

Abstract
Despite the many advances in our understanding of the genetic basis of Mendelian forms of Parkinson's disease (PD), a large number of early-onset cases still remain to be explained. Many of these cases, present with a form of disease that is identical to that underlined by genetic causes, but do not have mutations in any of the currently known disease-causing genes. Here, we hypothesized that de novo mutations may account for a proportion of these early-onset, sporadic cases. We performed exome sequencing in full parent–child trios where the proband presents with typical PD to unequivocally identify de novo mutations. This approach allows us to test all genes in the genome in an unbiased manner. We have identified and confirmed 20 coding de novo mutations in 21 trios. We have used publicly available population genetic data to compare variant frequencies and our independent in-house dataset of exome sequencing in PD (with over 1200 cases) to identify additional variants in the same genes. Of the genes identified to carry de novo mutations, PTEN, VAPB and ASNA1 are supported by various sources of data to be involved in PD. We show that these genes are reported to be within a protein–protein interaction network with PD genes and that they contain additional rare, case-specific, mutations in our independent cohort of PD cases. Our results support the involvement of these three genes in PD and suggest that testing for de novo mutations in sporadic disease may aid in the identification of novel disease-causing genes.
Introduction
Although the detailed aetiology of Parkinson's disease (PD) remains largely unknown, data suggest the disease may be triggered through different mechanisms: protein inclusions accumulation, diminished mitochondrial activity, proteasomal/lysosomal dysfunction and impaired dopamine production (1). An increasing number of publications show a strong genetic component for PD. Studies in familial forms of PD have allowed for the identification of disease-causing mutations in several genes, causing either dominant or recessive forms of disease inheritance. Genome-wide association studies (GWAS) have also significantly contributed to a more comprehensive knowledge of the risk loci involved in PD (2). Despite these results, there is still a large number of sporadic, early-onset cases that carry no mutation in the known PD genes. These individuals have, in many cases, a form of disease that is indistinguishable from genetically linked disease.
De novo mutations have been commonly studied in neurodevelopmental disorders such as autism (3–5) and schizophrenia (6,7) with only a few examples in neurodegenerative diseases. De novo mutations in the ATP1A3 gene have been found as the cause for rapid-onset dystonia parkinsonism (8), while Rosewich et al. (9) reported de novo mutations in this gene in alternating hemiplegia of childhood. De Carvalho Aguiar and colleagues (10) showed one de novo mutation in TOR1A in a primary torsion dystonia case. Two cases of static encephalopathy of childhood with neurodegeneration in adulthood showed mutations in WDR45 (11). In all these cases, patients start to show symptoms during childhood or adolescence.
Recently studies have suggested the involvement of de novo mutations in other neurodegenerative diseases, such as in early-onset Alzheimer's disease (AD) (12,13) and amyotrophic lateral sclerosis (ALS) (14–17).
There is also preliminary evidence supporting the role of de novo mutations in PD: Puschmann and colleagues (18) reported on a presumably de novo A53T mutation in SNCA and non-mendelian multiplications of the gene (19,20) have also been shown. More recently, Hansen and collaborators (21) described two mutations in the SLC6A3 gene (encoding the dopamine transporter, DAT1), one of which was presumed to be de novo. It should be noted however that in both the SNCA A53T and the SLC6A3 cases, the authors were not able to positively confirm the presence of de novo mutations, given the absence of parental DNA samples.
Here, we hypothesize that a subset of early-onset PD cases, with no mutations in any of the known PD-causing genes, may be due to the occurrence of de novo mutations. We used whole-exome sequencing in full parent–child PD trios to unequivocally identify these events.
Results
From the systematic analysis of the whole-exome sequencing data, 24 genes showed de novo mutations in the trios. We validated 20 of the 24 variants with Sanger sequencing methods (Table 1) comparing sequences from both parents with the proband (examples for the three genes of interest in Fig. 1). The four variants not validated were false positives from the exome sequencing. We have identified, on average, one de novo coding event per trio, which is in line with what would be expected for the human population (22) (Table 2). Only four of the variants identified have been previously described in population databases: the variants in EPPK1, COL12A1, PEPD and SLC52A1 have low frequencies ranging from 0.00003 to 0.003 in Exome Aggregation Consortium (ExAC). All other variants are presumably novel as they are absent from tested databases. PD cases did not show any potential pathogenic mutations in the known PD genes—all variants identified in those genes were either frequent in the general population, present in the unaffected parents, located in not conserved residues or predicted to be benign by PolyPhen and SIFT (23,24).
Table 1.
List of de novo variants identified in the PD trios
| Trio | Gene | Chromosome | Position | Transcript | Codon change | Amino acid change | Reference SNP | SIFT prediction | Polyphen prediction | CADD phred score | ESP6500 | 1000 Genomes | ExAC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | COL12A1 | 6 | 75 887 555 | NM_004370 | cGa/cAa | R754Q | rs377480187 | None | Probably_Damaging | 32 | 0.000084 | 0 | 0.00003311 |
| RUNDC3A | 17 | 42 390 571 | NM_001144825 | tGt/tTt | C108F | Deleterious | Probably_Damaging | 22.2 | 0 | 0 | 0 | ||
| VAPB | 20 | 56 9932 80 | NM_004738 | gaTGTt/gat | ΔV25 | None | None | None | 0 | 0 | 0 | ||
| 2 | ANKRD13A | 12 | 1 104 655 60 | NM_033121 | Ggt/Tgt | G312C | Deleterious | Possibly_Damaging | 22.8 | 0 | 0 | 0 | |
| MKS1 | 17 | 56 293 486 | NM_001165927 | aCc/aTc | T117I | Deleterious | Probably_Damaging | 33 | 0 | 0 | 0 | ||
| 3 | PAPD4 | 5 | 78 938 703 | NM_173797 | Tta/Gta | L241V | Tolerated | Benign | 15.53 | 0 | 0 | 0 | |
| VPS53 | 17 | 455 114 | NM_001128159 | Aag/Gag | K622E | Deleterious | Probably_Damaging | 29.7 | 0 | 0 | 0 | ||
| 4 | PMEL | 12 | 56 349 087 | NM_001200054 | atG/atA | M614I | Tolerated | Benign | 10.33 | 0 | 0 | 0 | |
| SLC5A9 | 1 | 48 695 007 | NM_001135181 | cCt/cTt | P152L | Deleterious | Probably_Damaging | 33 | 0 | 0 | 0 | ||
| 5 | ASNA1 | 19 | 12 858 398 | NM_004317 | Ctg/Gtg | L303V | Deleterious | Benign | None | 0 | 0 | 0 | |
| 6 | EPPK1 | 8 | 14 494 1903 | NM_031308 | aCg/aTg | T1840M | rs79961029 | Tolerated | Benign | 9.541 | 0.008705 | 0.00838658 | 0.003023 |
| 7 | FBXL17 | 5 | 10 770 3586 | NM_001163315 | gaC/gaA | D354E | Tolerated | Possibly_Damaging | 17.45 | 0 | 0 | 0 | |
| 8 | KCNV2 | 9 | 27 189 66 | NM_133497 | caG/caT | Q409H | Tolerated | Benign | 13.86 | 0 | 0 | 0 | |
| 9 | LCT | 2 | 13 654 8243 | NM_002299 | Aat/Cat | N1774H | Deleterious | Probably_Damaging | 21.9 | 0 | 0 | 0 | |
| 10 | MGA | 15 | 41 988 608 | NM_001164273 | cCa/cAa | P467Q | Deleterious | Probably_Damaging | 16.87 | 0 | 0 | 0 | |
| 11 | PEPD | 19 | 33 878 830 | NM_000285 | cGc/cAc | R437H | rs373297406 | Tolerated | Benign | 13.91 | 0.000329 | 0.000199681 | 0.00006709 |
| 12 | PML | 15 | 74 327 974 | NM_033250 | cgG/cgC | R676R | None | None | None | 0 | 0 | 0 | |
| 13 | PSD4 | 2 | 11 395 0082 | NM_012455 | aAc/aGc | N585S | Tolerated | Benign | 9.504 | 0 | 0 | 0 | |
| 14 | PTEN | 10 | 89 711 992 | NM_000314 | Cca/Tca | P204S | Tolerated | Possibly_Damaging | 32 | 0 | 0 | 0 | |
| 15 | SLC52A1 | 17 | 49 363 59 | NM_001104577 | Gcc/Acc | A414T | rs142353672 | Tolerated | Benign | 11.56 | 0.007 | 0 | 0.00005776 |
Figure 1.
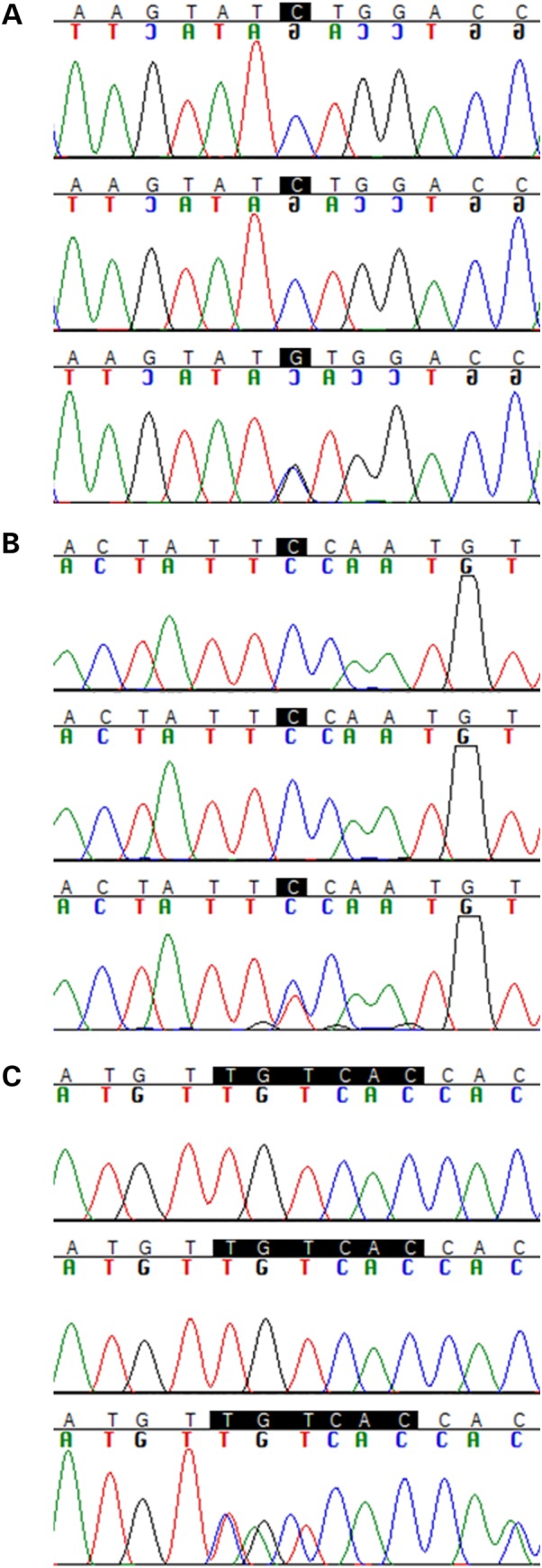
Sanger sequencing chromatograms for (A) ASNA1 (p.L303V); (B) PTEN (p.P204S); and (C) VAPB (p.ΔV25). Top: father; middle: mother; bottom: proband.
Table 2.
Sequencing metrics
| Proband | Total on target (bp) | Mean on target coverage | Fraction targeted bases >2x | Total de novo coding mutations |
|---|---|---|---|---|
| 1 | 1 445 598 290 | 27.12 | 0.97 | 3 |
| 2 | 1 421 276 651 | 26.52 | 0.97 | 2 |
| 3 | 1 146 375 416 | 21.47 | 0.97 | 2 |
| 4 | 7 152 790 854 | 135.14 | 0.94 | 2 |
| 5 | 8 974 607 696 | 167.43 | 0.94 | 1 |
| 6 | 7 933 070 487 | 147.95 | 0.94 | 1 |
| 7 | 1 376 003 145 | 26.44 | 0.97 | 1 |
| 8 | 1 914 732 654 | 36.21 | 0.98 | 1 |
| 9 | 4 286 967 024 | 76.61 | 0.95 | 1 |
| 10 | 6 133 197 922 | 112.73 | 0.97 | 1 |
| 11 | 3 381 480 354 | 61.73 | 0.94 | 1 |
| 12 | 6 529 240 671 | 121.95 | 0.94 | 1 |
| 13 | 4 485 511 060 | 87.10 | 0.99 | 1 |
| 14 | 1 843 089 171 | 33.41 | 0.98 | 1 |
| 15 | 6 364 816 268 | 119.29 | 0.94 | 1 |
| 16 | 3 347 839 519 | 59.15 | 0.95 | 0 |
| 17 | 1 171 269 433 | 22.19 | 0.96 | 0 |
| 18 | 2 110 891 218 | 39.22 | 0.98 | 0 |
| 19 | 4 145 185 493 | 80.55 | 0.99 | 0 |
| 20 | 3 649 693 000 | 70.18 | 0.99 | 0 |
| 21 | 1 461 050 157 | 28.87 | 0.93 | 0 |
| Average | 3 822 604 118 | 71.49 | 0.96 | 0.95 |
From the protein network analysis of the 20 validated de novo mutation carrying genes, PTEN, VAPB, ASNA1, PML and VPS53 showed interactions with the known PD genes (Fig. 2). However, VPS53 and PML did not show high interaction scores with disease-causing genes. Robust functional interactions with known PD genes were identified for the three remaining candidates. (Table 3 and Fig. 2).
Figure 2.

Action view from the STRING network analysis. Colours correspond to interactions according to the legend (top left); Evidence view (grey) corresponds mainly to interactions obtained from text-mining sources. The remaining interaction colours are based on published experimental results, with Green representing activation, Red inhibition, etc. If the directionality of the effect is known, this is indicated by the symbol at the end of the edge next to the protein that is acted upon. Down-regulation is a red bar and up-regulation is a green arrow. *denotes genes identified to carry de novo mutations.
Table 3.
Scores from the STRING protein–protein analysis restricted to Homo sapiens
| Gene | Interaction | Total score | Co-expression | Experimental | Knowledge | Text-mining |
|---|---|---|---|---|---|---|
| PTEN | PARK7 | 0.990 | 0 | 0.844 | 0 | 0.940 |
| SYNJ1 | 0.928 | 0 | 0.116 | 0.900 | 0.242 | |
| PINK1 | 0.699 | 0 | 0.172 | 0 | 0.643 | |
| LRRK2 | 0.667 | 0 | 0.295 | 0 | 0.537 | |
| PARK2 | 0.608 | 0 | 0 | 0 | 0.599 | |
| SNCA | 0.472 | 0 | 0 | 0 | 0.472 | |
| VAPB | STX1B | 0.919 | 0 | 0.730 | 0 | 0.697 |
| SYNJ1 | 0.510 | 0 | 0.363 | 0 | 0.245 | |
| VPS13C | 0.449 | 0 | 0 | 0 | 0.436 | |
| PML | HLA-DRB5 | 0.900 | 0 | 0 | 0.900 | 0 |
| PARK2 | 0.507 | 0 | 0 | 0 | 0.507 | |
| ASNA1 | VAPB | 0.887 | 0 | 0.413 | 0 | 0.812 |
| VPS53 | VPS35 | 0.502 | 0.111 | 0 | 0 | 0.451 |
| LRRK2 | 0.413 | 0 | 0.102 | 0 | 0.374 |
The total scores are computed by combining the probabilities from the different evidence channels (co-expression, experimental, knowledge, text-mining), correcting for the probability of randomly observing an interaction, as described in STRING′s documentation.
A total of 10 interactions were observed between proteins corresponding to genes containing de novo variants and proteins corresponding to known PD genes. This is significantly higher than expected by chance (P = 0.009), however, after correcting for the total numbers of interactions observed for each protein, the result does not retain statistical significance (P = 0.162). Five out of the ten interactions involved PTEN. Nevertheless, this number was not significantly higher than expected by chance (P = 0.226), after correcting for the large total number of interactions involving PTEN.
Seventy-four interactions were observed between the proteins corresponding to the 33 known PD genes. This is significantly higher than expected by chance (P < 0.000001). Several individual proteins showed a significant excess of interactions, even after correcting for the multiple testing of proteins. The interaction network between the proteins corresponding to PD genes is shown in Supplementary Material (see Supplementary Material, Table S1 and Fig. S1).
Interestingly, when taking into account the allele frequency from the ExAC database for all the genes studied here, we observed the lowest total number of variants normalized by protein size to be present in the three genes with higher interaction with known PD genes—PTEN, VAPB and ASNA1 (see Supplementary Material, Fig. S2), suggesting these may be under stronger selection pressure.
The International Parkinson's Disease Genomics Consortium (IPDGC) dataset shows 2 missense and 1 frameshift mutations in PTEN (all in PD cases, no rare variants were found in controls); 4 missense, 1 frameshift and 1 stop-gained mutations in ASNA1; and 5 missense and 1 deletion mutations in VAPB. With the exception of three common variants in VAPB, all others are exceedingly rare in the population (Table 4) and heterozygous in the IPDGC dataset.
Table 4.
List of IPDGC variants in the top candidate genes
| Gene | Chromosome | Position | rs_ids | Codon change | Amino acid change | MAF cases | MAF controls | MAF ESP6500 | MAF ExAC | ExAC alleles |
|---|---|---|---|---|---|---|---|---|---|---|
| ASNA1 | 19 | 12 848 341 | Frameshift (insG) | −8 | 0.039 | 0 | 0 | 0 | 0 | |
| 12 848 341 | Tgg/Ggg | W8G | 0.039 | 0 | 0 | 0 | 0 | |||
| 12 848 342 | rs138730527 | tGg/tAg | W8* (stop gained) | 0 | 0.099 | 0.0154 | 0.00001726 | 1 | ||
| 12 849 344 | Ctc/Gtc | L61V | 0.039 | 0 | 0 | 0 | 0 | |||
| 12 849 356 | Cgt/Tgt | R65C | 0.039 | 0 | 0 | 0 | 0 | |||
| 12 858 023 | Ctg/Gtg | L208V | 0.039 | 0 | 0 | 0 | 0 | |||
| 12 858 901 | rs200489378 | Ccc/Tcc | P344S | 0.039 | 0 | 0.0077 | 0.00007819 | 9 | ||
| PTEN | 10 | 89 685 310 | Aat/Gat | N69D | 0.04 | 0 | 0 | 0 | 0 | |
| 89 692 866 | aAt/aGt | N117S | 0.039 | 0 | 0 | 0.000008237 | 1 | |||
| 89 720 798 | Frameshift (delTACT) | −317 | 0.039 | 0 | 0 | 0 | 0 | |||
| VAPB | 20 | 56 964 571 | cGa/cTa | R19L | 0.041 | 0 | 0 | 0 | 0 | |
| 57 014 058 | Ttg/Atg | L125M | 0.039 | 0 | 0 | 0.00001661 | 2 | |||
| 57 014 075 | rs146459055 | gaT/gaG | D130E | 0.079 | 0 | 0.0692 | 0.001359 | 163 | ||
| 57 016 039 | agttct/agt (del) | SS160S | 0.313 | 0.298 | 0 | 0.001695 | 203 | |||
| 57 016 076 | rs143144050 | atG/atA | M170I | 0.313 | 0.099 | 0.1153 | 0.001373 | 165 | ||
| 57 016 117 | rs145483046 | cGg/cAg | R184Q | 0.039 | 0 | 0.0308 | 0.00007541 | 9 |
Synonymous mutations were not included.
MAF, minor allele frequency.
The frameshift mutation in PTEN (NM_000314:c.955_958delACTT:p.Thr319*) was identified in a case from the IPDGC dataset with positive, albeit limited, family history for PD: the proband's father developed the disease in his 50s. Samples from both parents and unaffected sibling were available. After testing for segregation in the limited family, we observed that the mutation in PTEN is, in all likelihood, de novo, since it is absent from either parent, and thus unlikely to cause the disease in this family. However, we should note that the age at onset is noticeably different between the affected father and the proband in this family (54 years in the father and 38 years in the proband).
When looking at the cases with confirmed de novo mutations in the genes with a functional interaction with known PD causing genes, these showed a typical presentation of PD.
The PTEN de novo mutation carrier, aged 23, is a British Caucasian male who noticed a left-sided thumb action tremor. Gradually, the tremor progressed to be present at rest and to involve his left arm. Four years later, he developed gait difficulties with some shuffling and stumbling. During the same period the tremor spread to involve his right arm as well, and at the age of 28 he was diagnosed with young-onset Parkinson's disease (YOPD). He was started on carbidopa/levodopa with significant improvement on his gait but no effect on the tremor. Only two years after treatment, he developed drug-induced dyskinesias. At the time his treatment had consisted of carbidopa/levodopa, rasagiline and ropinirole, and, thus, amantadine was added. Age 32 and due to clinical progression and particularly more severe dyskinesias, the patient underwent bilateral deep brain stimulation of the subthalamic nucleus with very good response. Currently, at the age of 36, he suffers from off-periods, has difficulties speaking, as well as increased urinary urgency. His medication consists of carbidopa/levodopa, entacapone, rasagiline, ropinirole and amantadine.
The VAPB de novo mutation carrier is a 44-year-old Caucasian man of British descent, who first noticed a left arm rest tremor at the age of 41. He also complained of shoulder stiffness on the same side and some difficulties in performing fine tasks with his left hand. Clinical examination revealed asymmetric parkinsonism (left>right side) and a subsequent dopamine transporter SPECT scan (DaTSCAN) confirmed the presence of a presynaptic dopaminergic deficit corresponding to the more affected body side. He was diagnosed with YOPD. Treatment was commenced with rasagiline and subsequently pramipexole was added.
The ASNA1 de novo mutation carrier developed PD at an age of 40 years. The first symptom was reduced right arm swing, soon followed by tremor, muscle stiffness, dexterity, gait and writing difficulties. At age 43 he experienced fatigue, constipation and freezing. The father is healthy and the mother suffers from rheumatoid arthritis. The family history for PD is negative.
Discussion
This is the first study to systematically screen for de novo mutations in early-onset sporadic PD using parent–proband trios. We performed exome sequencing in 21 trios and found 20 de novo mutations (19 single nucleotide variants and one in-frame deletion). All probands were heterozygous in those positions. The genes screened are involved in a variety of functions from poly(A) polymerase activity to ubiquitin-binding protein (see Supplementary Material, Table S2) and some are thought to be involved in known PD pathways.
PTEN, the phosphatase and tensin gene, has functions in neuronal migration, neuron number regulation and apoptosis in response to oxidative stress (25). PTEN somatic down-regulation has been linked to tumours, neural proliferation and is being proposed as a target for neuroprotection in PD cases (26,27), whereas PTEN mutations in the germline have been linked to developmental neurological diseases (28). Mutations in PTEN are mainly linked to macrocephaly, autism and ataxia (29–32).
We found novel variants in PTEN. One of our trio-based proband showed a de novo missense mutation in PTEN (p.P204S). Interestingly, mutations in PTEN seem to be rare: there is only one stop-gained and one frameshift in ExAC for a total of 149 coding variant alleles out of ∼120 000. Similarly, in our IPDGC dataset we found a relatively small number of variants in PTEN: four synonymous mutations, two missense mutations (p.N69D and p.N117S) and one frameshift, all of them very rare or non-existent in the population (Table 4). The two missense mutations are from patients with sporadic PD and the patient with the frameshift mutation has a reported positive familial history of PD. Although the frameshift mutation did not show segregation in this family, it is remarkable that it is another apparent de novo mutation in a case with a much lower age at onset than the affected father.
The delicate equilibrium of the various functions of PTEN means that it is often difficult to interpret genetic variability in this gene. PTEN transcriptionally activates PINK1, which is downregulated in the event of PTEN ablation (33,34). PINK1 variants are responsible for PD through a mechanism that is thought to involve oxidative stress caused by mitochondrial dysfunction. PTEN was also linked to the induction of cytochrome c oxidase activity and ATP production in mitochondria (34). On the other hand, PTEN induces cell death and inhibits the PI3K/Akt signalling that reduces inflammation and promotes cell proliferation and survival (26). Furthermore, DJ-1 physically binds to PTEN negatively regulating its activity which, in turn, upregulates PI3K/Akt pathway (35).
PTEN has a large range of activities and cellular locations in a variety of cells. Different mutations in this gene can affect PTEN activity and localization in different ways (36). Deletion of the N-terminal domain PBM (residues 1–15) impedes PTEN interaction with the plasma membrane [see (37) for PTEN protein structure]. When the phosphatase domain (residues 15–185) is mutated, there is a reduction in phosphatase activity and an increase in PI3K/Akt activity. The C2 domain (residues 185–351) inhibits cell migration. The C-tail domain (residues 351–401) is important for interactions with transmembrane proteins and a 2 bp C-terminal PDZ-binding domain (36).
Most of the mutations described for developmental anomalies are missense mutations and appear mainly on the phosphatase and C2 domains, e.g. (30,32,38,39). In addition, a variant can affect the gene's function only in specific subcellular localizations. In 2007 Trotman and colleagues (40) described a missense mutation p.K289E that did not affect the membrane localization of PTEN, but the protein was not present in the nucleus. Another nuclear mislocalization was apparent when mutations were present in the ATP binding motifs at the residues 60–73 and 122–136 (41). This nuclear mislocalization was observed in AD cases with the redistribution of PTEN to the cytoplasm into intracellular neurofibrillary tangles and senile plaques (42).
VAPB is involved in vesicular and endosomal trafficking and was identified in ALS cases (43–45). VAPB expression levels are significantly lower in the spinal cord of ALS cases (46). Furthermore, missense mutations in VAPB protein such as p.P56S and p.T46I cause intracellular ubiquitinated aggregates that predominantly affect motor neurons and induce motor neuron death (43,45).
We found a de novo in-frame deletion of a valine residue at position 25 (p.ΔV25) in VAPB. Only one other deletion in VAPB was documented, p.ΔS160, which is common in the population and any association with ALS was discarded (47). Interestingly, there are only two high impact variants in VAPB in the ExAC dataset: two nonsense variants each in a single sample, out of >60 000 individuals.
Structural studies indicate human VAPB protein is organized in three main conserved domains consisting of the N-terminal major sperm protein (MSP) domain from residue 1 to 125, the coiled coil (CC) domain from residue 151 to 195 and the C-terminal transmembrane (TM) domain including the remaining residues until residue 243 (48). The MSP is very important in VAPB as it is the domain that binds to the FFAT (two phenylalanines in an acidic tract) motif of lipid-binding-proteins and is involved in the ubiquitination pathway. VAPB binds through that domain to proteins such as STX1A and STX1B, p97 ATPase and ASNA1 (49). Misfolding of MSP domain induces the formation of the aggregates and the binding site becomes unavailable on the endoplasmic reticulum (50).
As Gupta et al. (48) noticed, the p.T46I and p.P56S missense mutations that cause ALS are located in the MSP. On the other hand, the p.ΔS160 deletion, located on the CC is common and does not cause ALS. Additionally, another study detected a missense variant p.D130E that showed no prevalence in cases (51) and falls between the MSP and the CC domains. Mutations in the MSP could be the leading cause for the dysfunction in the motor neurons. Nevertheless, some evidence in Drosophila is starting to show that mutations in the C-terminal TM can cause neurodegeneration in the corresponding variant p.V234I in humans (52).
Our results follow the same trend. The identified de novo p.ΔV25 deletion is absent in the general population and is located in the MSP. The mutations p.R19L and p.L125 M identified in the IPDGC dataset, both from sporadic cases, are also in the MSP (Table 4), are not present in the general population and only the latter shows two heterozygous cases in ExAC. When moving towards the CC in the IPDGC dataset, we find the frequent p.D130E and p.ΔS160 variants mentioned above, and two extra missense mutations (p.M170I and p.R184Q) with higher frequencies in the general population.
We found the VAPB de novo variant in a trio with two additional de novo variants in COL12A1 (p.R754Q) and RUNDC3A (p.C108F). COL12A1 was associated with myopathy (53) and the only association to a neurodegenerative disease is its decreased amount in the cervical spinal cord of patients with ALS (54). COL12A1 presented 96 variants in the IPDGC dataset (see Supplementary Material, Table S3). In contrast, RUNDC3A presented five mutations, three of them rare and observed only in patients. Due to the fact that this trio presented three confirmed de novo variants, it is not possible to say with certainty, which, if any, is associated with the disease. However the fact that VAPB directly interacts with known PD genes strongly argues in favour of the involvement of this gene.
The results at VAPB and PTEN, which have both been shown to be involved in disparate phenotypes, fit nicely with the growing body of evidence suggesting that pleiotropic events are a greatly underappreciated event in human disease (55).
For ASNA1, we found one de novo missense mutation p.L303V in one of the trios not present in any database. High impact variants in this gene seem rare, with mainly missense variants present in ExAC and only three frameshifts (1 allele each) and one stop-gain mutations. In the IPDGC dataset, we see a similar trend with only seven rare variants. The missense variants in IPDGC (Table 4) were identified in patients presenting with positive familial history of PD (variants: p.L61V and p.R65C); and early-onset sporadic PD (variant p.L208V). ASNA1 is known to have a function in transporting proteins as a part of the transmembrane recognition complex (TRC). Interestingly, ASNA1 has been shown to bind to VAPB (49), although a role in human neurodegenerative diseases has still to be investigated.
In conclusion, we have identified de novo mutations in early-onset, sporadic PD cases. We have used publicly available population genetic data and our in-house dataset of exome-sequenced PD cases and controls to replicate and confirm our findings. From our list of confirmed de novo mutations, three genes are particularly interesting, as they have been reported to interact with known PD genes and present rare, case-specific variants in our large cohort of PD cases.
It is plausible that some of the apparently sporadic PD cases are due to recessive or compound heterozygous mutations in genes yet to be associated with disease; however we did not identify any better recessive candidates in these trios.
The present study has some shortcomings that we are not able to immediately address. The sample size is small; unfortunately it is very difficult to identify PD cases with both parents available for genetic analyses. The methodology used, although appropriate and in line with what has been previously published for de novo mutations in disease, misses a significant amount of genetic variability, particularly in the form of non-coding variants. Additionally, apparently de novo variants can actually be inherited from a parent that has low-level mosaicism for that variant. This is a difficult issue to address [as recently shown by Acuna-Hidalgo and colleagues (56)] and would require very high sequencing coverage, perhaps even of multiple tissues, in all individuals and is thus beyond the scope of the present work. In addition, definitive proof of pathological significance for PD can only be achieved by one of the following: identifying the same mutation in complete segregation in large families, identifying multiple families with even limited segregation of the same variants or functional studies of the protein harbouring the mutations showing altered function.
On the other hand, we have been able to screen, in an unbiased manner, a cohort of over 1200 PD cases and 400 controls for mutations in all of the genes identified in our trio study, having confirmed the presence of rare, case-specific mutations in the same genes.
De novo mutations may be a vastly underappreciated cause of apparently sporadic forms of adult-onset disease, such as PD. We show de novo mutations in genes, which are known to interact with known PD-causing genes, and we suggest these may be responsible for the disease in these cases.
Material and Methods
We performed whole-exome sequencing in 21 full trios. Criteria for inclusion of cases were based on age at onset (<40 years), typical presentation of PD with negative family history, absence of pathogenic mutations in any of the known PD genes and availability of both parents for genetic studies. All cases included are of European descent.
Exomes for the 21 trios were captured using Illumina's Nextera Rapid Capture according to the manufacturer's recommendations. Indexed and pooled libraries were then sequenced on Illumina's HiSeq2000 (100 bp, paired-end) to a mean target coverage of 30×. Reads were mapped to GRCh37 using bwa-mem (0.7.12) and followed GATK Best Practices for v3 (57). Briefly, this consisted of flagging duplicate reads, realignment around indels, base recalibration and variant calling on all trios simultaneously using the HaplotypeCaller tool. Variant qualities were then recalibrated as described in DePristo et al. (58). After obtaining a high quality variant set, we performed a genotype-refinement protocol, as described in (http://gatkforums.broadinstitute.org/discussion/4723/genotype-refinement-workflow). This protocol uses population and pedigree priors to improve estimates of genotype likelihoods, providing higher quality variant calls.
To confirm all relationships and identify unreported familial relationships, we performed identity-by-descent (IBD) estimation for all pairs of individuals. The proportion of IBD was obtained with PLINK (59) and varied between 0.35–0.45 for the trio pairs of each parent with the descendant, and equal to zero between parents and all the remaining pairs. The lower end of the spectrum of IBD values is due to lower variant qualities in some samples.
We used three publicly available resources to extract genome-wide variant frequency data: the Exome Sequencing Project (ESP6500) (60), the 1000 Genomes Project (Oct 2014) (61) and the ExAC (62).
Sanger sequencing was used to confirm the presence of all de novo mutations identified by whole-exome sequencing. Primers designed for this validation are shown in the Supplementary Material (see Supplementary Material, Table S4; PCR conditions used are available upon request). Table 1 shows each of the transcripts used as basis for annotation of the variants identified.
Protein–protein interactions and functional protein associations were defined with STRING v10 (63). The input consisted of a list of known PD genes (64), and genes containing validated de novo variants. We considered total scores above 0.400 (medium confidence) that correspond to the combination of four different scores: co-expression, experimental, knowledge and text-mining. The total number of interactions between proteins corresponding to genes containing de novo variants and proteins corresponding to known PD genes was calculated, as was the number of such interactions involving the protein corresponding to de novo gene in turn. The significance of these quantities was assessed by randomly positioning the proteins corresponding to de novo and known PD genes on the interaction map such that each protein is mapped to a protein with the same total number of interactions (to avoid potential bias caused by the genes of interest having large numbers of interactions, thus being likely to interact with each other by chance). The number of interactions between each de novo protein and PD proteins, and the total number of such interactions, was calculated on the randomized map and compared with that observed in the actual data. This process was repeated 1 000 000 times to obtain P-values. Correction for multiple testing of proteins was performed by comparing the maximum number of interactions for any single protein in each simulated dataset to the number of interactions observed for each protein in the real data. A similar procedure was used to test for an excess of interactions between the proteins corresponding to known PD genes. It should be noted that throughout the text, interactions are defined as per STRING's definitions and in many cases these are only predicted, without experimental evidence.
Finally, we extracted information for genes of interest from in-house data, produced within the IPDGC. This dataset consisted of a total of 1715 samples exome-sequenced with 1243 of diagnosed PD cases and 472 controls. Details from this dataset have been previously published (65).
Supplementary Material
Funding
Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust and the Medical Research Council.
Supplementary Material
Acknowledgements
This work was supported by a Research Grant from the Bachmann-Strauss Foundation. J.B. is supported by a Research Fellowship from the Alzheimer's Society. C.G. receives academic research support from the DFG (GA2031/1–2) and has received support in the form of a travel grant from the Guarantors of Brain. R.G. is supported by a Research Fellowship from the Alzheimer's Research UK. S.A.S. was supported by the Else Kroener-Fresenius Stiftung and received intramural funding from the University of Kiel. She received royalties from Oxford University Press and Springer.
Conflict of Interest statement. The authors report no conflicts of interest.
References
- 1.Lill C.M., Bertram L. (2011) Towards unveiling the genetics of neurodegenerative diseases. Semin. Neurol., 31, 531–541. [DOI] [PubMed] [Google Scholar]
- 2.Bras J., Guerreiro R., Hardy J. (2012) Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat. Rev. Neurosci., 13, 453–464. [DOI] [PubMed] [Google Scholar]
- 3.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. et al. (2007) Strong association of de novo copy number mutations with autism. Science, 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Roak B.J., Deriziotis P., Lee C., Vives L., Schwartz J.J., Girirajan S., Karakoc E., Mackenzie A.P., Ng S.B., Baker C. et al. (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet., 43, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. (2008) Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet., 40, 880–885. [DOI] [PubMed] [Google Scholar]
- 7.Girard S.L., Gauthier J., Noreau A., Xiong L., Zhou S., Jouan L., Dionne-Laporte A., Spiegelman D., Henrion E., Diallo O. et al. (2011) Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet., 43, 860–863. [DOI] [PubMed] [Google Scholar]
- 8.Harbo H.F., Finsterer J., Baets J., Van Broeckhoven C., Di Donato S., Fontaine B., De Jonghe P., Lossos A., Lynch T., Mariotti C. et al. (2009) EFNS guidelines on the molecular diagnosis of neurogenetic disorders: general issues, Huntington's disease, Parkinson's disease and dystonias. Eur. J. Neurol., 16, 777–785. [DOI] [PubMed] [Google Scholar]
- 9.Rosewich H., Thiele H., Ohlenbusch A., Maschke U., Altmuller J., Frommolt P., Zirn B., Ebinger F., Siemes H., Nurnberg P. et al. (2012) Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol., 11, 764–773. [DOI] [PubMed] [Google Scholar]
- 10.De Carvalho Aguiar P., Fuchs T., Borges V., Lamar K.M., Silva S.M., Ferraz H.B., Ozelius L. (2010) Screening of Brazilian families with primary dystonia reveals a novel THAP1 mutation and a de novo TOR1A GAG deletion. Mov. Disord., 25, 2854–2857. [DOI] [PubMed] [Google Scholar]
- 11.Saitsu H., Nishimura T., Muramatsu K., Kodera H., Kumada S., Sugai K., Kasai-Yoshida E., Sawaura N., Nishida H., Hoshino A. et al. (2013) De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet., 45, 445–449, 449e441. [DOI] [PubMed] [Google Scholar]
- 12.Portet F., Dauvilliers Y., Campion D., Raux G., Hauw J.J., Lyon-Caen O., Camu W., Touchon J. (2003) Very early onset AD with a de novo mutation in the presenilin 1 gene (Met 233 Leu). Neurology, 61, 1136–1137. [DOI] [PubMed] [Google Scholar]
- 13.Golan M.P., Styczynska M., Jozwiak K., Walecki J., Maruszak A., Pniewski J., Lugiewicz R., Filipek S., Zekanowski C., Barcikowska M. (2007) Early-onset Alzheimer's disease with a de novo mutation in the presenilin 1 gene. Exp. Neurol., 208, 264–268. [DOI] [PubMed] [Google Scholar]
- 14.Alexander M.D., Traynor B.J., Miller N., Corr B., Frost E., McQuaid S., Brett F.M., Green A., Hardiman O. (2002) ‘True’ sporadic ALS associated with a novel SOD-1 mutation. Ann. Neurol., 52, 680–683. [DOI] [PubMed] [Google Scholar]
- 15.DeJesus-Hernandez M., Kocerha J., Finch N., Crook R., Baker M., Desaro P., Johnston A., Rutherford N., Wojtas A., Kennelly K. et al. (2010) De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum. Mutat., 31, E1377–E1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chio A., Calvo A., Moglia C., Ossola I., Brunetti M., Sbaiz L., Lai S.L., Abramzon Y., Traynor B.J., Restagno G. (2011) A de novo missense mutation of the FUS gene in a ‘true’ sporadic ALS case. Neurobiol. Aging, 32, 553 e523–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chesi A., Staahl B.T., Jovicic A., Couthouis J., Fasolino M., Raphael A.R., Yamazaki T., Elias L., Polak M., Kelly C. et al. (2013) Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat. Neurosci., 16, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puschmann A., Ross O.A., Vilarino-Guell C., Lincoln S.J., Kachergus J.M., Cobb S.A., Lindquist S.G., Nielsen J.E., Wszolek Z.K., Farrer M. et al. (2009) A Swedish family with de novo alpha-synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat. Disord., 15, 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishioka K., Hayashi S., Farrer M.J., Singleton A.B., Yoshino H., Imai H., Kitami T., Sato K., Kuroda R., Tomiyama H. et al. (2006) Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson's disease. Ann. Neurol., 59, 298–309. [DOI] [PubMed] [Google Scholar]
- 20.Ahn T.B., Kim S.Y., Kim J.Y., Park S.S., Lee D.S., Min H.J., Kim Y.K., Kim S.E., Kim J.M., Kim H.J. et al. (2008) alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology, 70, 43–49. [DOI] [PubMed] [Google Scholar]
- 21.Hansen F.H., Skjorringe T., Yasmeen S., Arends N.V., Sahai M.A., Erreger K., Andreassen T.F., Holy M., Hamilton P.J., Neergheen V. et al. (2014) Missense dopamine transporter mutations associate with adult parkinsonism and ADHD. J. Clin. Invest., 124, 3107–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neale B.M., Kou Y., Liu L., Ma'ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P., Henikoff S., Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 24.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L., Liu F., Salmonsen R.A., Turner T.K., Litofsky N.S., Di Cristofano A., Pandolfi P.P., Jones S.N., Recht L.D., Ross A.H. (2002) PTEN in neural precursor cells: regulation of migration, apoptosis, and proliferation. Mol. Cell. Neurosci., 20, 21–29. [DOI] [PubMed] [Google Scholar]
- 26.Diaz-Ruiz O., Zapata A., Shan L., Zhang Y., Tomac A.C., Malik N., de la Cruz F., Backman C.M. (2009) Selective deletion of PTEN in dopamine neurons leads to trophic effects and adaptation of striatal medium spiny projecting neurons. PLoS One, 4, e7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domanskyi A., Geissler C., Vinnikov I.A., Alter H., Schober A., Vogt M.A., Gass P., Parlato R., Schutz G. (2011) Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson's disease models. FASEB J., 25, 2898–2910. [DOI] [PubMed] [Google Scholar]
- 28.Chang N., El-Hayek Y.H., Gomez E., Wan Q. (2007) Phosphatase PTEN in neuronal injury and brain disorders. Trends Neurosci., 30, 581–586. [DOI] [PubMed] [Google Scholar]
- 29.Gary D.S., Mattson M.P. (2002) PTEN regulates Akt kinase activity in hippocampal neurons and increases their sensitivity to glutamate and apoptosis. Neuromolecular Med., 2, 261–269. [DOI] [PubMed] [Google Scholar]
- 30.Butler M.G., Dasouki M.J., Zhou X.P., Talebizadeh Z., Brown M., Takahashi T.N., Miles J.H., Wang C.H., Stratton R., Pilarski R. et al. (2005) Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet., 42, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herman G.E., Butter E., Enrile B., Pastore M., Prior T.W., Sommer A. (2007) Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am. J. Med. Genet. A, 143A, 589–593. [DOI] [PubMed] [Google Scholar]
- 32.O'Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K. et al. (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science, 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Unoki M., Nakamura Y. (2001) Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene, 20, 4457–4465. [DOI] [PubMed] [Google Scholar]
- 34.Liang H., He S., Yang J., Jia X., Wang P., Chen X., Zhang Z., Zou X., McNutt M.A., Shen W.H. et al. (2014) PTENalpha, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab., 19, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y.C., Kitaura H., Taira T., Iguchi-Ariga S.M., Ariga H. (2009) Oxidation of DJ-1-dependent cell transformation through direct binding of DJ-1 to PTEN. Int. J. Oncol., 35, 1331–1341. [PubMed] [Google Scholar]
- 36.Kreis P., Leondaritis G., Lieberam I., Eickholt B.J. (2014) Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders. Front. Mol. Neurosci., 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song M.S., Salmena L., Pandolfi P.P. (2012) The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol., 13, 283–296. [DOI] [PubMed] [Google Scholar]
- 38.Longy M., Coulon V., Duboue B., David A., Larregue M., Eng C., Amati P., Kraimps J.L., Bottani A., Lacombe D. et al. (1998) Mutations of PTEN in patients with Bannayan-Riley-Ruvalcaba phenotype. J. Med. Genet., 35, 886–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Celebi J.T., Tsou H.C., Chen F.F., Zhang H., Ping X.L., Lebwohl M.G., Kezis J., Peacocke M. (1999) Phenotypic findings of Cowden syndrome and Bannayan-Zonana syndrome in a family associated with a single germline mutation in PTEN. J. Med. Genet., 36, 360–364. [PMC free article] [PubMed] [Google Scholar]
- 40.Trotman L.C., Wang X., Alimonti A., Chen Z., Teruya-Feldstein J., Yang H., Pavletich N.P., Carver B.S., Cordon-Cardo C., Erdjument-Bromage H. et al. (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell, 128, 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lobo G.P., Waite K.A., Planchon S.M., Romigh T., Nassif N.T., Eng C. (2009) Germline and somatic cancer-associated mutations in the ATP-binding motifs of PTEN influence its subcellular localization and tumor suppressive function. Hum. Mol. Genet., 18, 2851–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sonoda Y., Mukai H., Matsuo K., Takahashi M., Ono Y., Maeda K., Akiyama H., Kawamata T. (2010) Accumulation of tumor-suppressor PTEN in Alzheimer neurofibrillary tangles. Neurosci Lett., 471, 20–24. [DOI] [PubMed] [Google Scholar]
- 43.Nishimura A.L., Mitne-Neto M., Silva H.C., Richieri-Costa A., Middleton S., Cascio D., Kok F., Oliveira J.R., Gillingwater T., Webb J. et al. (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet., 75, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Millecamps S., Salachas F., Cazeneuve C., Gordon P., Bricka B., Camuzat A., Guillot-Noel L., Russaouen O., Bruneteau G., Pradat P.F. et al. (2010) SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J. Med. Genet., 47, 554–560. [DOI] [PubMed] [Google Scholar]
- 45.Chen H.J., Anagnostou G., Chai A., Withers J., Morris A., Adhikaree J., Pennetta G., de Belleroche J.S. (2010) Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem., 285, 40266–40281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anagnostou G., Akbar M.T., Paul P., Angelinetta C., Steiner T.J., de Belleroche J. (2010) Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging, 31, 969–985. [DOI] [PubMed] [Google Scholar]
- 47.Landers J.E., Leclerc A.L., Shi L., Virkud A., Cho T., Maxwell M.M., Henry A.F., Polak M., Glass J.D., Kwiatkowski T.J. et al. (2008) New VAPB deletion variant and exclusion of VAPB mutations in familial ALS. Neurology, 70, 1179–1185. [DOI] [PubMed] [Google Scholar]
- 48.Gupta G., Qin H., Song J. (2012) Intrinsically unstructured domain 3 of hepatitis C Virus NS5A forms a ‘fuzzy complex’ with VAPB-MSP domain which carries ALS-causing mutations. PLoS One, 7, e39261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baron Y., Pedrioli P.G., Tyagi K., Johnson C., Wood N.T., Fountaine D., Wightman M., Alexandru G. (2014) VAPB/ALS8 interacts with FFAT-like proteins including the p97 cofactor FAF1 and the ASNA1 ATPase. BMC Biol., 12, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim S., Leal S.S., Ben Halevy D., Gomes C.M., Lev S. (2010) Structural requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-associated VAP-B(P56S) neurotoxicity. J. Biol. Chem., 285, 13839–13849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conforti F.L., Sprovieri T., Mazzei R., Ungaro C., Tessitore A., Tedeschi G., Patitucci A., Magariello A., Gabriele A., Labella V. et al. (2006) Sporadic ALS is not associated with VAPB gene mutations in Southern Italy. J. Negat. Results Biomed., 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanhueza M., Zechini L., Gillespie T., Pennetta G. (2014) Gain-of-function mutations in the ALS8 causative gene VAPB have detrimental effects on neurons and muscles. Biol. Open, 3, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou Y., Zwolanek D., Izu Y., Gandhy S., Schreiber G., Brockmann K., Devoto M., Tian Z., Hu Y., Veit G. et al. (2014) Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum. Mol. Genet., 23, 2339–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satoh J., Yamamoto Y., Kitano S., Takitani M., Asahina N., Kino Y. (2014) Molecular network analysis suggests a logical hypothesis for the pathological role of c9orf72 in amyotrophic lateral sclerosis/frontotemporal dementia. J. Cent. Nerv. Syst. Dis., 6, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guerreiro R., Bras J., Hardy J., Singleton A. (2014) Next generation sequencing techniques in neurological diseases: redefining clinical and molecular associations. Hum. Mol. Genet., 23, R47–R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Acuna-Hidalgo R., Bo T., Kwint M.P., van de Vorst M., Pinelli M., Veltman J.A., Hoischen A., Vissers L.E., Gilissen C. (2015) Post-zygotic point mutations are an underrecognized source of de novo genomic variation. Am. J. Hum. Genet., 97, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. et al. (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics, 11, 11 10 11–11 10 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Exome Variant Server. (2014) NHLBI GO Exome Sequencing Project (ESP), Seattle, WA URL: http://evs.gs.washington.edu/EVS/ (October 2014). [Google Scholar]
- 61.Genomes Project C., Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. (2012) An integrated map of genetic variation from 1092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Exome Aggregation Consortium. (2015) (ExAC), Cambridge, MA URL: http://exac.broadinstitute.org (June 2015).
- 63.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. et al. (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res., 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bras J., Guerreiro R., Hardy J. (2015) SnapShot: genetics of Parkinson's disease. Cell, 160, 570–570 e571. [DOI] [PubMed] [Google Scholar]
- 65.Jansen I.E., Bras J.M., Lesage S., Schulte C., Gibbs J.R., Nalls M.A., Brice A., Wood N.W., Morris H., Hardy J.A. et al. (2015) CHCHD2 and Parkinson's disease. Lancet Neurol., 14, 678–679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.