Caldwell K.A., Yue Z., Cookson M.R., Klein C., Vila M., Bezard E. (2013) Lysosomal impairment in Parkinson's disease. Movement Disord., 28, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy K.E., Halliday G.M. (2014) Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy, 10, 1350–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parnetti L., Chiasserini D., Persichetti E., Eusebi P., Varghese S., Qureshi M.M., Dardis A., Deganuto M., De Carlo C., Castrioto A. et al. (2014) Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson's disease. Movement Disord., 29, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gegg M.E., Burke D., Heales S.J., Cooper J.M., Hardy J., Wood N.W., Schapira A.H. (2012) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol., 72, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sardi S.P., Clarke J., Viel C., Chan M., Tamsett T.J., Treleaven C.M., Bu J., Sweet L., Passini M.A., Dodge J.C. et al. (2013) Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl Acad. Sci. U.S.A., 110, 3537–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flinn L., Mortiboys H., Volkmann K., Koster R.W., Ingham P.W., Bandmann O. (2009) Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain, 132, 1613–1623. [DOI] [PubMed] [Google Scholar]
- 47.Bui H.H., Leohr J.K., Kuo M.S. (2012) Analysis of sphingolipids in extracted human plasma using liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Biochem., 423, 187–194. [DOI] [PubMed] [Google Scholar]
- 48.Wilkinson R.N., Elworthy S., Ingham P.W., van Eeden F.J. (2013) A method for high-throughput PCR-based genotyping of larval zebrafish tail biopsies. BioTechniques, 55, 314–316. [DOI] [PubMed] [Google Scholar]
- 49.Rink E., Wullimann M.F. (2001) The teleostean (zebrafish) dopaminergic system ascending to the subpallium (striatum) is located in the basal diencephalon (posterior tuberculum). Brain Res., 889, 316–330. [DOI] [PubMed] [Google Scholar]
- 50.Rink E., Wullimann M.F. (2002) Connections of the ventral telencephalon and tyrosine hydroxylase distribution in the zebrafish brain (Danio rerio) lead to identification of an ascending dopaminergic system in a teleost. Brain Res. Bull., 57, 385–387. [DOI] [PubMed] [Google Scholar]
- 51.Sallinen V., Torkko V., Sundvik M., Reenila I., Khrustalyov D., Kaslin J., Panula P. (2009) MPTP and MPP+ target specific aminergic cell populations in larval zebrafish. J. Neurochem., 108, 719–731. [DOI] [PubMed] [Google Scholar]
- 52.Ellett F., Pase L., Hayman J.W., Andrianopoulos A., Lieschke G.J. (2011) mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood, 117, e49–e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bai Q., Parris R.S., Burton E.A. (2014) Different mechanisms regulate expression of zebrafish myelin protein zero (P0) in myelinating oligodendrocytes and its induction following axonal injury. J. Biol. Chem., 289, 24114–24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bai Q., Sun M., Stolz D.B., Burton E.A. (2011) Major isoform of zebrafish P0 is a 23.5 kDa myelin glycoprotein expressed in selected white matter tracts of the central nervous system. J. Comp. Neurol., 519, 1580–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.