

Abstract
Glycogen storage disease type IV (GSD IV) is a rare autosomal recessive disorder caused by deficiency of the glycogen-branching enzyme (GBE). The diagnostic hallmark of the disease is the accumulation of a poorly branched form of glycogen known as polyglucosan (PG). The disease is clinically heterogeneous, with variable tissue involvement and age at onset. Complete loss of enzyme activity is lethal in utero or in infancy and affects primarily the muscle and the liver. However, residual enzyme activity as low as 5–20% leads to juvenile or adult onset of a disorder that primarily affects the central and peripheral nervous system and muscles and in the latter is termed adult polyglucosan body disease (APBD). Here, we describe a mouse model of GSD IV that reflects this spectrum of disease. Homologous recombination was used to knock in the most common GBE1 mutation p.Y329S c.986A > C found in APBD patients of Ashkenazi Jewish decent. Mice homozygous for this allele (Gbe1ys/ys) exhibit a phenotype similar to APBD, with widespread accumulation of PG. Adult mice exhibit progressive neuromuscular dysfunction and die prematurely. While the onset of symptoms is limited to adult mice, PG accumulates in tissues of newborn mice but is initially absent from the cerebral cortex and heart muscle. Thus, PG is well tolerated in most tissues, but the eventual accumulation in neurons and their axons causes neuropathy that leads to hind limb spasticity and premature death. This mouse model mimics the pathology and pathophysiologic features of human adult-onset branching enzyme deficiency.
Introduction
Glycogen is a highly branched glucose polymer synthesized by two enzymes: (i) glycogen synthase (GYS), which attaches glucose to nascent linear chains of glycogen, and (ii) the glycogen-branching enzyme (GBE), which attaches a short branch of ∼4 glucose units to the linear chain. The absence of GBE causes glycogen storage disease type IV (GSD IV, OMIM 232500). GSD IV is an autosomal recessive disorder typically diagnosed histologically by the accumulation of a poorly branched form of glycogen known as polyglucosan (PG). In GSD IV, PG mainly accumulates in the liver, heart, skeletal muscle and central nervous system, tissues that have high metabolic activity. However, GSD IV is a very heterogeneous disorder, affecting different organs at different ages, with visceral and/or neuromuscular involvement. Based primarily upon the amount of residual GBE activity, there are clinically distinguishable forms of the disease, including an early-onset form associated with a complete loss of enzyme activity, a juvenile or adult-onset form associated with partial activity and a clinically distinct adult-onset disease that is known as adult polyglucosan body disease (APBD). The infantile form of the disease is known as Andersen's disease and is typically due to the absence of GBE activity. The clinical features include failure to thrive, hepatosplenomegaly and progressive liver cirrhosis, typically leading to death in early childhood (1). Residual GBE activity causes different disease symptoms at different ages from fetus to adult; therefore, presentation of the disease can be separated into four groups based upon the age at onset: perinatal, presenting as fetal akinesia and perinatal death; congenital, with hypotonia and death in early infancy; childhood, with myopathy and/or cardiomyopathy; and adult, with isolated myopathy or APBD.
APBD was first described in 1971 (2) and is characterized clinically by progressive upper and lower motor neuron dysfunction, marked distal sensory loss (mainly in the lower extremities), early neurogenic bladder, cerebellar dysfunction and cognitive problems (3–8). The typical neuropathologic findings are numerous large PG bodies (PGB) in the peripheral nerves, cerebral hemispheres, basal ganglia, cerebellum and spinal cord (5,7,9,10). Isolated cases of PG myopathy without peripheral nerve involvement have also been described (11,12).
Currently, no treatment is available for GSD IV, although liver transplantation has been performed in patients with apparently isolated liver involvement (13). Two naturally occurring animal models of GBE deficiency, American quarter horses and Norwegian forest cats, are not practical laboratory animals (14,15). Mouse models generated either by homologous recombination or chemical germline mutagenesis that leads to the complete elimination of GBE activity only exhibit early-onset GSD IV (16). Currently, the best mouse model of GBE deficiency for adult-onset GSD IV is a mouse with reduced Gbe1 expression due to insertion of a gene cassette into the otherwise intact gene (17). We have developed a new mouse model of GBE deficiency by knocking in via homologous recombination the most common human GBE1 mutation (p.Y329S) found in Ashkenazi Jewish patients. We developed this model in order to better understand the pathogenesis of the disease and to test therapeutic strategies.
Results
Generation of mice with p.Y329S mutation in GBE
A mouse model of GBE deficiency was generated by knocking in the p.Y329S mutation into exon 7 and introducing flippase recognition target (FRT) recombination sequences upstream and downstream of the mouse Gbe1 exon 7 via homologous recombination (Fig. 1A). Since the p.Y329S mutation is linked to an adjacent 50-base-pair (bp) FRT sequence (indicated by open triangles in Fig. 1A), we used this size difference to genotype by polymerase chain reaction (PCR) wild-type, heterozygous or homozygous mice. Gbe1+/ys-neo animals were first bred to Cre-expressing mice in order to remove the PGK-neomycin cassette, or they were intercrossed to generate Gbe1ys-neo/ys-neo homozygous mice harboring both the p.Y329S mutation and the PGK-neomycin cassette (flanked by the gray triangles that represent LoxP consensus sequences in Fig. 1A). PCR analysis using primers that bind upstream and downstream of exon 7 amplifies a 270 bp wild-type DNA fragment, or a 320 bp DNA fragment containing the FRT consensus sequence was that knocked in linked to the point mutation. A representative PCR analysis of genotyping is shown in Figure 1A. Elimination of exon 7 by breeding to mice expressing an flp-e transgene yielded a severe Andersen disease model, as we reported previously (18). Excision of PGK-neomycin cassette was detected by PCR analysis using primers that can amplify either a 400 bp-long cassette and part of intron 7 or a 300 bp-long fragment containing part of intron 7, and FRT and lox-p consensus sequences (data not shown). In order to confirm the homozygous p.Y329S mutation in exon 7, total muscle RNA isolated from Gbe1ys/ys mice was reverse transcribed and amplified by PCR. The PCR primers were designed to amplify an 810 bp wild-type Gbe1 mRNA fragment spanning exons 4–10 that are 54 kb apart in genomic DNA. Reverse transcriptase-polymerase chain reaction (RT-PCR) products were sequenced to demonstrate the homozygous p.Y329S mutation (Fig. 1C).
Figure 1.

Gene targeting and molecular characterization of a p.Y329S GBE1 knock-in mouse model. A partial map of the Gbe1 locus and the targeting vector containing FRT sites flanking exon 7 with c.986A > C change before and after homologous recombination in ES cells. Crossing Gbe1+/ys-neo mice with ROSA 26-iCre mice excises PGK-neomycin cassette from one allele of the Gbe1 gene by Cre recombinase. Subsequent breeding of heterozygous mice indicated by Gbe1+/ys creates a Gbe1 knock-in mouse (Gbe1ys/ys). (B) Agarose gel electrophoresis shows the genotyping of p.Y329S-positive and p.Y329S-negative mice, primers used for the PCR are indicated in Panel A as F and R. (C) RT-PCR analysis of the mRNA extracted from muscle tissue, using primers spanning exons 5–10. The sequence analysis from exons 6 to 8 from the RT-PCR product shown.
GBE-deficient embryos are stillborn after a normal gestation
Removing exon 7 by Flpe recombination leads to loss of GBE activity (18). Complete loss of GBE activity causes death in utero and still birth in humans (16,19,20). Gbe1−/− mice likewise die at or soon after birth. The mice are comparable with heterozygous and wild-type littermates in terms of size and appearance after birth (Fig. 2A). Segregation of the deletion follows the expected Mendelian distribution: out of 62 pups, 16 were wild type, 31 were heterozygous and 15 homozygous, reflecting no in utero lethality. Western blot analysis of muscle extracts obtained from newborn pups demonstrated the absence of GBE protein, while Gbe1+/− and Gbe1+/+ animals express detectable GBE protein. In contrast, adult animals harboring the PGK-neomycin cassette (Gbe1ys-neo/ys-neo) have a significantly reduced but still detectable amount of GBE protein in muscle (Fig. 2B). Reduced GBE protein due to the p.Y329S mutation and the presence of the PGK-neomycin cassette causes early death in homozygous mice. The survival of 20 Gbe1ys-neo/ys-neo, 47 Gbe1ys/ys and 29 Gbe1+/+ mice was analyzed by MedCalc Software (MedCalc® Version 14.12.0 Copyright© 1993–2014), and a Kaplan–Meier curve was plotted (Fig. 2C). We did not observe any Gbe1ys-neo/ys-neo mice survive beyond 32 weeks. Their maximum life span is 7 weeks, shorter than that of mice harboring the PGK-neomycin cassette but lacking the point mutation (18). However, mice homozygous for the p.Y329S mutation alone can survive up to 80 weeks (Fig. 2C). As GBE deficiency causes weakness, we tested Gbe1ys/ys animals for their grip strength. Six 1-year-old wild-type or Gbe1ys/ys mice were used to test grip strength. Both male and female Gbe1ys/ys mice had significant weakness compared with wild-type littermates (Fig. 2D). After 1 year, mice start to have spastic paraplegia and cannot use their hind limbs (Supplementary Material, Fig. S2)
Figure 2.

Lack of GBE does not affect embryonic development, but low enzyme activity decreases strength and life span. (A) Neonates from the same litter are shown before being genotyped. According to their genotype, they are indicated by Gbe1−/−, Gbe1+/− and Gbe1+/+. Note no evidence of hydrops fetalis or malformations, except Gbe1−/− mice are stillborn. Mice homozygous for the p.Y329S knock-in mutation in addition to the PGK-neomycin cassette can live up to 8 months. Without the PGK-neomycin cassette, mice can survive up to 20 months with paralysis of hind limbs. Heterozygous or wild-type littermates can survive beyond 24 months. (B) Western blot analysis of GBE, GYS1 and GAPDH in muscle extracts obtained from Gbe1−/−. Gbe1+/−, Gbe1+/+ embryos and Gbe1ys-neo/ys-neo Gbe1ys/ys and Gbe1+/+ mice are shown in panel. (C) Kaplan–Meier plot illustrates the incidence of death in Gbe1ys-neo/ys-neo (n = 20), Gbe1ys/ys (n = 47) versus Gbe1+/+ mice (n = 29) P < 0.0001. (D) Gbe1ys/ys mice have weaker grip strength than wild-type littermates (n = 16 P < 0.001).
Effect of the p.Y329S mutation on GBE and glycogen content
It has previously been shown in humans that the p.Y329S substitution decreases GBE activity to 20% of healthy controls and leads to reduced branching of glycogen (8). The murine GBE protein is 98% identical to human GBE; therefore, we measured GBE activity in wild-type, Gbe1ys-neo/ys-neo and Gbe1ys/ys animals. Unlike exon 7 knockout animals, both Gbe1ys-neo/ys-neo and Gbe1ys/ys animals have a low but detectable amount of residual GBE activity, while removal of the PGK-neomycin cassette increases the residual GBE activity almost 2 fold in tissues studied (Table 1). Next, we determined the glycogen content in muscle tissue among the different genotypes of Gbe1 mice. Although GBE takes part in the synthesis of glycogen and mediates the proper branching, in all GBE-deficient mice, we found that brain, heart, liver and muscle glycogen content was significantly greater than in controls, reflecting a greater accumulation of the abnormal PG (Table 1) despite a comparable amount of GYS protein in muscles, as determined by western blotting (Fig. 2B).
Table 1.
GBE activity in Gbe1+/+, Gbe1+/ys-neo, Gbe1+/ys, Gbe1ys/ys mice and glycogen content in Gbe1+/+ and Gbe1ys/ys mice
| Percent GBE activity |
Glycogen (μg/g wet tissue) | |||||
|---|---|---|---|---|---|---|
| WT | YS-neo/+ | Ys/+ | YS/YS | WT | YS/YS | |
| Brain | 100 sd: ± 5 | 43 sd: ± 9 | 66 sd: ± 13 | 21 sd: ± 14 | 5 sd: ± 1 | 85 sd: ± 49 |
| Heart | 100 sd: ± 17.6 | 51 sd: ± 14 | 67 sd: ± 20 | 21 sd: ± 4 | 20 sd: ± 21 | 1809 sd: ± 113 |
| Liver | 100 sd: ± 10 | 50 sd: ± 15 | 62 sd: ± 14 | 37 sd: ± 14 | 1585 sd: ± 664 | 2414 sd: ± 483 |
| Muscle | 100 sd: ± 9 | 47 sd: ± 8 | 59 sd: ± 12 | 16 sd: ± 42 | 68 sd: ± 5 | 2170 sd: ± 281 |
Values represent the mean ± SD, P < 0.001 (n = 6 for each group).
Low GBE activity leads to PG formation in tissues
Clinically, GSD IV is diagnosed by the presence of poorly branched glycogen that is resistant to diastase (alpha amylase) digestion. Therefore, we examined the accumulation of diastase-resistant PG in multiple organs from 1-year-old Gbe1ys/ys mice. Because the juvenile and adult forms of GSD IV are typically a neuromuscular disorder, we examined brain and muscle samples first. Brain sections of Gbe1ys/ys mice have PGB located in the neuropil (dendrites and axons). Although there is considerable PG accumulation in the brain, the cell bodies of neurons do not harbor detectable PG. No cell loss was observed in either the cerebral cortex or spinal cord (Fig. 3A–D, Supplementary Material, Figs S2 and S3). Skeletal muscle and liver sections also contained PGB, which were absent in the tissues of wild-type mice (Fig. 3E–H); likewise, PGB also accumulate in cardiac muscle of Gbe1ys/ys mice (Fig. 5). In addition, in the liver, moderate perisinusoidal/pericellular dense collagen bundles (in Liver Zone 3) were observed in Gbe1ys/ys mice, but there was no portal or periportal fibrosis. To score the fibrosis, the non-alcoholic fatty liver disease fibrosis scoring system was used (21). According to this scoring system, the fibrosis was compatible with a Stage 1B in the Gbe1ys/ys mice (Fig. 3I and J).
Figure 3.
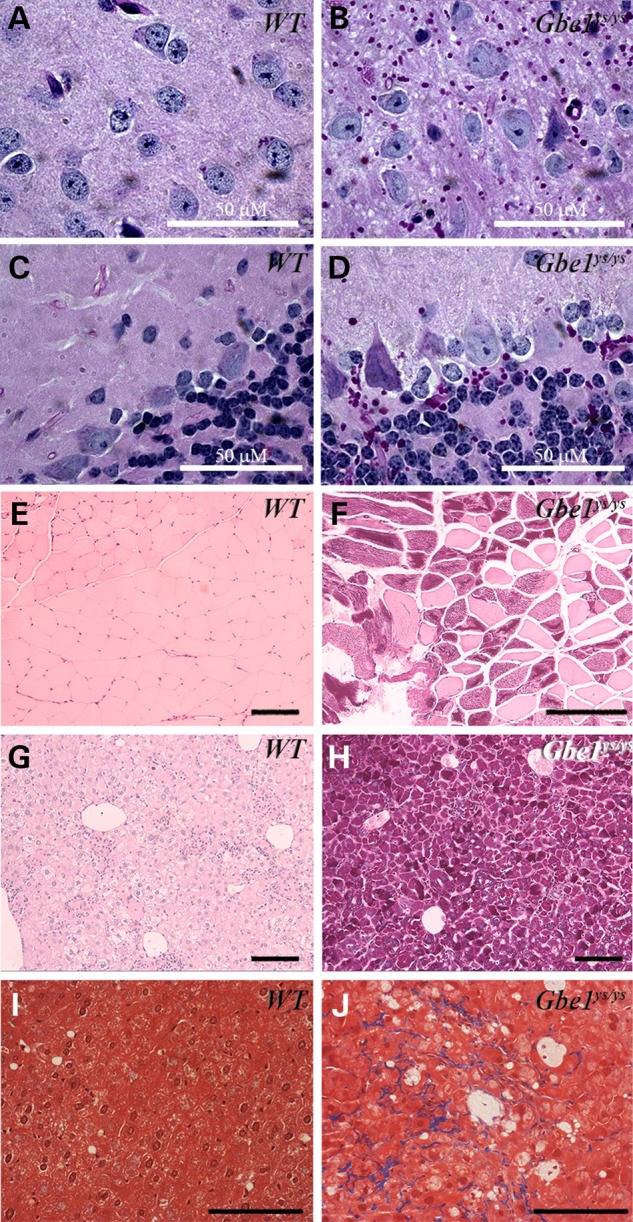
Decreased GBE activity leads to widespread PGB accumulation and fibrosis in the liver. PGB (red stain) are detected in brain (A–D), skeletal muscle (E and F) and liver (G and H) sections from adult Gbe1ys/ys mice. Gomori trichrome staining of the liver (I and J) shows fibrosis, and blue deposits indicating fibrosis in Gbe1ys/ys liver. Images are 200× magnification, except brain (A–D) and liver (I and J) sections are 1000× and 400× magnifications, respectively. Solid bars represents 100 μm or as indicated.
Figure 5.

PGB are ubiquitinylated. Formalin-fixed and paraffin-embedded control liver (A and B), Gbe1ys/ys liver (C and D), heart (E and F) and muscle (G and H) have been diastase treated and PAS stained (A, C, E and G) on the left hand side, while same sections were hybridized with pan ubiquitin antibody and stained by immunohistochemistry (B, D, F and H). Area in frame has been magnified to show the ubiquitin-free PGB (H). Liver sections were examined in 200×, and heart and muscle sections were examined in 400× magnifications. Solid bar represents 100 μm or as indicated.
Serum creatine kinase is increased in Gbe1ys/ys mice
Serum analytes were examined in Gbe1ys/ys mice. As we had previously observed in Gbeneo/neo mice (17), fasting glucose levels were lower in Gbe1ys/ys mice when compared with controls [50 versus 73 g/dl; P < 0.0024 (n = 6 and 6)]. We did not observe a significant change in alanine or aspartate transaminases. Excess accumulation of PGB adversely affects muscle homeostasis and causes the release of the muscle enzyme creatine kinase (CK). Gbe1ys/ys mice have elevated serum CK activity when compared with control animals [2947 U/l ± 361 versus 1312 U/l ± 624; P < 0.000013 (n = 4 for Gbe1ys/ys and 5 for wild type)].
Gbe1ys/ys neonates contain detectable PG
The pathogenic mechanism of GBE deficiency is still a subject of debate. Since we generated an animal model with the most common human GBE1 mutation, we examined whether younger animals that do not show any outward signs of the disease still have detectable PGB in tissues. Therefore, we tested whether glycogen is present in fetal tissues. Due to size limitations, we used whole-mount embryo sections and assessed the glycogen content by histochemistry. Whole embryos were fixed in 10% buffered formaldehyde, embedded in paraffin, sectioned and briefly treated with diastase to digest normal glycogen. Sections were then stained with periodic Schiff base (PAS). Limited digestion with diastase degrades almost all structurally normal glycogen, while longer and poorly branched PG remains intact and thus stains with PAS. Whole cross sections were examined by light microscopy. Sections of choroid plexus, brain stem, muscle, and dorsal root ganglion cells, liver, and epithelial cells of the brain had PGB (Fig. 4A–H, indicated by arrows). However, the heart (not shown) and the brain cerebral cortex did not exhibit any accumulation of PGB (Fig. 4I and J).
Figure 4.
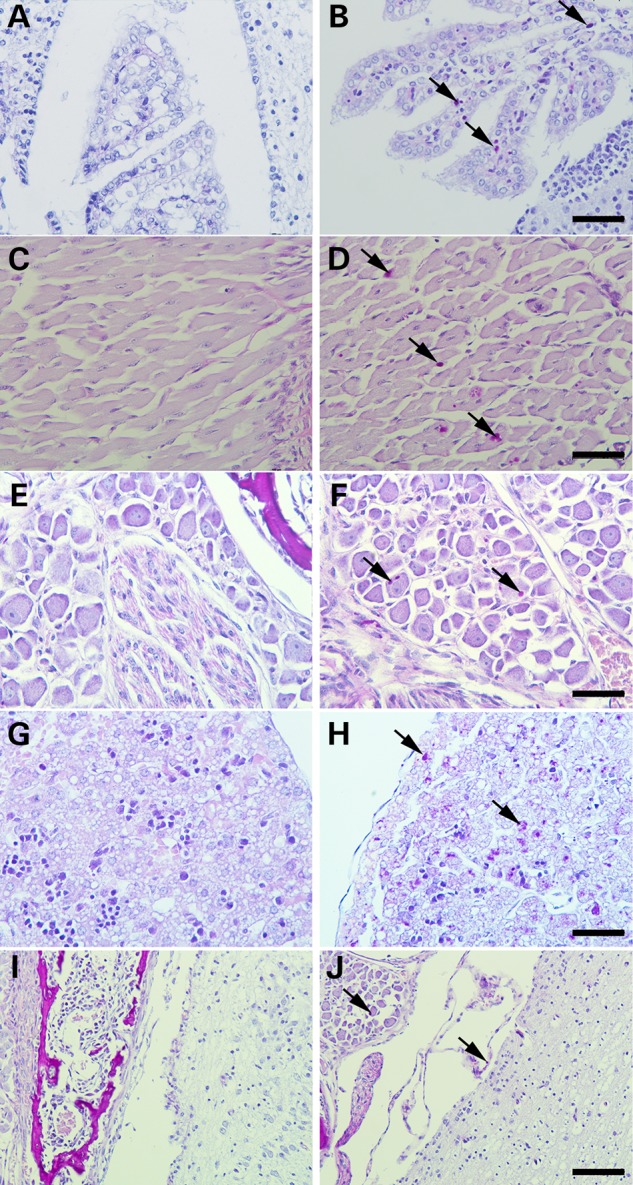
Newborn mice have significant accumulation of PGB in tissues except the heart. Formalin-fixed and paraffin-embedded wild-type (on the left) and Gbe1ys/ys neonates (on the right) were sectioned, treated with diastase and stained with PAS. Epithelial cells of choroid plexus (A and B 400×), muscle (C and D 400×), dorsal root ganglion cells (E and F 400×), liver (G and H 400×) and brain stem (I and J 200×). PGB are indicated by arrows.
Accumulated PGB are ubiquitinylated
Ubiquitinylation is the intracellular targeting mechanism that marks molecules or organelles for proteosomal or lysosomal degradation (22). Since PG is a poorly branched molecule, its abnormal structure may allow it to escape from the surveillance of ubiquitinylating enzymes that mark glycogen and its protein scaffold for lysosomal degradation, similar to the interruption of glycogen particle ubiquitinylation observed in Lafora's disease (LD) (23). In order to study the ubiquitinylation status of PG in GBE-deficient mice, heart and skeletal muscle samples were prepared from 1-year-old mice and examined for PGB accumulation as described above, together with immunohistochemistry. Liver, heart and skeletal muscle sections exhibit strong staining of PGB with anti-ubiquitin antibodies, which is absent in control liver samples. However, not all PGB co-stained with ubiquitin in the muscle and the heart (Fig. 5E–H), suggesting that PG-associated proteins can selectively be ubiquitinylated but can also remain intact in the cell protected from lysosomal degradation.
Discussion
GBE deficiency is a rare disorder with a very heterogeneous clinical presentation that appears to be determined in part by the degree of residual enzyme activity. Complete loss of activity in humans is lethal either in the third trimester of pregnancy or in infancy (Andersen's disease), while mutations that reduce enzyme activity cause juvenile- or adult-onset disease. Juvenile-onset disease typically exhibits liver and/or heart involvement, while adult-onset disease is mainly a neuromuscular disorder and may be misdiagnosed as amyotrophic lateral sclerosis, multiple sclerosis or Alzheimer's disease (5,7,8,24). Here, we describe a series of mouse models of GSD IV that accurately recapitulate the histological, biochemical and clinical spectrum of disease. Deletion of exon 7 eliminates enzyme activity in all tissues and thus is a model of Andersen's disease. In contrast, decreasing the expression of the Gbe1 via transcriptional interference by the reverse-oriented PGK-neomycin cassette and/or introduction of the p.Y329S mutation found in the Ashkenazi Jewish population both lead to hypomorphic alleles with residual enzyme activity and to a later onset disease (Fig. 2A, and Supplementary Material, Fig. S1), but with pronounced accumulation of PGB. Exon 7 of Gbe1 contains the c-terminus of the alpha amylase domain and the linker region that connects the alpha amylase domain to the glycosyl-transferase domain. Since exon 7 encodes an in-frame 70-amino-acid polypeptide, we have shown that its deletion leads to an unstable protein. These pups are born full term but fail to survive. Could this be due to pathogenic features of PG or, as has been proposed (25), to secondary reduction of GYS activity? If PG has a toxic effect, we should observe the same severe phenotype in animals with the p.Y329S mutation since their tissues contain more PGB than exon 7-deleted animals. We can potentially explain the lack of viability by the absence of digestible glycogen in the complete GBE deficiency state. On the other hand, the p.Y329S mutation decreases enzyme activity but still allows for the formation of poorly branched glycogen, which can provide free glucose better than unbranched glycogen and presumably enough glucose for the newborn after birth. However, a lack of glycogen in tissues has been reported previously in the absence of GYS (26,27), which, unlike GBE, has two isoforms: the liver-specific GYS2 and GYS1 expressed in other tissues. GYS2-deficient mice have near-normal development, whereas 90% of GYS1-deficient mice die after birth. Thus, given that there is a single Gbe1 locus, Gbe1−/− mice mimic the course of GYS1-deficient mice, but, like GYS2-deficient mice, they also lack significant amounts of liver glycogen in the presence of readily detectable PGB. Hence, the early death of GBE-deficient mice is not likely to be due to the absence of structurally normal glycogen in the liver, but rather to its deficiency in other tissues. Although we did not observe hydropic embryos or left ventricular non-compaction, the hearts of Gbe1−/− mice appear to have disrupted myocardial architecture in association with widespread PGB accumulation (17). Future studies in Gbe1−/− mice will address the molecular basis for this structurally abnormal myocardium.
Although low GBE activity caused by the PGK-neomycin cassette and the p.Y329S mutation in newborn pups does not cause complications immediately after birth, the residual enzyme activity determines the life span of the mice. This clearly indicates that accumulated PG begins to affect muscle and nerve functions causing premature death, similar to cases with more severe mutations causing the juvenile form of GSD IV (19).
The PGK-neomycin cassette used for positive selection in mouse embryonic stem (ES) cells alters the expression of Gbe1. Both GBE activity assays and western blotting have shown that transcription of the Gbe1 gene is reduced. This has generated a phenotype similar to the juvenile and adult forms of GSD IV. Point mutations that decrease enzyme activity, such as Y329S and R545H, cause amylopectinosis (aka APBD) as a recessive trait. Thus, in order to manifest the disease, the enzyme activity must be <50% (28). Previously there were rare case reports of manifesting heterozygotes, with clinical features occurring in the sixth decade of life (29); however, these patients were recently shown to have a deep intronic mutation not detected by standard DNA sequencing (30). In aged humans and animals, PG-like bodies known as corpora amylacea can be seen in white matter and axons, but the relationship to GBE activity remains unexplored (31). Gbe1+/ys mice are being systematically aged to determine whether a reduction in GBE can lead to the formation of PGB in the brain.
PG is also a component of the Lafora bodies that lead to juvenile myoclonic epilepsy (Lafora's disease). Lafora's disease is an autosomal recessive disorder with typical disease onset in teenage years and progressive myoclonic epilepsy leading to death within a decade. The Lafora bodies are found in the perikarya of neurons and other cell types. The pathogenic mechanism of LD does not involve the glycogenolytic enzymes per se but is caused by the functional loss of regulatory enzymes that may indirectly affect glycogenolytic enzymes (32). Two genes causing LD, EPM2A and NHLRC1 (EPM2B), have been identified (33,34). An animal model of LD has also been described; however, the exact mechanism of PG formation is not well understood (35–37). Interestingly, breeding GYS1-deficient mice to LD mice did prevent LD and decrease the pathogenicity of PG in disease (38).
Although we have shown that PGB accumulating in the liver, heart and muscle are ubiquitinylated, the PG is not degraded, perhaps because the degradation mechanisms are overwhelmed or impaired. Ubiquitination is a signal for proteosomal or lysosomal degradation, and ubiquitin-rich inclusions are seen in other diseases (39). In Gbe1ys/ys mice heart and brain tissues, all PGB were ubiquitinylated; however, skeletal muscle tissue showed some PGB free of ubiquitin. This suggests that either there is selective ubiquitinylation in muscle or that newly synthesized PG has not yet been ubiquitinylated. The former model, selective ubiquitinylation of such large molecules, is not likely. Second, as muscle has a very high turnover of glycogen, the latter model, that newly synthesized PG are not ubiquitinylated, appears more tenable. Although ubiquitination was identified as a marking mechanism in order to degrade molecules or organelles, it also activates or inactivates marked proteins similar to phosphorylation. Ubiquitylated proteins in PGB may have different functions, including intracellular trafficking of glycogen and/or activation/deactivation of associated proteins. We will use this new model to further study the mechanism of ubiquitylation of PGB.
In summary, we have generated a novel mouse model of APBD using the most common mutation found in the Ashkenazi Jewish population. Similar to APBD patients, PGB accumulate in muscle, brain, heart and liver and cause premature death. Stiffness in the hind limbs after 1 year of age is consistent with the natural history of the disease (please see the Supplementary Material, Video S1) (8). As affected mice cannot use their hind limbs, further studies are needed to address whether the problem is a combination of upper and/or lower motor neuron dysfunction. This model can also be used to develop possible treatment strategies that enhance the physiologic degradation of PG. The recent report that enzyme replacement therapy with recombinant acid α-glucosidase may benefit glycogen debranching deficient patients (40) suggests the possibility of a similar approach for GBE deficiency. Therefore, this mouse model will serve as a useful tool for examining the biology of PGB formation and its degradation, and as a means for testing possible therapeutic approaches.
Materials and Methods
Knock-in of p.Y329S mutation into Gbe1
The Gbe1 targeting vector was assembled by PCR-amplified and restriction enzyme-digested fragments from ES 129Sv mouse DNA. A 1020 bp NheI fragment containing exon 7 was cloned in pUC18 vector and modified by inserting FRT oligos upstream and downstream of exon 7. TAC to TCC change corresponding to 329th codon was introduced by site-directed mutagenesis. A phosphoglycerate kinase (PGK) promoter-neomycin resistance cassette flanked by loxP sites was cloned 5′ to the FRT site downstream of seventh exon for positive drug selection. The final construct contained a 2.1 kb 5′ short recombination arm composed of a portion of intron 6, the upstream FRT site followed by exon 7, the PGK-neomycin resistance cassette for positive selection flanked by loxP sites, and the FRT site on the 3′ end, followed by a 6.5 kb-long recombination arm spanning intron 7. Finally, a MC1-driven thymidine kinase-1 gene was incorporated into the plasmid for negative selection against non-homologous recombination (see Fig. 1). All of the junctions in the final construct were confirmed by sequencing and restriction enzyme digestion. The NotI-linearized vector was electroporated into AB2.2 ES cells by the mouse transgenic core facility at Baylor College of Medicine, Houston, TX. Selection of ES cells used Ganciclovir® as a negative selection drug and G418 for positive selection. Cells surviving in the presence of G418 were screened by PCR for appropriately targeted integration. Positive ES cell clones were confirmed by additional PCR and Southern analyses to contain the two FRT sites to be correctly targeted at both the 5′ and 3′ ends of exon 7. The ES cells were used to generate chimeric mice by blastocyst microinjection, and these mice bred to confirm transmission of the targeted allele.
Genotyping wild-type and Cre-recombined alleles
Tail DNA was isolated by standard techniques and the Gbe1 locus PCR amplified using primers, 5′ AGC TTT GGT TAT AGA CGA ATC ACT, b, 5′ GTC TAT GTC CAG CAC AGT ATT AAG GA and c.-5′ TCC TGA AAT GGG ATA TAT GGG ATA TG. PCR reaction was prepared as described in the vendor's protocol (New England BioLabs) Thermal cycles were programmed for touchdown PCR as follows: initial 5 min denaturation at 95°C followed by touchdown PCR cycles with the following steps: denaturation at 95°C for 30 min, annealing at 65°C decreasing 0.5°C every cycle for the next 20 cycle and 72°C extension. After the touchdown protocol, 20 cycles of regular PCR was carried out starting with 95°C denaturation for 30 s, annealing at 55°C for 30 s and primer extension at 72°C for 45 s. Amplified fragments are separated on 2% agarose gel and photographed.
Preparation of samples for biochemical analyses
Four to six months old adult mice and 4-month-old female mice on the 17th day of pregnancy were anesthetized by IsoThesia™ (Butler Animal Health Supply Dublin, OH) inhalation and sacrificed by cervical dislocation. The embryos and the tissues of adult animals were immediately frozen in liquid nitrogen and stored at −80°C for further analyses. Tissues were cut, weighed and homogenized in the assay buffers as described below.
Western analysis
For western analysis, 30 µg of tissue homogenate was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes and incubated with monoclonal antibodies raised against human GBE and GYS1 (Origene Rockville, MD). Detection was achieved with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence.
Quantification of glycogen
Glycogen content was estimated by measuring glucose released by amyloglucosidase digestion of ethanol-precipitated glycogen from muscle or liver tissue, as described (4,1). Samples of frozen muscle and liver tissue (∼30–60 mg) were boiled in 200 µl of 30% (wt/vol) KOH for 30 min with occasional shaking. After cooling, 67 µl of 0.25 M Na2SO4 and 535 µl of ethanol were added. Next, samples were centrifuged at 14 500g for 20 min at 4°C to collect glycogen. The glycogen pellet was suspended in water (100 µl), 200 µl of ethanol was added and centrifugation as described above was used to harvest glycogen. This ethanol precipitation step was repeated, and the glycogen pellet was dried in a Speed-Vac. Dried glycogen pellets were suspended in 100 µl of amyloglucosidase [0.3 mg/ml in 0.2 M sodium acetate (pH 4.8)] and incubated at 37°C for 3 h to digest glycogen. To determine the glucose concentration in the samples, an aliquot (5 µl) of digested glycogen was added to 95 µl of a solution containing 0.3 M triethanolamine (pH 7.6), 0.4 mm MgCl2, 0.9 mm nicotinamide adenine dinucleotide phosphate, 1 mm ATP and 0.1 µg/ml glucose-6-phosphate dehydrogenase. The absorbance at 340 nm was read before and after the addition of 0.1 µg of hexokinase.
Alanine and aspartate transaminase and creatine kinase assays
Five microliters of serum, obtained from six 1-year-old male wild-type and Gbe1ys/ys mice, were used in ALT, AST (Genzyme corp USA) and CPK (Pointe Scientific, Canton, MI) enzyme assays. Serum was separated from the blood clot. Blood was drawn by cardiac puncture right after carbon dioxide asphyxiation. Enzyme activities were measured by spectrophotometry as described by the supplier.
Glycogen-branching enzyme activity
GBE activity was assayed as described by Tay et al. (20). Briefly, frozen tissue samples were homogenized in all-glass homogenizers in nine volumes of 5 mm Tris, 1 mm EDTA, 5 mm mercaptoethanol, pH 7.2 and centrifuged at 9200g for 10 min. Branching enzyme activity was measured by an indirect assay based upon incorporation of radioactive glucose-1-phosphate (PerkinElmer Life and Analytical Sciences, Boston, MA) into glycogen by the reverse activity of phosphorylase a (Sigma St. Lois, MO) as an auxiliary enzyme. At 30, 45 and 60 min time intervals, 5 µl of reaction mix was spotted on Whatman® Number 5 qualitative filter paper (Maidstone, UK). Unincorporated glucose-1-phosphate was washed for 15 min using three changes of 66% v/v ethanol. Glycogen-bound radioactive glucose-1-phosphate was quantified by liquid scintillation counter (Packard Instruments, Boston, MA).
Tissue staining and histochemistry
Tissue sections were prepared from 4-month-old mice, fixed in 10% formalin and embedded in paraffin. Slices of 5 µm were deparaffinized, and one slide of each sectioned block was treated with 40 ml of 5 µg/ml α-amylase (Sigma) for 25 s in a microwave oven set to 600 watts, the slides washed with deionized water, and oxidized with 0.5% periodic acid for 5 min, stained with Schiff reagent for 15 min and then counterstained in hematoxylin for 15 min and rinsed in tap water. The slides were then examined by light microscopy (Nikon Eclipse90i, Melville, NY). Gomori trichrome staining was performed at the Columbia University pathology services.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by a Muscular Dystrophy Association Development Grant (MDA10027); the Adult Polyglucosan Body Disease Research Foundation; The Keith B. Hayes Foundation and the Intellectual and Developmental Disabilities Research Center and Digestive Diseases Center at Baylor College of Medicine. The Intellectual and Developmental Disabilities Research Center is funded by award number P30HD024064 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Human Development or the National Institutes of Health.
Supplementary Material
References
- 1.Andersen D.H. (1956) Familial cirrhosis of the liver with storage of abnormal glycogen. Lab. Invest., 5, 11–20. [PubMed] [Google Scholar]
- 2.Suzuki K., David E., Kutschman B. (1971) Presenile dementia with ‘Lafora-like’ intraneuronal inclusions. Arch. Neurol., 25, 69–80. [DOI] [PubMed] [Google Scholar]
- 3.Cafferty M.S., Lovelace R.E., Hays A.P., Servidei S., Dimauro S., Rowland L.P. (1991) Polyglucosan body disease. Muscle Nerve, 14, 102–107. [DOI] [PubMed] [Google Scholar]
- 4.Gray F., Gherardi R., Marshall A., Janota I., Poirier J. (1988) Adult polyglucosan body disease (APBD). J. Neuropathol. Exp. Neurol., 47, 459–474. [DOI] [PubMed] [Google Scholar]
- 5.McDonald T.D., Faust P.L., Bruno C., DiMauro S., Goldman J.E. (1993) Polyglucosan body disease simulating amyotrophic lateral sclerosis. Neurology, 43, 785–790. [DOI] [PubMed] [Google Scholar]
- 6.Peress N.S., DiMauro S., Roxburgh V.A. (1979) Adult polysaccharidosis. Clinicopathological, ultrastructural, and biochemical features. Arch. Neurol., 36, 840–845. [DOI] [PubMed] [Google Scholar]
- 7.Robitaille Y., Carpenter S., Karpati G., DiMauro S.D. (1980) A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora's disease and normal ageing. Brain, 103, 315–336. [DOI] [PubMed] [Google Scholar]
- 8.Mochel F., Schiffmann R., Steenweg M.E., Akman H.O., Wallace M., Sedel F., Laforet P., Levy R., Powers J.M., Demeret S. et al. (2012) Adult polyglucosan body disease: natural history and key magnetic resonance imaging findings. Ann. Neurol., 72, 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson N.P., Wharton S., Anderson J., Scolding N.J. (1998) Adult polyglucosan body disease associated with an extrapyramidal syndrome. J. Neurol. Neurosurg. Psychiatry, 65, 788–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bit-Ivan E.N., Lee K.H., Gitelman D., Weintraub S., Mesulam M., Rademakers R., Isaacs A.M., Hatanpaa K.J., White C.L. III, Mao Q. et al. (2014) Adult polyglucosan body disease with GBE1 haploinsufficiency and concomitant frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol., 40, 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malfatti E., Nilsson J., Hedberg-Oldfors C., Hernandez-Lain A., Michel F., Dominguez-Gonzalez C., Viennet G., Akman H.O., Kornblum C., Van den Bergh P. et al. (2014) A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Ann. Neurol., 76, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nilsson J., Schoser B., Laforet P., Kalev O., Lindberg C., Romero N.B., Davila Lopez M., Akman H.O., Wahbi K., Iglseder S. et al. (2013) Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann. Neurol., 74, 914–919. [DOI] [PubMed] [Google Scholar]
- 13.Ban H.R., Kim K.M., Jang J.Y., Kim G.H., You H.W., Kim K., Yu E., Kim D.Y., Kim K.H., Lee Y.J. et al. (2009) Living donor liver transplantation in a Korean child with glycogen storage disease type IV and a GBE1 mutation. Gut Liver, 3, 60–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fyfe J.C., Giger U., Van Winkle T.J., Haskins M.E., Steinberg S.A., Wang P., Patterson D.F. (1992) Glycogen storage disease type IV: inherited deficiency of branching enzyme activity in cats. Pediatr. Res., 32, 719–725. [DOI] [PubMed] [Google Scholar]
- 15.Valberg S.J., Ward T.L., Rush B., Kinde H., Hiraragi H., Nahey D., Fyfe J., Mickelson J.R. (2001) Glycogen branching enzyme deficiency in quarter horse foals. J. Vet. Intern. Med., 15, 572–580. [DOI] [PubMed] [Google Scholar]
- 16.Lee Y.C., Chang C.J., Bali D., Chen Y.T., Yan Y.T. (2011) Glycogen-branching enzyme deficiency leads to abnormal cardiac development: novel insights into glycogen storage disease IV. Hum. Mol. Genet., 20, 455–465. [DOI] [PubMed] [Google Scholar]
- 17.Akman H.O., Sheiko T., Tay S.K., Finegold M.J., Dimauro S., Craigen W.J. (2011) Generation of a novel mouse model that recapitulates early and adult onset glycogenosis type IV. Hum. Mol. Genet., 20, 4430–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akman H.O., Raghavan A., Craigen W.J. (2011) Animal models of glycogen storage disorders. Prog. Mol. Biol. Transl. Sci., 100, 369–388. [DOI] [PubMed] [Google Scholar]
- 19.Bruno C., van Diggelen O.P., Cassandrini D., Gimpelev M., Giuffre B., Donati M.A., Introvini P., Alegria A., Assereto S., Morandi L. et al. (2004) Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology, 63, 1053–1058. [DOI] [PubMed] [Google Scholar]
- 20.Tay S.K., Akman H.O., Chung W.K., Pike M.G., Muntoni F., Hays A.P., Shanske S., Valberg S.J., Mickelson J.R., Tanji K., DiMauro S. (2004) Fatal infantile neuromuscular presentation of glycogen storage disease type IV. Neuromuscul. Disord., 14, 253–260. [DOI] [PubMed] [Google Scholar]
- 21.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A. et al. (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology, 41, 1313–1321. [DOI] [PubMed] [Google Scholar]
- 22.Jiang S., Wells C.D., Roach P.J. (2011) Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem. Biophys. Res. Commun., 413, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiberia E., Turnbull J., Wang T., Ruggieri A., Zhao X.C., Pencea N., Israelian J., Wang Y., Ackerley C.A., Wang P., Liu Y., Minassian B.A. (2012) Increased laforin and laforin binding to glycogen underlie Lafora body formation in malin-deficient Lafora disease. J. Biol. Chem., 287, 25650–25659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abel T.J., Hebb A.O., Keene C.D., Born D.E., Silbergeld D.L. (2010) Parahippocampal corpora amylacea: case report. Neurosurgery, 66, E1206–E1207. [DOI] [PubMed] [Google Scholar]
- 25.Kakhlon O., Glickstein H., Feinstein N., Liu Y., Baba O., Terashima T., Akman H.O., Dimauro S., Lossos A. (2013) Polyglucosan neurotoxicity caused by glycogen branching enzyme deficiency can be reversed by inhibition of glycogen synthase. J. Neurochem., 127, 101–113. [DOI] [PubMed] [Google Scholar]
- 26.Pederson B.A., Schroeder J.M., Parker G.E., Smith M.W., DePaoli-Roach A.A., Roach P.J. (2005) Glucose metabolism in mice lacking muscle glycogen synthase. Diabetes, 54, 3466–3473. [DOI] [PubMed] [Google Scholar]
- 27.Irimia J.M., Meyer C.M., Peper C.L., Zhai L., Bock C.B., Previs S.F., McGuinness O.P., DePaoli-Roach A., Roach P.J. (2010) Impaired glucose tolerance and predisposition to the fasted state in liver glycogen synthase knock-out mice. J. Biol. chem., 285, 12851–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao Y., Kishnani P., Wu J.Y., Chen Y.T. (1996) Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J. Clin. Invest., 97, 941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ubogu E.E., Hong S.T., Akman H.O., Dimauro S., Katirji B., Preston D.C., Shapiro B.E. (2005) Adult polyglucosan body disease: a case report of a manifesting heterozygote. Muscle Nerve, 32, 675–681. [DOI] [PubMed] [Google Scholar]
- 30.Akman H.O., Kakhlon O., Coku J., Peverelli L., Rosenmann H., Rozenstein-Tsalkovich L., Turnbull J., Meiner V., Chama L., Lerer I. et al. (2015) Deep Intronic GBE1 mutation in manifesting heterozygous patients with adult polyglucosan body disease. JAMA Neurol., 72, 441–445. [DOI] [PubMed] [Google Scholar]
- 31.Sinadinos C., Valles-Ortega J., Boulan L., Solsona E., Tevy M.F., Marquez M., Duran J., Lopez-Iglesias C., Calbo J., Blasco E. et al. (2014) Neuronal glycogen synthesis contributes to physiological aging. Aging Cell, 13, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ianzano L., Zhang J., Chan E.M., Zhao X.C., Lohi H., Scherer S.W., Minassian B.A. (2005) Lafora progressive myoclonus epilepsy mutation database-EPM2A and NHLRC1 (EPM2B) genes. Hum. Mutat., 26, 397. [DOI] [PubMed] [Google Scholar]
- 33.Chan E.M., Omer S., Ahmed M., Bridges L.R., Bennett C., Scherer S.W., Minassian B.A. (2004) Progressive myoclonus epilepsy with polyglucosans (Lafora disease): evidence for a third locus. Neurology, 63, 565–567. [DOI] [PubMed] [Google Scholar]
- 34.Minassian B.A., Lee J.R., Herbrick J.A., Huizenga J., Soder S., Mungall A.J., Dunham I., Gardner R., Fong C.Y., Carpenter S. et al. (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet., 20, 171–174. [DOI] [PubMed] [Google Scholar]
- 35.Ganesh S., Delgado-Escueta A.V., Sakamoto T., Avila M.R., Machado-Salas J., Hoshii Y., Akagi T., Gomi H., Suzuki T., Amano K. et al. (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet., 11, 1251–1262. [DOI] [PubMed] [Google Scholar]
- 36.Ianzano L., Young E.J., Zhao X.C., Chan E.M., Rodriguez M.T., Torrado M.V., Scherer S.W., Minassian B.A. (2004) Loss of function of the cytoplasmic isoform of the protein laforin (EPM2A) causes Lafora progressive myoclonus epilepsy. Hum. Mutat., 23, 170–176. [DOI] [PubMed] [Google Scholar]
- 37.Tagliabracci V.S., Roach P.J. (2010) Insights into the mechanism of polysaccharide dephosphorylation by a glucan phosphatase. Proc. Natl. Acad. Sci. USA, 107, 15312–15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pederson B.A., Turnbull J., Epp J.R., Weaver S.A., Zhao X., Pencea N., Roach P.J., Frankland P.W., Ackerley C.A., Minassian B.A. (2013) Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann. Neurol., 74, 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter-Landsberg C., Leyk J. (2013) Inclusion body formation, macroautophagy, and the role of HDAC6 in neurodegeneration. Acta Neuropathol., 126, 793–807. [DOI] [PubMed] [Google Scholar]
- 40.Sun B., Fredrickson K., Austin S., Tolun A.A., Thurberg B.L., Kraus W.E., Bali D., Chen Y.T., Kishnani P.S. (2013) Alglucosidase alfa enzyme replacement therapy as a therapeutic approach for glycogen storage disease type III. Mol. Genet. Metab., 108, 145–147. [DOI] [PubMed] [Google Scholar]
- 41.Pederson B.A., Chen H., Schroeder J.M., Shou W., DePaoli-Roach A.A., Roach P.J. (2004) Abnormal cardiac development in the absence of heart glycogen. Mol. Cell. Biol., 24, 7179–7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.