

Abstract
Cav1 channels mediate L-type Ca2+ currents that trigger the exocytotic release of glutamate from the specialized “ribbon” synapse of retinal photoreceptors (PRs) and cochlear inner hair cells (IHCs). Genetic evidence from animal models and humans support a role for Cav1.3 and Cav1.4 as the primary Cav channels in IHCs and PRs, respectively. Because of the unique features of transmission at ribbon synapses, Cav1.3 and Cav1.4 exhibit unusual properties that are well-suited for their physiological roles. These properties may be intrinsic to the channel subunit(s) and/or may be conferred by regulatory interactions with synaptic signaling molecules. This review will cover advances in our understanding of the function of Cav1 channels at sensory ribbon synapses, and how dysregulation of these channels leads to disorders of vision and hearing.
Graphical abstract
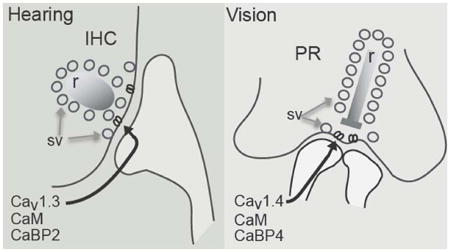
Cav1.3 and Cav1.4 channel complexes of inner hair cells (IHC) and photoreceptor (PR) cells, respectively, share a number of biophysical properties and modulatory proteins at ribbon synapses (r = ribbon, sv = synaptic vesicles).
Introduction
The perception of light and sound is initiated by photoreceptors (PRs) in the retina and inner hair cells (IHCs) in the cochlea, respectively. Both PRs and IHCs convert an environmental stimulus into an electrical signal that is communicated to second-order neurons and ultimately to the brain. An important structural feature of both PRs and IHCs is the synaptic ribbon. Associated with the presynaptic active zone, the ribbon tethers thousands of synaptic vesicles and allows for rapid and sustained release of glutamate in response to graded changes in membrane potential [1]. Voltage-gated Ca2+ channels are positioned within nanometers of the ribbon [2], allowing for tight coupling of depolarization-dependent Ca2+ influx and the molecular machinery involved in exocytosis. In contrast to the dominant role of Cav2 channels at most synapses in the central nervous system, Cav1 channels are required for exocytosis at ribbon synapses [3-5]. Visual and acoustic stimuli must be encoded continuously with high fidelity and sensitivity. This review will highlight the unique features and regulatory functions of Cav1 channels at the PR and IHC synapse, and their importance for faithful transmission of sensory stimuli.
Cav1.3 channels and hearing
In the cochlea, sound-induced vibrations displace hair bundles rooted in the apical surface of IHCs. The subsequent modulation of mechanoelectrical transduction currents causes a graded receptor potential that regulates the opening of Cav1 channels clustered near the presynaptic ribbon. Ca2+ influx through these channels triggers the exocytotic release of glutamate onto postsynaptic spiral ganglion neuron afferents, which transmit auditory information into the central nervous system. Even in the absence of sensory stimulation, IHC synapses are tonically active, and support spontaneous afferent firing rates that can exceed 100 Hz [6]. The intensity and timing of sound is encoded by an increase in steady-state afferent firing rates, and by the ability of IHCs to phase-lock transmitter release to frequencies up to 5 kHz, respectively.
Current evidence favors a crucial role for Cav1.3 channels as the major Cav channel in IHCs. For example, in mice lacking Cav1.3 channels (Cav1.3 KO), IHCs are incapable of evoked exocytosis and exhibit whole-cell Ca2+ currents that are ∼90% of that in IHCs from wild-type mice [7, 8]. As a consequence, Cav1.3 KO mice are deaf, with no evidence of sound-evoked afferent activity in auditory brainstem responses (ABR) [7, 9]. Other components of the Cav1.3 complex in IHCs include the auxiliary Cav β [10] and α2δ subunits.
Analyses of Cav1.3 channels in heterologous expression systems indicate a number of properties consistent with those of native Cav channels and their physiological demands in IHCs. First, Cav1.3 activates rapidly compared to the related Cav1.2 channel abundant in the brain and heart [11, 12]. Whole-cell and single-channel recordings of Ca2+ currents in IHCs indicate time constants for activation and latency to first opening in the submillisecond range [13-17]. Rapid activation kinetics of Cav1.3 channels are likely important for temporal aspects of sound coding, such as the rapid onset of sound and the ability to accurately trigger firing of the auditory nerve to reflect sound frequency (i.e., phase locking).
Second, Cav1.3 channels activate at relatively negative voltages [11, 12]. In single-channel recordings of immature mouse IHCs, channel openings were observed at voltages as negative as -70 mV [14]. Since the resting potential of these cells is ∼ -60 mV [18], the negative activation threshold of Cav1.3 would be able to support tonic transmitter release at rest. Moreover, the stronger activation of these channels in response to sound-evoked alterations in the receptor potential could encode increased sound intensity with greater rates of transmitter release.
Third, Cav1.3 channels in IHCs exhibit little Ca2+- or voltage- dependent inactivation (CDI, VDI), in contrast to their behavior in heterologous expression systems and in the heart [11, 16, 17, 19-23]. CDI is a negative feedback regulation mediated by permeating Ca2+ ions and relies on calmodulin (CaM) interacting with an IQ domain in the cytoplasmic C-terminal domain of the Cav1.3 α1 subunit. CDI is not seen with Ba2+ as the charge carrier since Ba2+ binds poorly to CaM, so Ba2+ currents exhibit VDI, which is generally slower than CDI, (reviewed in [24]). The low levels of CDI in IHCs are thought to support the continuous encoding of sound information through sustained transmitter release. Possible factors contributing to the limited CDI of Cav1.3 channels in IHCs include the expression of Cav1.3 variants lacking a functional IQ domain [25]. In addition, the synaptic scaffolding protein, Rab3-interacting molecule 2 (RIM2), inhibits CDI and VDI of Cav1.3 channels in transfected tsA-201 cells. However, RIM2 is primarily expressed in the immature cochlea [26], such that other factors must contribute to the slow CDI and VDI in mature IHCs.
One possibility concerns CaBPs, a family of CaM-like Ca2+ binding proteins that are highly expressed in the brain, retina, and inner ear [20, 27-31]. Like CaM, CaBPs have 4 EF-hand Ca2+ binding domains, at least one of which is non-functional [27]. Antibodies against CaBP1, CaBP2, CaBP4, and CaBP5 label both immature and mature IHCs [20, 28]. In transfected HEK293T cells, CaBPs suppress CDI of Cav1.3 [20, 28, 32]. The mechanism may involve displacement of CaM by CaBPs from the Cav1 α1 IQ domain [33-35]. However, CaBPs bind to additional sites in Cav1 α1, and so may allosterically modulate CaM interactions with the channel [36-38]. CaBP2 may be an important physiological regulator of Cav1.3 in IHCs since a mutation in the CaBP2 gene causes moderate-to severe sensorineural hearing loss. The mutation causes premature truncation of CaBP2, such that the mutant CaBP2 lacks the C-terminal EF-hands. In transfected HEK293T cells, the mutant CaBP2 is less able to suppress CDI of Cav1.3 channels [39]. While additional research is required, increased CDI of Cav1.3 currents may be insufficient to support sustained neurotransmitter release from IHCs required for proper sound coding.
The requirement for Cav1.3 for hearing is clearly illustrated by the profound deafness seen in individuals with a mutation in CACNA1D [40]. This mutation results in a glycine insertion into the transmembrane helix IS6 in Cav1.3 variants containing exon 8B. In transfected tsA-201 cells, mutant channels are trafficked to the plasma membrane but do not conduct Ca2+ currents. The mechanism may involve reduced ability of voltage-sensors to couple to pore opening, or some failure of conductance downstream of pore-opening. In addition to deafness, the affected individuals exhibit severe sinus bradycardia [40], consistent with the role of Cav1.3 in regulating sinoatrial pacemaking and with the cardiac phenotype of Cav1.3 KO mice [7, 19].
Dysregulation of Cav1.3 channels is implicated in additional forms of hearing impairment. For example, severe hearing loss can result from thyroid hormone (TH) deficiency, mutations in thyroid hormone receptor β, or iodine deficiency [41]. Studies of TH-deficient rodents indicate abnormal maturation of IHC ionic currents and physiology. In normal mouse IHCs, Cav1.3 currents and BK K+ currents normally undergo a developmental decline or increase, respectively, prior to the onset of hearing in mice (postnatal day (P)12) [42, 43]. However, Cav1.3 currents remain elevated, and BK currents are undetectable, in TH-deficient rats and mice [44, 45]. The abnormally elevated Cav1.3 current density may result from alterations in Cav1.3 trafficking or turnover, since the corresponding mRNA levels are not affected by TH deficiency [44]. Due to the maintained high levels of Cav1.3, spontaneous Ca2+-dependent action potentials, which normally disappear around P13 [8, 46], are still evident in TH-deficient mice [44, 45]. IHCs from these mice also exhibit abnormalities in afferent and efferent innervation that suggest that IHCs have not transitioned at the molecular or morphological level to support tonic, graded changes in neurotransmitter release in response to sound stimulation [44]. TH is required for multiple aspects of auditory development, including maturation of the tectorial membrane and the generation of the endocochlear potential [41]. The extent to which the synaptic defects in IHC maturation contribute to deafness due to TH deficiency remains an open question.
A failure to regulate Cav1.3 channels also is found in a mouse model of Usher syndrome – the most common cause of combined deafness and blindness in humans [47]. “Deaf-circler” mice (dfcr) bear a mutation in the harmonin gene, which is a target of human mutations causing Usher syndrome [48]. Harmonin contains PDZ-domains, which are well-established motifs for protein-protein interactions [49]. Harmonin binds to a consensus PDZ binding site at the C-terminus of the Cav1.3 α1 subunit, and this interaction inhibits Cav1.3 current density in transfected HEK293T cells. The mechanism involves enhanced proteosomal degradation of Cav1.3, which can be reversed by the proteosomal inhibitor MG132. The dfcr mutation prevents harmonin binding to Cav1.3 and causes increased Cav1.3 current density in dfcr compared to control IHCs [50]. Immunolabeling for harmonin at IHC synapses increases after hearing onset (P12) [50]. These results support a model in which harmonin promotes developmental trimming of Cav1.3 channels, which may be important for IHC maturation. Harmonin also enhances voltage-dependent facilitation of Cav1.3 channels [51], which may be important for boosting Cav1.3 function during sound stimulation. The primary cause of deafness associated with harmonin mutations is likely not to involve Cav1.3 channels given that harmonin is also a key regulatory of mechanosensory channels in IHC hair bundles [52]. However, synaptic abnormalities due to altered regulation of Cav1.3 trafficking and function could exacerbate the mechanosensory defect.
Cav1.4 channels and vision
In the retina, rod PRs mediate vision in dim light, while cone PRs mediate daytime vision and color perception. At the relatively depolarized membrane potential of PRs in darkness (∼ -40 mV), the opening of Cav1 channels clustered near the presynaptic ribbon triggers the sustained release of glutamate, which inhibits postsynaptic ON bipolar cells. Light-dependent hyperpolarization suppresses Cav1-dependent glutamate release, which depolarizes ON bipolar cells, ultimately enhancing the activity of third-order retinal ganglion cells that transmit visual information into the brain. Cone PRs form synapses with multiple types of ON or OFF bipolar neurons, which are either depolarized or hyperpolarized, respectively, by light stimulation of cones.
The major Cav1 channel at PR synapses is Cav1.4, which localizes in rod and cone PR terminals in the outer plexiform layer of the retina [53-57]. In mice with genetic inactivation of the Cav1.4 α1 subunit (Cav1.4 KO), Ca2+ entry into PRs in response to depolarization is substantially impaired [58]. In electroretinograms, which measure visual function, Cav1.4 KO mice show no evidence of synaptic transmission between PRs and bipolar cells [56, 58]. However, Cav1.4 KO mice also exhibit PR degeneration with age [59] and defects in PR synapse morphology [58, 60] due to a failure in synapse maturation [56, 59, 61]. In addition to Cav1.4 α1, the Cav1.4 complex at PR synapses likely includes auxiliary Cavβ2 and α2δ4 subunits. Mice with reduced expression of Cavβ2 and α2δ4 exhibit a similar visual and synaptic phenotype as in Cav1.4 KO mice [62, 63]. Immunochemical studies support the association of Cav1.4 α1 with Cavβ2 and α2δ4 in mouse retina [64].
Cav1.4 channels in heterologous expression systems exhibit properties similar to native Cav1 channels in PRs [65]. Compared to Cav1.2 channels, Cav1.4 channels activate at more negative voltages, which allows for tonic transmitter release at the dark membrane potential [66-68]. Although the IQ domain is conserved in Cav1.4 α1, a C-terminal modulatory domain (CTM) of this channel inhibits CDI. The mechanism involves an intramolecular interaction between the CTM and a proximal region of the Cav1.4 α1 C-terminal domain, which inhibits Ca2+-free (apo) CaM interactions with the IQ domain [69-72]. As for Cav1.3 channels at the IHC synapse, limited CDI of Cav1.4 channels at PR synapses supports the continuous release of glutamate that is required for sensory encoding.
In contrast to the presence of multiple CaBPs in IHCs, CaBP4 is the only CaBP family member expressed in PRs. CaBP4 interacts with and colocalizes with Cav1.4 in the outer plexiform layer of the mouse retina [29, 56, 64]. While CaBP4 does not regulate CDI of full-length Cav1.4 channels, deletion of the ICDI allows CaBP4 suppression of Cav1.4 CDI [73]. CaBP4 binds to the IQ domain in Cav1.4 α1 and inhibits the intermolecular binding of the CTM with the proximal Cav1.4 α1 C-terminal site [73]. In transfected HEK293T cells, CaBP4 enhances voltage-dependent activation and reduces VDI of Cav1.4 channels [29, 73]. The net increase in Cav1.4 channel availability due to CaBP4 may be important for facilitating glutamate release from PRs in darkness. Consistent with this hypothesis, mice lacking CaBP4 (CaBP4 KO) exhibit visual and retinal phenotypes similar to those in Cav1.4 KO mice. In addition to significantly impaired light-modulated PR transmission, the morphology of PR synapses is abnormal [29, 56].
A requirement for Cav1.4 in human vision is supported by the multiple visual disorders associated with mutations in the gene encoding Cav1.4 α1, CACNA1F. The visual phenotypes associated with loss-of function of Cav1.4 are generally less severe in humans than in mouse models of the disease, which may be due to the normal or compensatory expression of Cav1.3 channels in human PRs. These disorders include congenital stationary night blindness type 2 (CSNB2), which causes heterogeneous symptoms that may include low visual acuity, myopia, and nystagmus [74-77]. Despite the name, night blindness is not always associated with CSNB2 phenotypes [78]. Other disorders associated with CACNA1F mutations include Åland island eye disease [79, 80] and cone-rod dystrophy (CORDX) [74, 81, 82]. Visual phenotypes of these disorders are similar to those in CSNB2, although there is early and more severe loss of cone function in CORDX compared to in CSNB2 [83]. An autosomal recessive form of cone-rod dystrophy is caused by mutations in the CACNA2D4 gene encoding α2δ4 [82], consistent with its importance as a Cav1.4 subunit [64].
The clinical variability associated with CACNA1F mutations may result from the functional impact of a particular mutation on Cav1.4 channels. In electrophysiological analyses in heterologous expression systems, CSNB2 mutant Cav1.4 channels may exhibit a loss-of function, gain-of function, and sometimes no observable difference compared to wild-type channels [68, 84]. For example, one CSNB2 mutation causes substitution of a proline for leucine860 (L860P), which is located in a conserved amphipathic helix in the loop connecting domains II and III. L860P reduces the number of function Cav1.4 channels at the cell surface by increasing the turnover of the protein [85]. While this would conceivably reduce Ca2+ signals supporting PR glutamate release in darkness, it is also possible that PR synapse development would be impaired, given that PR synapses do not form in Cav1.4 KO mice [56, 59, 61]. More complex mechanisms may result from truncating mutations that delete the CTM from Cav1.4 (K1591X [86], R1827X [87]). These mutations cause a loss-of function by increasing CDI, but also a gain-of function through enhance voltage-dependent activation, a process which the CTM normally suppresses[71, 85]. A mouse model of a gain-of function CSNB2 mutation (I745T) has provided some insights into the underlying pathophysiological mechanisms. This mutation causes an unusually severe form of CSNB2, and a large (-30 mV) shift in the voltage-dependence of Cav1.4 activation [88]. I745T knock-in mice exhibit abnormal PR synapse morphology, reduced cone and rod transmission, and degeneration of PRs due to apoptosis [55, 59].
Two mutations in the gene encoding CaBP4 also contribute to visual phenotypes similar to those in patients with CACNA1F mutations. One mutation is a valine substitution for glutamate and a frameshift that elongates the protein (E267fs). The second is a truncating mutation (R216X) that deletes the C-terminal EF-hands of CaBP4 [89, 90], which is essentially identical in nature to the CaBP2 mutation that causes autosomal recessive hearing loss [32]. These mutations do not affect the ability of CaBP4 to bind to Cav1.4, but prevent effects of CaBP4 on voltage-dependent activation and inactivation [73]. Decreased Cav1.4 channel availability would be expected to inhibit levels of PR glutamate release, thus reducing the dynamic range of PR signaling in these patients.
Summary
In IHCs and PRs, respectively, Cav1.3 and Cav1.4 share a number of biophysical properties that are important for their roles in mediating tonic neurotransmitter release at relatively negative membrane potentials, which is modulated by sensory stimuli. The growing number of visual and auditory disorders linked to altered Cav1.3 and Cav1.4 function emphasizes the need to better understand the roles and regulation of these channels in their native cell-types. Complicating matters is the fact that both Cav1.3 and Cav1.4 are expected to interact with a variety of proteins [91, 92]. Defining how genetic alterations in Cav1.3 and Cav1.4, and their interacting proteins, affect the localization and function of these channels in IHCs and PR remains an important challenge for understanding the pathophysiology of the associated channelopathies, as well as for the development of novel therapies.
References
- 1.Logiudice L, Matthews G. The role of ribbons at sensory synapses. Neuroscientist. 2009;15(4):380–391. doi: 10.1177/1073858408331373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tachibana M, Okada T, Arimura T, Kobayashi K, Piccolino M. Dihydropyridine-sensitive calcium current mediates neurotransmitter release from bipolar cells of the goldfish retina. J Neurosci. 1993;13(7):2898–2909. doi: 10.1523/JNEUROSCI.13-07-02898.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitz Y, Witkovsky P. Dependence of photoreceptor glutamate release on a dihydropyridine-sensitive calcium channel. Neuroscience. 1997;78:1209–1216. doi: 10.1016/s0306-4522(96)00678-1. [DOI] [PubMed] [Google Scholar]
- 5.Zhang SY, Robertson D, Yates G, Everett A. Role of L-type Ca(2+) channels in transmitter release from mammalian inner hair cells I. Gross sound-evoked potentials. J Neurophysiol. 1999;82(6):3307–1335. doi: 10.1152/jn.1999.82.6.3307. [DOI] [PubMed] [Google Scholar]
- 6.Liberman MC. Auditory-nerve response from cats raised in a low-noise chamber. J Acoust Soc Am. 1978;63(2):442–455. doi: 10.1121/1.381736. [DOI] [PubMed] [Google Scholar]
- 7.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102(1):89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 8.Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23(34):10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dou H, Vazquez AE, Namkung Y, Chu H, Cardell EL, Nie L, Parson S, Shin HS, Yamoah EN. Null mutation of alpha1D Ca2+ channel gene results in deafness but no vestibular defect in mice. J Assoc Res Otolaryngol. 2004;5(2):215–226. doi: 10.1007/s10162-003-4020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neef J, Gehrt A, Bulankina AV, Meyer AC, Riedel D, Gregg RG, Strenzke N, Moser T. The Ca2+ channel subunit beta2 regulates Ca2+ channel abundance and function in inner hair cells and is required for hearing. J Neurosci. 2009;29(34):10730–10740. doi: 10.1523/JNEUROSCI.1577-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. α1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276(25):22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 12.Xu W, Lipscombe D. Neuronal Cav1.3 α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21(16):5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zampini V, Johnson SL, Franz C, Knipper M, Holley MC, Magistretti J, Masetto S, Marcotti W. Burst activity and ultrafast activation kinetics of CaV1.3 Ca(2)(+) channels support presynaptic activity in adult gerbil hair cell ribbon synapses. J Physiol. 2013;591(Pt 16):3811–3820. doi: 10.1113/jphysiol.2013.251272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zampini V, Johnson SL, Franz C, Lawrence ND, Munkner S, Engel J, Knipper M, Magistretti J, Masetto S, Marcotti W. Elementary properties of CaV1.3 Ca(2+) channels expressed in mouse cochlear inner hair cells. J Physiol. 2010;588(1):187–199. doi: 10.1113/jphysiol.2009.181917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zampini V, Valli P, Zucca G, Masetto S. Single-channel L-type Ca2+ currents in chicken embryo semicircular canal type I and type II hair cells. J Neurophysiol. 2006;96(2):602–612. doi: 10.1152/jn.01315.2005. [DOI] [PubMed] [Google Scholar]
- 16.Inagaki A, Lee A. Developmental alterations in the biophysical properties of Ca(v) 1.3 Ca(2+) channels in mouse inner hair cells. Channels. 2013;7(3):171–181. doi: 10.4161/chan.24104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnee ME, Ricci AJ. Biophysical and pharmacological characterization of voltage-gated calcium currents in turtle auditory hair cells. J Physiol. 2003;549(3):697–717. doi: 10.1113/jphysiol.2002.037481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcotti W, Johnson SL, Holley MC, Kros CJ. Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J Physiol. 2003;548(Pt 2):383–400. doi: 10.1113/jphysiol.2002.034801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100(9):5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA, Yue DT. Switching of Ca2+-dependent inactivation of Cav1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26(42):10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant L, Fuchs P. Calcium- and calmodulin-dependent inactivation of calcium channels in inner hair cells of the rat cochlea. J Neurophysiology. 2008;99(5):2183–2193. doi: 10.1152/jn.01174.2007. [DOI] [PubMed] [Google Scholar]
- 22.Lee S, Briklin O, Hiel H, Fuchs P. Calcium-dependent inactivation of calcium channels in cochlear hair cells of the chicken. J Physiol. 2007;583(Pt 3):909–922. doi: 10.1113/jphysiol.2007.135582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson SL, Marcotti W. Biophysical properties of CaV1.3 calcium channels in gerbil inner hair cells. J Physiol. 2008;586(4):1029–1042. doi: 10.1113/jphysiol.2007.145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ben-Johny M, Yue DT. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol. 2014;143(6):679–692. doi: 10.1085/jgp.201311153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen Y, Yu D, Hiel H, Liao P, Yue DT, Fuchs PA, Soong TW. Alternative splicing of the Cav1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J Neurosci. 2006;26(42):10690–10699. doi: 10.1523/JNEUROSCI.2093-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gebhart M, Juhasz-Vedres G, Zuccotti A, Brandt N, Engel J, Trockenbacher A, Kaur G, Obermair GJ, Knipper M, Koschak A, Striessnig J. Modulation of Cav1.3 Ca2+ channel gating by Rab3 interacting molecule. Mol Cell Neurosci. 2010;44(3):246–259. doi: 10.1016/j.mcn.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Haeseleer F, Sokal I, Verlinde CL, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem. 2000;275(2):1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui G, Meyer AC, Calin-Jageman I, Neef J, Haeseleer F, Moser T, Lee A. Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. The Journal of Physiology. 2007;585(3):791–803. doi: 10.1113/jphysiol.2007.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca(2+)-binding protein 4, a Ca(v)1.4 channel regulator, in photoreceptor synaptic function. Nature Neuroscience. 2004;7(10):1079–1087. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim KY, Scholl ES, Liu X, Shepherd A, Haeseleer F, Lee A. Localization and expression of CaBP1/caldendrin in the mouse brain. Neuroscience. 2014;268:33–47. doi: 10.1016/j.neuroscience.2014.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seidenbecher CI, Langnaese K, Sanmarti-Vila L, Boeckers TM, Smalla KH, Sabel BA, Garner CC, Gundelfinger ED, Kreutz MR. Caldendrin, a novel neuronal calcium-binding protein confined to the somato-dendritic compartment. J Biol Chem. 1998;273(33):21324–21331. doi: 10.1074/jbc.273.33.21324. [DOI] [PubMed] [Google Scholar]
- 32.Schrauwen I, Helfmann S, Inagaki A, Predoehl F, Tabatabaiefar MA, Picher MM, Sommen M, Seco CZ, Oostrik J, Kremer H, Dheedene A, Claes C, Fransen E, Chaleshtori MH, Coucke P, Lee A, Moser T, Van Camp G. A mutation in CABP2, expressed in cochlear hair cells, causes autosomal-recessive hearing impairment. Am J Hum Genet. 2012;91(4):636–645. doi: 10.1016/j.ajhg.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou H, Kim SA, Kirk EA, Tippens AL, Sun H, Haeseleer F, Lee A. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24(19):4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Findeisen F, Rumpf CH, Minor DL., Jr Apo states of calmodulin and CaBP1 control CaV1 voltage-gated calcium channel function through direct competition for the IQ domain. J Mol Biol. 2013;425(17):3217–3234. doi: 10.1016/j.jmb.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oz S, Benmocha A, Sasson Y, Sachyani D, Almagor L, Lee A, Hirsch JA, Dascal N. Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1.2 L-type Ca2+ channels by calmodulin and Ca2+-binding protein 1. J Biol Chem. 2013;288(18):12680–12691. doi: 10.1074/jbc.M113.460949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280(33):29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 37.Yang PS, Johny MB, Yue DT. Allostery in Ca(2)(+) channel modulation by calcium-binding proteins. Nat Chem Biol. 2014;10(3):231–238. doi: 10.1038/nchembio.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oz S, Tsemakhovich V, Christel CJ, Lee A, Dascal N. CaBP1 regulates voltage-dependent inactivation and activation of Cav1.2 (L-type) calcium channels. J Biol Chem. 2011;286(16):13945–13953. doi: 10.1074/jbc.M110.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schrauwen I, Helfmann S, Inagaki A, Predoehl F, Tabatabaiefar MA, Picher MM, Sommen M, Seco CZ, Oostrik J, Kremer H, Dheedene A, Claes C, Fransen E, Chaleshtori MH, Coucke P, Lee A, Moser T, Van-Camp G. A Mutation in CABP2, Expressed in Cochlear Hair Cells, Causes Autosomal-Recessive Hearing Impairment. The American Journal of Human Genetics. 2012;91(4):636–645. doi: 10.1016/j.ajhg.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, Ali A, Ahmad I, Sinnegger-Brauns MJ, Brandt N, Engel J, Mangoni ME, Farooq M, Khan HU, Nurnberg P, Striessnig J, Bolz HJ. Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14(1):77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- 41.Ng L, Kelley MW, Forrest D. Making sense with thyroid hormone--the role of T(3) in auditory development. Nat Rev Endocrinol. 2013;9(5):296–307. doi: 10.1038/nrendo.2013.58. [DOI] [PubMed] [Google Scholar]
- 42.Johnson SL, Marcotti W, Kros CJ. Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol. 2005;563(1):177–191. doi: 10.1113/jphysiol.2004.074740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kros CJ, Ruppersberg JP, Rusch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998;394(6690):281–284. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- 44.Sendin G, Bulankina AV, Riedel D, Moser T. Maturation of ribbon synapses in hair cells is driven by thyroid hormone. J Neurosci. 2007;27(12):3163–3173. doi: 10.1523/JNEUROSCI.3974-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandt N, Kuhn S, Munkner S, Braig C, Winter H, Blin N, Vonthein R, Knipper M, Engel J. Thyroid hormone deficiency affects postnatal spiking activity and expression of Ca2+ and K+ channels in rodent inner hair cells. J Neurosci. 2007;27(12):3174–3186. doi: 10.1523/JNEUROSCI.3965-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marcotti W, Johnson SL, Rusch A, Kros CJ. Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol. 2003;552(3):743–761. doi: 10.1113/jphysiol.2003.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petit C. Usher syndrome: from genetics to pathogenesis. Annu Rev Genomics Hum Genet. 2001;2:271–297. doi: 10.1146/annurev.genom.2.1.271. [DOI] [PubMed] [Google Scholar]
- 48.Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet. 2003;12(23):3075–3086. doi: 10.1093/hmg/ddg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5(10):771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 50.Gregory FD, Bryan KE, Pangrsic T, Calin-Jageman IE, Moser T, Lee A. Harmonin inhibits presynaptic Cav1.3 Ca2+ channels in mouse inner hair cells. Nat Neurosci. 2011;14(9):1109–1111. doi: 10.1038/nn.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gregory FD, Pangrsic T, Calin-Jageman IE, Moser T, Lee A. Harmonin enhances voltage-dependent facilitation of Cav1.3 channels and synchronous exocytosis in mouse inner hair cells. J Physiol. 2013;591(Pt 13):3253–3269. doi: 10.1113/jphysiol.2013.254367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, Dumont RA, Hintermann E, Sczaniecka A, Schwander M, Williams D, Kachar B, Gillespie PG, Muller U. Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron. 2009;62(3):375–387. doi: 10.1016/j.neuron.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morgans CW, Gaughwin P, Maleszka R. Expression of the alpha1F calcium channel subunit by photoreceptors in the rat retina. Mol Vis. 2001;7:202–209. [PubMed] [Google Scholar]
- 54.Morgans CW. Localization of the alpha(1F) calcium channel subunit in the rat retina. Invest Ophthalmol Vis Sci. 2001;42(10):2414–2418. [PubMed] [Google Scholar]
- 55.Knoflach D, Kerov V, Sartori SB, Obermair GJ, Schmuckermair C, Liu X, Sothilingam V, Garrido MG, Baker SA, Glösmann M, Schicker K, Seeliger M, Lee A, Koschak A. Cav1.4 IT mouse as model for vision impairment in human congenital stationary night blindness type 2. Channels. 2013;7(6):503–513. doi: 10.4161/chan.26368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, Kerov V, Haeseleer F, Majumder A, Artemyev N, Baker SA, Lee A. Dysregulation of Cav1.4 channels disrupts the maturation of photoreceptor synaptic ribbons in congenital stationary night blindness type 2. Channels. 2013;7(6):514–523. doi: 10.4161/chan.26376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Specht D, Wu SB, Turner P, Dearden P, Koentgen F, Wolfrum U, Maw M, Brandstatter JH, tom Dieck S. Effects of presynaptic mutations on a postsynaptic Cacna1s calcium channel colocalized with mGluR6 at mouse photoreceptor ribbon synapses. Invest Ophthalmol Vis Sci. 2009;50(2):505–515. doi: 10.1167/iovs.08-2758. [DOI] [PubMed] [Google Scholar]
- 58.Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE, Bech-Hansen NT. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet. 2005;14(20):3035–3046. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- 59.Regus-Leidig H, Atorf J, Feigenspan A, Kremers J, Maw MA, Brandstatter JH. Photoreceptor degeneration in two mouse models for congenital stationary night blindness type 2. PLoS One. 2014;9(1):e86769. doi: 10.1371/journal.pone.0086769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raven MA, Orton NC, Nassar H, Williams GA, Stell WK, Jacobs GH, Bech-Hansen NT, Reese BE. Early afferent signaling in the outer plexiform layer regulates development of horizontal cell morphology. J Comp Neurol. 2008;506(5):745–758. doi: 10.1002/cne.21526. [DOI] [PubMed] [Google Scholar]
- 61.Zabouri N, Haverkamp S. Calcium channel-dependent molecular maturation of photoreceptor synapses. PLoS One. 2013;8(5):e63853. doi: 10.1371/journal.pone.0063853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wycisk KA, Budde B, Feil S, Skosyrski S, Buzzi F, Neidhardt J, Glaus E, Nürnberg P, Ruether K, Berger W. Structural and Functional Abnormalities of Retinal Ribbon Synapses due to Cacna2d4 Mutation. Investigative Ophthalmology & Visual Science. 2006;47(8):3523–3530. doi: 10.1167/iovs.06-0271. [DOI] [PubMed] [Google Scholar]
- 63.Ball SL, Powers PA, Shin HS, Morgans CW, Peachey NS, Gregg RG. Role of the b2 subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci. 2002;43(5):1595–1603. [PubMed] [Google Scholar]
- 64.Lee A, Wang S, Williams B, Hagen J, Scheetz T, Haeseleer F. Characterization of Cav1.4 Complexes (α11.4, β2, α2δ4) in HEK293T cells and in the Retina. J Biol Chem. 2015:1505–1521. doi: 10.1074/jbc.M114.607465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Corey DP, Dubinsky JM, Schwartz EA. The calcium current in inner segments of rods from the salamander (Ambystoma tigrinum) retina. J Physiol. 1984;354:557–575. doi: 10.1113/jphysiol.1984.sp015393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baumann L, Gerstner A, Zong X, Biel M, Wahl-Schott C. Functional characterization of the L-type Ca2+ channel Cav1.4alpha1 from mouse retina. Invest Ophthalmol Vis Sci. 2004;45(2):708–713. doi: 10.1167/iovs.03-0937. [DOI] [PubMed] [Google Scholar]
- 67.Koschak A, Reimer D, Walter D, Hoda JC, Heinzle T, Grabner M, Striessnig J. Cav1.4alpha1 Subunits Can Form Slowly Inactivating Dihydropyridine-Sensitive L-Type Ca2+ Channels Lacking Ca2+-Dependent Inactivation. The Journal of Neuroscience. 2003;23(14):6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McRory JE, Hamid J, Doering CJ, Garcia E, Parker R, Hamming K, Chen L, Hildebrand M, Beedle AM, Feldcamp L, Zamponi GW, Snutch TP. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J Neurosci. 2004;24(7):1707–1718. doi: 10.1523/JNEUROSCI.4846-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Griessmeier K, Cuny H, Roetzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C. Calmodulin is a functional regulator of CAV1.4 L-type Ca2+ channels. J Biol Chem. 2009;284(43):29809–29816. doi: 10.1074/jbc.M109.048082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wahl-Schott C, Baumann L, Cuny H, Eckert C, Griessmeier K, Biel M. Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc Natl Acad Sci U S A. 2006;103(42):15657–15662. doi: 10.1073/pnas.0604621103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh A, Hamedinger D, Hoda JC, Gebhart M, Koschak A, Romanin C, Striessnig J. C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat Neurosci. 2006;9(9):1108–1116. doi: 10.1038/nn1751. [DOI] [PubMed] [Google Scholar]
- 72.Liu X, Yang PS, Yang W, Yue DT. Enzyme-inhibitor-like tuning of Ca(2+) channel connectivity with calmodulin. Nature. 2010;463(7283):968–972. doi: 10.1038/nature08766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shaltiel L, Paparizos C, Fenske S, Hassan S, Gruner C, Rotzer K, Biel M, Wahl-Schott CA. Complex regulation of voltage-dependent activation and inactivation properties of retinal voltage-gated Cav1.4 L-type Ca2+ channels by Ca2+-binding protein 4 (CaBP4) J Biol Chem. 2012;287(43):36312–36321. doi: 10.1074/jbc.M112.392811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jalkanen R, Mantyjarvi M, Tobias R, Isosomppi J, Sankila EM, Alitalo T, Bech-Hansen NT. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J Med Genet. 2006;43(8):699–704. doi: 10.1136/jmg.2006.040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boycott K, Maybaum T, Naylor M, Weleber R, Robitaille J, Miyake Y, Bergen A, Pierpont M, Pearce W, Bech-Hansen N. A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants. Human Genetics. 2001;108(2):91–97. doi: 10.1007/s004390100461. [DOI] [PubMed] [Google Scholar]
- 76.Boycott KM, Pearce WG, Bech-Hansen NT. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can J Ophthalmol. 2000;35(4):204–213. doi: 10.1016/s0008-4182(00)80031-9. [DOI] [PubMed] [Google Scholar]
- 77.Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. Loss-of-function mutations in a calcium-channel [alpha]1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19(3):264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 78.Bijveld MM, van Genderen MM, Hoeben FP, Katzin AA, van Nispen RM, Riemslag FC, Kappers AM. Assessment of night vision problems in patients with congenital stationary night blindness. PLoS One. 2013;8(5):e62927. doi: 10.1371/journal.pone.0062927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vincent A, Wright T, Day MA, Westall CA, Heon E. A novel p.Gly603Arg mutation in CACNA1F causes Aland island eye disease and incomplete congenital stationary night blindness phenotypes in a family. Molecular Vision. 2011;17:3262–3270. [PMC free article] [PubMed] [Google Scholar]
- 80.Jalkanen R, Bech-Hansen NT, Tobias R, Sankila EM, Mantyjarvi M, Forsius H, de la Chapelle A, Alitalo T. A novel CACNA1F gene mutation causes Aland Island eye disease. Invest Ophthalmol Vis Sci. 2007;48(6):2498–2502. doi: 10.1167/iovs.06-1103. [DOI] [PubMed] [Google Scholar]
- 81.Hauke J, Schild A, Neugebauer A, Lappa A, Fricke J, Fauser S, Rösler S, Pannes A, Zarrinnam D, Altmüller J, Motameny S, Nürnberg G, Nürnberg P, Hahnen E, Beck BB. A novel large in-frame deletion within the CACNA1F gene associates with a cone-rod dystrophy 3-like phenotype. PLoS ONE. 2013;8(10):e76414. doi: 10.1371/journal.pone.0076414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wycisk KA, Zeitz C, Feil S, Wittmer M, Forster U, Neidhardt J, Wissinger B, Zrenner E, Wilke R, Kohl S, Berger W. Mutation in the Auxiliary Calcium-Channel Subunit CACNA2D4 Causes Autosomal Recessive Cone Dystrophy. American Journal of Human Genetics. 2006;79(5):973–977. doi: 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29(5):335–375. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 84.Striessnig J, Bolz HJ, Koschak A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. 2010;460(2):361–374. doi: 10.1007/s00424-010-0800-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burtscher V, Schicker K, Novikova E, Pöhn B, Stockner T, Kugler C, Singh A, Zeitz C, Lancelot ME, Audo I, Leroy BP, Freissmuth M, Herzig S, Matthes J, Koschak A. Spectrum of Cav1.4 dysfunction in congenital stationary night blindness type 2. Biochimica et Biophysica Acta. 2014;1838(8):2053–2065. doi: 10.1016/j.bbamem.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kraner SD, Filatov GN, Sun WJ, Bannerman P, Lindstrom J, Barchi RL. Analysis of local structure in the D2/S1-S2 region of the rat skeletal muscle type 1 sodium channel using insertional mutagenesis. J Neurochem. 1998;70:1628–1635. doi: 10.1046/j.1471-4159.1998.70041628.x. [DOI] [PubMed] [Google Scholar]
- 87.Wutz K, Sauer C, Zrenner E, Lorenz B, Alitalo T, Broghammer M, Hergersberg M, de la Chapelle A, Weber BH, Wissinger B, Meindl A, Pusch CM. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur J Hum Genet. 2002;10(8):449–456. doi: 10.1038/sj.ejhg.5200828. [DOI] [PubMed] [Google Scholar]
- 88.Hemara-Wahanui A, Berjukow S, Hope CI, Dearden PK, Wu SB, Wilson-Wheeler J, Sharp DM, Lundon-Treweek P, Clover GM, Hoda JC, Striessnig J, Marksteiner R, Hering S, Maw MA. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc Natl Acad Sci U S A. 2005;102(21):7553–7558. doi: 10.1073/pnas.0501907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeitz C, Kloeckener-Gruissem B, Forster U, Kohl S, Magyar I, Wissinger B, Matyas G, Borruat FX, Schorderet DF, Zrenner E, Munier FL, Berger W. Mutations in CABP4, the Gene Encoding the Ca2+-Binding Protein 4, Cause Autosomal Recessive Night Blindness. Am J Hum Genet. 2006;79(4):657–667. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Littink KW, van Genderen MM, Collin RW, Roosing S, de Brouwer AP, Riemslag FC, Venselaar H, Thiadens AA, Hoyng CB, Rohrschneider K, den Hollander AI, Cremers FP, van den Born LI. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest Ophthalmol Vis Sci. 2009;50(5):2344–2350. doi: 10.1167/iovs.08-2553. [DOI] [PubMed] [Google Scholar]
- 91.Lee A, Fakler B, Kaczmarek LK, Isom LL. More than a pore: ion channel signaling complexes. J Neurosci. 2014;34(46):15159–15169. doi: 10.1523/JNEUROSCI.3275-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Calin-Jageman I, Lee A. Cav1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem. 2008;105(3):573–583. doi: 10.1111/j.1471-4159.2008.05286.x. [DOI] [PubMed] [Google Scholar]