

Abstract
Historically, most vaccines have been based on killed or live‐attenuated infectious agents. Although very successful at immunizing populations against disease, both approaches raise safety concerns and often have limited production capacity. This has resulted in increased emphasis on the development of subunit vaccines. Several recombinant systems have been considered for subunit vaccine manufacture, including plants, which offer advantages both in cost and in scale of production. We have developed a plant expression system utilizing a ‘launch vector’, which combines the advantageous features of standard agrobacterial binary plasmids and plant viral vectors, to achieve high‐level target antigen expression in plants. As an additional feature, to aid in target expression, stability and purification, we have engineered a thermostable carrier molecule to which antigens are fused. We have applied this launch vector/carrier system to engineer and express target antigens from various pathogens, including, influenza A/Vietnam/04 (H5N1) virus.
Keywords: Carrier molecule, influenza vaccine, launch vector, lichenase, plant viral expression, subunit vaccine
Introduction
Most vaccines against infectious agents have a similar goal, that of priming the immune system so that the specific pathogen is destroyed before it can multiply and cause disease. Historically, this priming has been achieved using vaccines based on whole viruses or bacteria that have been killed or on live‐attenuated strains. Although these vaccines have been proved to be highly effective against some pathogens, these approaches have not yielded efficacious vaccines against several other major targets. In addition, there are also some safety concerns associated with the use of attenuated strains and killed pathogens. For these reasons, there has been increasing interest in the development of subunit vaccines that are predominantly based on antigenic surface proteins or polysaccharides. Recombinant DNA technology has facilitated the development of subunit vaccines, such as hepatitis B, 1 and has led to a pan‐genomic approach to vaccine development. 2 Recent events, however, have demonstrated the need for the development and implementation of alternative technologies to overcome limitations in capacity, speed and costs of vaccine production.
Plant‐based production systems
In recent years, there has been increasing interest in using plants as production systems for therapeutics, vaccines, and antibodies, 3 , 4 , 5 with several vaccine candidates completing phase I clinical trials. 6 , 7 , 8 , 9 , 10 , 11 Plants afford several advantages as production systems, including, safety from contamination with animal pathogens, high production capacity, and relatively low capital investment. To date, the following strategies have been used to produce recombinant proteins, including vaccine antigens, in plants: nuclear transformation, 12 chloroplast transformation, 13 and infection with plant RNA viral vectors. 14 When tested in animal models, several plant‐produced antigens have induced cellular and humoral immune responses conferring protection against challenge. 4
In spite of these encouraging reports, there are disadvantages to each of these plant‐based approaches. Nuclear transformants often show low levels of expression, and are subject to long lead times. Chloroplast transformation, while giving high levels of target expression, 13 is limited to few plant species, has limited potential for post‐translational modification (e.g. no glycosylation), and is also subject to long lead times. As with chloroplast transformation, engineered plant viruses allow for high levels of recombinant protein expression. 14 Plant viruses have the added advantages of short lead times, rapid bulk‐up capacity, and the potential to fuse peptides to the viral coat protein (CP), so facilitating production and enhancing immunogenicity. 8 However, the potential instability of the plant viral expression vector is a concern, and may limit the range of targets.
Development of a ‘launch vector’ for high‐level protein expression in plants
To address some of the limitations of the above plant‐based approaches, we have developed an expression system based on a ‘launch vector’, pBID4 (Figure 1), which consists of two key components, an Agrobacterium binary plasmid, pBI121 15 and a plant virus expression vector. 16 Agrobacterium binary plasmids, the standard tools for generating stable transgenic plants 17 are based on the tumour‐inducing plasmid of Agrobacterium tumefaciens. Target sequences to be expressed, along with regulatory elements, are incorporated between the left border (LB) and right border (RB) of the binary plasmid. Upon introduction of agrobacteria into plant tissue, multiple single‐stranded DNA (ssDNA) copies of sequence between LB and RB are generated and released. It is this feature of the binary vector, to generate multiple ssDNA copies, which we use to launch our viral vector into plant tissue. The viral vector we have used is based on tobacco mosaic virus (TMV). TMV is the most extensively studied plant virus, and has been most widely used for the expression of foreign proteins in plants. 18 The single‐stranded positive‐sense RNA genome of TMV encodes the viral replicase, the cell‐to‐cell movement protein, and the CP that encapsidates the viral RNA and mediates viral spread throughout the entire plant. In pBID4, the CP sequence is replaced with the target gene sequence to be expressed. In the absence of CP, the resultant viral vector is a naked self‐replicating RNA that is less subject to recombination than CP‐containing vectors, and that cannot effectively spread and survive in the environment.
Figure 1.

Schematic diagram of the launch vector pBID4. The vector contains the 35S promoter of cauliflower mosaic virus (a DNA plant virus) that drives initial transcription of the recombinant viral genome following introduction into plants, and the nos terminator, the transcriptional terminator of Agrobacterium nopaline synthase. The tobacco mosaic virus genome contains genes for virus replication (126/183k) and cell‐to‐cell movement (MP), and the cloned target gene, inserted into a unique cloning site and under the transcriptional control of the coat protein subgenomic mRNA promoter (arrow). Left and right border sequences (LB and RB) of the Agrobacterium binary plasmid delimit the region of the launch vector that is transferred into plant cells following infiltration of plants with the recombinant Agrobacterium.
The launch vector is delivered by agroinfiltration, during which, bacteria are artificially forced into the intercellular spaces of plant tissues, either by injection or vacuum infiltration. Following infiltration, multiple ssDNA copies of the viral vector sequences are launched into plant cells in a matter of minutes. The introduced viral sequences, along with the cloned targets, are then amplified through replication. Translation of these recombinant viral mRNAs typically results in the accumulation of 100 mg quantities of target protein per kg of plant tissue in less than a week. The launch vector‐based protein production system is depicted in Figure 2.
Figure 2.
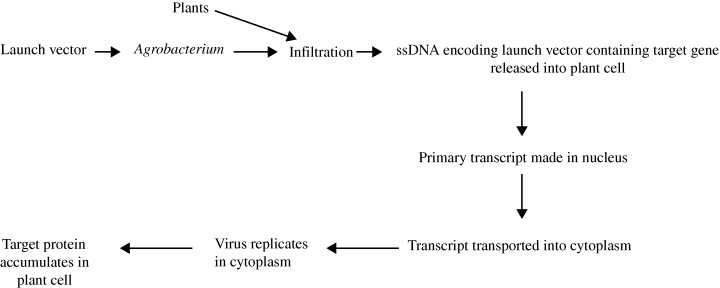
Schematic representation of recombinant protein production in plants using the launch vector system.
We have expressed a wide range of proteins in plants using the launch vector system. Figure 3(A–C) depict the expression of the green fluorescent protein in Nicotiana benthamiana plants following the introduction of Agrobacterium‐containing recombinant launch vector by injection (Figure 3A,B) or vacuum infiltration (Figure 3C). Vacuum infiltration has the particular advantage over injection as a mode of introducing the launch vector into all exposed plant tissues, therefore eliminating the need for a virus capable of spreading through the entire plant, and allowing for the use of a CP‐deficient viral vector. High‐level expression of target proteins can be achieved when the launch vector is used (Figure 3D). Accumulation of an engineered thermostable protein LicKM (described below) of up to 0·5 g/kg fresh leaf tissue has been achieved within 3 days postinfiltration. This expression level is c. 50‐fold higher than has been achieved with this target cloned into a standard binary plasmid. Gleba et al. 19 have reported a broadly similar approach, combining the features of agrobacterial and viral vectors to express several targets in plants.
Figure 3.
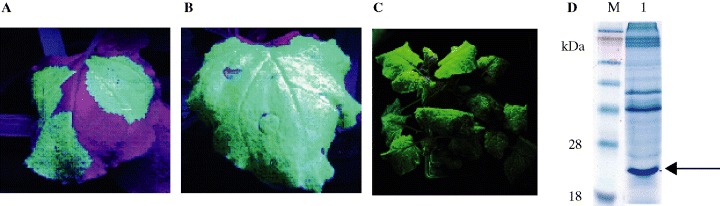
Expression of target proteins in Nicotiana benthamiana plants following infiltration with Agrobacterium‐containing pBID4. (A) Green fluorescent protein (GFP) expression 4 days post‐infiltration achieved by injection. (B) GFP expression 6 days postinfiltration achieved by injection. (C) GFP expression following vacuum infiltration of whole plants. (D) Expression of LicKM at up to 0·5 g/kg fresh plant tissue. Lane 1 shows extract from leaf tissue expressing LicKM. The arrow indicates the position of LicKM; M corresponds to molecular weight marker.
Carrier proteins for the expression and delivery of target antigens
For the development of effective subunit vaccines, target antigens are often fused or conjugated to carrier proteins to enhance expression, stability, and/or immunogenicity. Fusion of targets to carrier molecules can also aid in purification during vaccine production. For example, to obtain effective vaccines against Haemophilus influenza B, Neisseria meningitidis and Streptococcus pneumoniae target polysaccharides were covalently attached to carrier proteins. 20 , 21 , 22 , 23 Similarly, a variety of peptides have been fused to the receptor‐binding subunits of cholera toxin and Escherichia coli heat‐labile enterotoxin. 24 , 25 , 26 Fusion of peptides and proteins to viral structural proteins has also been shown to enhance stability and immunogenicity, 27 , 28 , 29 and to aid in target purification. 30
Here, we describe a carrier molecule for engineering and expressing target antigens. This carrier molecule is based on a thermostable enzyme, β‐1,3‐1,4‐glucanase (lichenase), from Clostridium thermocellum, 31 engineered versions of which have been used to express reporter genes in prokaryotic and eukaryotic systems. 32 Full‐length lichenase (c. 35 kDa) consists of a signal peptide, a catalytic domain, a Pro‐Thr‐rich box, and a docking domain (Figure 4A). For our purpose, we modified the molecule to replace the native signal peptide with the signal peptide from Nicotiana tabacum pathogenesis‐related protein PR1a, 33 and deleted the Pro‐Thr‐rich box and the docking domain. The catalytic domain has a loop structure dividing it into two regions, the N‐terminal region (N: amino acids 32–84) and the C‐terminal region (C: amino acids 85–246). These two regions can be split at the loop structure and circularly permuted to make the molecule more receptive to insertions without affecting enzymatic activity. A multiple cloning site was introduced at the junction between these two regions, and a 6xHis tag and the sequence KDEL were placed at the 3′‐end of the permuted carrier to facilitate purification and retention in the endoplasmic reticulum, respectively. Figure 4(B) shows a schematic diagram of the modified lichenase (LicKM; GenBank accession number: DQ776900) that has a molecular weight of c. 27 kDa. Target sequences can be expressed as N or C terminal fusions and/or as internal fusions, a feature that allows for the simultaneous expression of multiple targets.
Figure 4.
The lichenase carrier molecule. (A) Schematic representation of β‐1,3‐1,4‐glucanase (lichenase), from Clostridium thermocellum. (B) Schematic representation of the circularly permuted LicKM carrier. In each panel, the vertical hatch corresponds to the N‐terminal region of the catalytic domain, and the dotted box corresponds to the C‐terminal region of the catalytic domain.
LicKM allows for fusions to molecules ranging in size from small peptides to full‐length proteins of up to 100 kDa. We have engineered and successfully produced more than 30 target antigens from different pathogens, including respiratory syncytial virus, human immunodeficiency virus, plague, anthrax, influenza, and Trypanosoma brucei as fusions with LicKM. LicKM maintains its enzymatic activity at high temperatures (65°C), a property that also generally applies to fusions. This allows for easy and cost‐effective recovery of target proteins because a 10‐min heat treatment at 65°C removes up to 50% of contaminating plant proteins (Figure 5A). The fusion proteins can be tracked during purification by monitoring lichenase activity. The use of LicKM as a carrier molecule contributes additional advantages, including the potential for enhanced expression (Figure 5B), and incorporation of multiple vaccine determinants (Figure 5C).
Figure 5.
Features of LicKM as a carrier molecule. (A) Thermostability: equal amounts of tissue from uninoculated plants or plants expressing recombinant LicKM or LicKM fused to human papillomavirus (HPV) target protein were homogenized and equally split between two tubes. One tube from each pair was subjected to heat treatment. Samples in lanes 2, 4, and 6 were incubated at 65°C for 10 min, whereas samples in lanes 1, 3, and 5 were not heat treated. All samples were centrifuged to remove debris containing denatured proteins prior to analysis by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS‐PAGE) followed by Coomassie staining. Lanes 1 and 2 show uninoculated plant extract, lanes 3 and 4 show extract from plants expressing LicKM, and lanes 5 and 6 show extract from plants expressing LicKM fused to an antigen from HPV. Arrows indicate positions of LicKM (lanes 3 and 4), and LicKM‐HPV antigen (lanes 5 and 6). (B) Enhanced expression: domain 4 of protective antigen of Bacillus anthracis expressed alone (lane 3) or as a fusion to LicKM (lane 2) in Escherichia coli using the pQE30 expression system. Lane 1 corresponds to ‘empty vector’ control. Protein extracts were analyzed by SDS‐PAGE followed by Western blotting using anti‐PA antibody. (C) Multiple target expression: LicKM carrying influenza A/Vietnam/04 hemagglutinin stem domain as an internal fusion and A/Vietnam/04 neuraminidase as a C‐terminal fusion (lane 1), LicKM carrying influenza A/Vietnam/04 hemagglutinin globular domain as an internal fusion and A/Vietnam/04 neuraminidase as a C‐terminal fusion (lane 2). Protein extracts were analyzed by SDS‐PAGE followed by Western blotting using polyclonal anti‐lichenase antibody. In all panels, M corresponds to molecular weight marker, Magic Mark for Western blots, and BenchMark for Coomassie‐stained gels.
Expression of influenza target antigens as lichenase fusions in plants using pBID4
The influenza A virus presents one of the major infectious disease threats to human populations. 34 The almost annual epidemics of influenza are associated with point mutations in the envelope antigens hemagglutinin (HA) and neuraminidase (NA), enabling the virus to evade immune responses induced by prior infections or vaccination. 35 Of the greatest risks to public health is the emergence of strains of influenza A virus, with pandemic potential, that have new HA and NA antigens as a result of recombination between strains. 36 Our main defense against influenza is vaccination. Most of the world's influenza vaccine supply is manufactured using embryonated eggs, which has insufficient capacity to immunize fully the world's population against annual epidemics. 37 The current potential for a pandemic, caused by emerging H5N1 strains, emphasizes the urgent need to develop new manufacturing platforms for influenza vaccines, including recombinant systems for subunit vaccines. HA is the immunodominant antigen that is believed to induce protective immunity, and is a key target for subunit vaccines. The efficacy of vaccination against influenza is predominantly determined by the amount of immunogenic HA that is present, 38 and the globular domain of HA contains the majority of the antigenic sites and neutralizing epitopes. 39 NA has been shown to contribute to immunity when delivered alongside HA, 40 and thus is a further potential candidate for subunit vaccine development. A recombinant HA‐based vaccine candidate, produced in serum‐free insect cell cultures, has been successfully tested in a phase II clinical trial. 41 However, despite promising data with subunit vaccine candidates, production capacity remains a major issue.
Some safety concerns and capacity limitations of current influenza vaccine production could be addressed using the launch vector/lichenase carrier system for plant‐based production as described above. We have applied this system to express antigens from influenza type A/Vietnam/04 (H5N1) virus. We engineered the stem domain (H5SD), amino acids 17–58 followed by a triple glycine linker and amino acids 293–535, and the globular domain (H5GD), amino acids 59–292, of influenza A/Vietnam/04 virus HA (GenBank accession number: AAX83397) as internal in‐frame fusions with LicKM to give LicKM‐H5SD and LicKM‐H5GD, respectively, and inserted the fusions into pBID4. We also inserted NA (N1), amino acids 29–449 of influenza A/Vietnam/04 virus NA (GenBank accession number: AAT73327) into pBID4, although not as a fusion with LicKM. The expression of each target was evaluated in N. benthamiana plants following agroinfiltration. Tissue samples were collected 4–7 days following infiltration, and each target protein was enriched to over 80% purity by affinity chromatography.
The sero‐identity of each target was determined by Western blot and enzyme‐linked immunosorbent assay using reference polyclonal sheep sera. The plant‐produced LicKM‐H5SD and LicKM‐H5GD were recognized by serum raised against HA purified from influenza A/Vietnam/04 virus (Figure 6A,B), but not by serum raised against HA purified from influenza A/Wyoming/3/03 virus (Figure 6B). The plant‐produced NA was recognized by serum raised against the reassorted virus NIBRG‐17 (anti‐H7N1) (Figure 6C,D) but not by serum raised against the reassorted virus NIBRG‐18 (anti‐H7N2) (Figure 6D). Furthermore, the plant‐produced NA showed NA activity that was specifically inhibited by serum raised against NIBRG‐17, but not by serum raised against NIBRG‐18 (Figure 6E).
Figure 6.
In vitro characterization of plant‐produced influenza antigens. (A) The plant‐produced LicKM‐H5SD (Lane 2) and LicKM‐H5GD (Lane 3) analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS‐PAGE) followed by Western blotting using serum raised against purified hemagglutinin (HA) of influenza A/Vietnam/04 virus. Lane 1 shows influenza A/Vietnam/04 virus, and M corresponds to molecular weight marker. (B) The plant‐produced LicKM‐H5SD and LicKM‐H5GD analyzed by enzyme‐linked immunosorbent assay (ELISA) using serum raised against purified HA of either influenza A/Vietnam/04 virus (anti‐H5 PAb) or influenza A/Wyoming/3/03 virus (anti‐H3 PAb). (C) The plant‐produced neuraminidase (NA) (N1) analyzed by SDS‐PAGE followed by Western blotting using serum raised against reassorted virus NIBRG‐17 (anti‐H7N1). (D) The plant‐produced NA (N1) analyzed by ELISA using serum raised against either reassorted virus NIBRG‐17 (anti‐H7N1 PAb) or reassorted virus NIBRG‐18 (anti‐H7N2 PAb). (E) The NA activity of plant‐produced NA (N1) following pre‐incubation with serum raised against either reassorted virus NIBRG‐17 (anti‐H7N1 PAb) or reassorted virus NIBRG‐18 (anti‐H7N2 PAb). In all panels of (A) and (C), M corresponds to MagicMark molecular weight marker.
In summary, expression of target influenza H5N1 antigens in plants using our launch vector system has successfully been achieved, and procedures have been established to enrich these proteins to at least 80% purity. In the case of HA, the target antigens were expressed as fusions to our engineered lichenase carrier molecule. The expressed influenza targets showed antigen‐specific reactivity against standard sheep sera. These plant‐produced targets have now been advanced into animal studies in mice and ferrets to assess their immunogenicity and protective efficacy.
Conclusions
We have developed a plant‐based expression system utilizing a ‘launch vector’, pBID4, to produce target vaccine antigens. The launch vector system combines the desirable features of agrobacterial and plant viral expression vectors to allow for consistent high‐level production of recombinant proteins under contained conditions. The launch vector described here retains the positive features of plant viral vectors, without the associated disadvantages of instability and potential environmental release. Following inoculation, multiple copies of the vector are delivered to each recipient plant cell and then each copy is further amplified by viral replicases. This results in high levels of target protein accumulation in the cell. In addition, as the launch vector can be introduced into all aerial plant tissue by vacuum infiltration, there is no requirement for CP to facilitate viral vector movement, and consequently, the expression vectors are more stable, and the virus cannot survive in the environment. As with stable nuclear transgenic and plant viral expression systems, antigens expressed using the launch vector can be post‐translationally modified. Target antigens can be engineered into pBID4 and produced in bulk quantities in the time frame of a few months. Furthermore, as large‐scale production depends on non‐transgenic seeds, which can be prepared and stored in large quantities, the system has the potential for high capacity. These features make the launch vector system highly suitable for the production of vaccine targets, such as influenza antigens, that demand a short lead time and high capacity. As an additional optional feature for the expression of vaccine candidates, we have engineered a thermostable carrier molecule, LicKM, to which target antigens can be fused. Protein targets of up to 100 kDa have been successfully fused to LicKM, and furthermore, multiple targets can be fused to the same carrier molecule, allowing for the development of combination vaccines. Fusion to LicKM has enhanced target antigen expression. Moreover, LicKM simplifies the purification process, as heat treatment removes most contaminating plant proteins while leaving LicKM fusions unaffected. The combination of the launch vector and LicKM makes for a highly attractive system for vaccine antigen engineering and production.
Acknowledgements
We thank Caroline Nicholson and John Wood of the National Institute for Biological Standards and Control for providing target antigen sequence information and standard influenza reagents, respectively. We also thank Geoffrey Schild for advice for antigen design and assay development, Sylvia Maasa for providing LicKM‐HPV samples, and Margaret Shillingford for greenhouse management. Funds for work presented here were provided by InB: Biotechnologies, Inc., and Fraunhofer USA, Inc.
References
- 1. Petre J, Van Wijnendaele F, De Neys B, et al. Development of a hepatitis B vaccine from transformed yeast cells. Postgrad Med J 1987;63(Suppl. 2):73–81. [PubMed] [Google Scholar]
- 2. Tettelin H, Masignani V, Cieslewicz MJ, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ‘‘pan‐genome’’. Proc Natl Acad Sci USA 2005;102:13950–13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Twyman RM, Schillberg S, Fischer R. Transgenic plants in the biopharmaceutical market. Expert Opin Emerg Drugs 2005;10:185–218. [DOI] [PubMed] [Google Scholar]
- 4. Streatfield SJ, Howard JA. Plant‐based vaccines. Int J Parasitol 2003;33:479–493. [DOI] [PubMed] [Google Scholar]
- 5. Stoger E, Sack M, Fischer R, Christou P. Plantibodies: applications, advantages and bottlenecks. Curr Opin Biotechnol 2002;13:161–166. [DOI] [PubMed] [Google Scholar]
- 6. Tacket CO, Mason HS, Losonsky G, Clements JD, Levine MM, Arntzen CJ. Immunogenicity in humans of a recombinant bacterial antigen delivered in a transgenic potato. Nat Med 1998;4:607–609. [DOI] [PubMed] [Google Scholar]
- 7. Tacket CO, Mason HS, Losonsky G, Estes MK, Levine MM, Arntzen CJ. Human immune responses to a novel Norwalk virus vaccine delivered in transgenic potatoes. J Infect Dis 2000;182:302–305. [DOI] [PubMed] [Google Scholar]
- 8. Yusibov V, Hooper DC, Spitsin SV, et al. Expression in plants and immunogenicity of plant virus‐based experimental rabies vaccine. Vaccine 2002;20:3155–3164. [DOI] [PubMed] [Google Scholar]
- 9. Reddy SA, Czerwinski D, Rajapaksa R, et al. Plant derived single‐chain Fv idiotype vaccines are safe and immunogenic in patients with follicular lymphoma: results of a phase I study; in: Proceedings of the 44th Annual Conference of the American Society of Hematology, Philadelphia. Blood 2002;100:163a. [Google Scholar]
- 10. Tacket CO, Pasetti MF, Edelman R, Howard JA, Streatfield S. Immunogenicity of recombinant LT‐B delivered orally to humans in transgenic corn. Vaccine 2004;22:4385–4389. [DOI] [PubMed] [Google Scholar]
- 11. Thanavala Y, Mahoney M, Pal S, et al. Immunogenicity in humans of an edible vaccine for hepatitis B. Proc Natl Acad Sci USA 2005;102:3378–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hood EE, Jilka JM. Plant‐based production of xenogenic proteins. Curr Opin Biotechnol 1999;10:382–386. [DOI] [PubMed] [Google Scholar]
- 13. Kumar S, Daniell H. Engineering the chloroplast genome for hyperexpression of human therapeutic proteins and vaccine antigens. Methods Mol Biol 2004;267:365–383. [DOI] [PubMed] [Google Scholar]
- 14. Yusibov V, Rabindran S, Commandeur U, Twyman RM, Fischer R. The potential of plant virus vectors for vaccine production. Drugs R D 2006;7:203–217. [DOI] [PubMed] [Google Scholar]
- 15. Chen PY, Wang CK, Soong SC, To KY. Complete sequence of the binary vector pBI121 and its application in cloning T‐DNA insertion from transgenic plants. Mol Breed 2003;11:287–293. [Google Scholar]
- 16. Shivprasad S, Pogue GP, Lewandowski DJ, et al. Heterologous sequences greatly affect foreign gene expression in tobacco mosaic virus‐based vectors. Virology 1999;255:312–323. [DOI] [PubMed] [Google Scholar]
- 17. Gelvin SB. Agrobacterium and plant genes involved in T‐DNA transfer and integration. Annu Rev Plant Physiol Plant Mol Biol 2000;51:223–256. [DOI] [PubMed] [Google Scholar]
- 18. Okada Y. Historical overview of research on the tobacco mosaic virus genome: genome organization, infectivity and gene manipulation. Philos Trans R Soc Lond B, Biol Sci 1999;354:569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gleba Y, Klimyuk V, Marillonnet S. Magnifection – a new platform for expressing recombinant vaccines in plants. Vaccine 2005;23:2042–2048. [DOI] [PubMed] [Google Scholar]
- 20. Vella PP, Ellis RW. Haemophilus B conjugate vaccines. Biotechnology 1992;20:1–22. [DOI] [PubMed] [Google Scholar]
- 21. Kelly DF, Moxon ER, Pollard AJ. Haemophilus influenzae type b conjugate vaccines. Immunology 2004;113:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buttery JP, Moxon ER. Designing meningitis vaccines. J R Coll Physicians Lond 2000;34:163–168. [PMC free article] [PubMed] [Google Scholar]
- 23. Jacobson RM, Poland GA. The pneumococcal conjugate vaccine. Minerva Pediatr 2002;54:295–303. [PubMed] [Google Scholar]
- 24. Dertzbaugh MT, Elson CO. Comparative effectiveness of the cholera toxin B subunit and alkaline phosphatase as carriers for oral vaccines. Infect Immun 1993;61:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nashar TO, Amin T, Marcello A, Hirst TR. Current progress in the development of the B subunits of cholera toxin and Escherichia coli heat‐labile enterotoxin as carriers for the oral delivery of heterologous antigens and epitopes. Vaccine 1993;11:235–240. [DOI] [PubMed] [Google Scholar]
- 26. Liljeqvist S, Stahl S, Andreoni C, Binz H, Uhlen M, Murby M. Fusions to the cholera toxin B subunit: influence on pentamerization and GM1 binding. J Immunol Methods 1997;210:125–135. [DOI] [PubMed] [Google Scholar]
- 27. Stahl SJ, Murray K. Immunogenicity of peptide fusions to hepatitis B virus core antigen. Proc Natl Acad Sci USA 1989;86:6283–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ulrich R, Nassal M, Meisel H, Kruger DH. Core particles of hepatitis B virus as carrier for foreign epitopes. Adv Virus Res 1998;50:141–182. [DOI] [PubMed] [Google Scholar]
- 29. Smith ML, Lindbo JA, Dillard‐Telm S, et al. Modified tobacco mosaic virus particles as scaffolds for display of protein antigens for vaccine applications. Virology 2006;348:475–488. [DOI] [PubMed] [Google Scholar]
- 30. Turpen TH, Reinl SJ, Charoenvit Y, Hoffman SL, Fallarme V, Grill LK. Malarial epitopes expressed on the surface of recombinant tobacco mosaic virus. Biotechnology (N Y) 1995;13:53–57. [DOI] [PubMed] [Google Scholar]
- 31. Schimming S, Schwarz WH, Staudenbauer WL. Structure of the Clostridium thermocellum gene licB and the encoded beta‐1,3‐1,4‐glucanase. A catalytic region homologous to Bacillus lichenases joined to the reiterated domain of clostridial cellulases. Eur J Biochem 1992;204:13–19. [DOI] [PubMed] [Google Scholar]
- 32. Goldenkova IV, Musiichuk KA, Piruzian ES. A thermostable Clostridium thermocellum lichenase‐based reporter system for studying the gene expression regulation in prokaryotic and eukaryotic cells. Mol Biol (Mosk) 2002;36:868–876. [PubMed] [Google Scholar]
- 33. Pfitzner UM, Goodman HM. Isolation and characterization of cDNA clones encoding pathogenesis‐related proteins from tobacco mosaic virus infected tobacco plants. Nucleic Acids Res 1987;15:4449–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cox NJ, Subbarao K. Global epidemiology of influenza: past and present. Annu Rev Med 2000;51:407–421. [DOI] [PubMed] [Google Scholar]
- 35. Hampson AW. Influenza virus antigens and ‘antigenic drift’; in Potter CW. (ed): Influenza. Amsterdam: Elsevier, 2002; 49–86. [Google Scholar]
- 36. Scholtissek C. Pandemic influenza: antigenic shift; in Potter CW. (ed): Influenza. Amsterdam: Elsevier, 2002; 87–100. [Google Scholar]
- 37. Emanuel EJ, Wertheimer A. Who should get influenza vaccine when not all can? Science 2006;312:854–855. [DOI] [PubMed] [Google Scholar]
- 38. Brown F, Haaheim LR, Wood JM, Schild GC, (eds). Laboratory correlates of immunity to influenza ‐ a reassessment. Developments in biologicals, vol. 115 Basel: Karger, 2004. [Google Scholar]
- 39. Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody‐binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981;289:373–378. [DOI] [PubMed] [Google Scholar]
- 40. Johansson BE, Matthews JT, Kilbourne ED. Supplementation of conventional influenza A vaccine with purified viral neuraminidase results in a balanced and broadened immune response. Vaccine 1998;16:1009–1015. [DOI] [PubMed] [Google Scholar]
- 41. Treanor JJ, Schiff GM, Couch RB, et al. Dose‐related safety and immunogenicity of a trivalent baculovirus‐expressed influenza‐virus hemagglutinin vaccine in elderly adults. J Infect Dis 2006;193:1223–1228. [DOI] [PubMed] [Google Scholar]