

Summary
Generation, transformation, and utilization of organic molecules in support of cellular differentiation, growth, and maintenance are basic tenets that define life. In eukaryotes, mitochondrial oxygen consumption plays a central role in these processes. During the process of oxidative phosphorylation, mitochondria utilize oxygen to generate ATP from organic fuel molecules but in the process also produce reactive oxygen species (ROS). While ROS have long been appreciated for their damage-promoting, detrimental effects, there is now a greater understanding of their roles as signaling molecules. Here, we review mitochondrial ROS-mediated signaling pathways with an emphasis on how they are involved in various basal and adaptive physiological responses that control organismal homeostasis.
Mitochondria and Associated Homeostatic and Stress Signaling Pathways
Mitochondria are essential organelles present in all but a few mammalian cell types, where they perform multiple functions. They are the sites of the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS), through which large amounts of ATP are generated using the electrochemical gradient generated across the inner of two membranes by the electron transport chain (ETC). However, their critical roles in metabolism go far beyond glucose oxidation via OXPHOS and include fatty acid and amino acid metabolism and biosynthesis of hormones, heme, and iron sulfur clusters. Furthermore, in addition to metabolism, mitochondria are involved in apoptosis, ion homeostasis, and innate immunity, with new roles in cell and organismal biology being discovered at an unprecedented rate.
Mitochondria are complex in composition, form, and function. Though often depicted as small round or oval structures, they are instead usually dynamic, branched networks that constantly fuse and divide under control of specific fission and fusion machineries (Mishra and Chan, 2014). Proteomic analyses indicate that mammalian mitochondria contain ∼1,200 proteins, with the precise composition varying significantly between cell and tissue types (Calvo and Mootha, 2010). Thirteen of these proteins are encoded by the maternally inherited mitochondrial DNA (mtDNA) located in the matrix, while the rest are encoded by nuclear genes and targeted to the organelle by specific protein import pathways (Shadel and Clayton, 1997). Thus, mitochondrial biogenesis and homeostasis, including mtDNA expression and maintenance, are under strict control of nuclear gene expression programs (Scarpulla, 2008).
The overall status of mitochondria is constantly monitored, allowing their number, morphology, distribution, and activity to be modulated by developmental, physiological, and environmental cues. This requires bidirectional signaling pathways that meditate crosstalk between mitochondria and the nucleus. Pioneering studies in budding yeast revealed that mitochondrial dysfunction leads to so-called “retrograde signaling” events that result in adaptive changes in nuclear gene expression and metabolism mediated by specific transcription factors (Butow and Avadhani, 2004). Mitochondrial retrograde signaling pathways also exist in mammals and are now receiving considerable attention because they drive both beneficial and pathogenic adaptive responses.
Given their complicated nature, mitochondrial stress can manifest in many forms that elicit different stress signals. Reduced OXPHOS capacity or altered ETC function results in energy deprivation (e.g., reduced ATP/energy charge), altered mitochondrial ROS (mtROS) production, or loss of mitochondrial membrane potential that elicit specific stress-signaling responses (Butow and Avadhani, 2004; Sena and Chandel, 2012). Reduced mitochondrial protein import, improper assembly of large enzymatic complexes (e.g., OXPHOS and ribosomes), and altered chaperone activity can cause proteotoxic stress and mitochondrial unfolded protein responses (Haynes et al., 2013; Rugarli and Langer, 2012). As major sites of ROS production, mitochondria are also prone to oxidative damage and stress. Damage, mutation, or depletion of mtDNA causes distinct forms of mitochondrial stress and downstream signaling (Scheibye-Knudsen et al., 2015; West et al., 2015). Finally, altered morphology, dynamics, and distribution can lead to distinct forms of stress and are linked to mitochondrial turnover by autophagy or mitophagy (Labbé et al., 2014). Changes in these parameters and associated stress-signaling responses can occur downstream of physiological (e.g., nutrient limitations, substrate availability, exercise), environmental (exposure to drugs or toxins), and genetic cues. With regard to genetics, the importance of these pathways is underscored by the fact that inherited mitochondrial diseases are caused by mutations in genes encoding proteins involved in each of these processes that can be inherited maternally (mtDNA mutations) or in a Mendelian fashion (nuclear gene mutations) (Nunnari and Suomalainen, 2012). However, much remains to be learned about these stress pathways. For example, the specific sensors and signaling responses to different forms of mitochondrial stress and how they are manifested in different cells and tissues remain largely unknown. Furthermore, the degree of crosstalk between different mitochondrial stress pathways and what determines whether they elicit beneficial or maladaptive responses are not clear.
Mitochondrial ROS Signaling
ROS are formed from one-electron transfers from a redox donor to molecular oxygen (O2). This initially forms the anionic free-radical superoxide that can be converted to hydrogen peroxide by superoxide dismutase enzymes (Figure 1). Hydroxyl radical is another ROS that can be formed (e.g., by metal-catalyzed oxidation of hydrogen peroxide), but in this Review, “ROS” refers to superoxide and hydrogen peroxide unless otherwise noted. In mitochondria, the orderly flow of electrons down the mitochondrial ETC to complex IV results in their final deposition into molecular oxygen to form water. However, electrons can also react prematurely with oxygen at sites in the ETC to form superoxide/hydrogen peroxide (Murphy, 2009). Complexes I and III are often regarded as the major sites of mtROS production, but more recent studies indicate that at least ten other mitochondrial enzymes also contribute, including complex II (Quinlan et al., 2013). That different sites of mtROS production have distinct signaling roles and the primary production sites likely change under different physiological conditions is likely (Quinlan et al., 2013; Sena and Chandel, 2012).
Figure 1. Mitochondrial ROS Signaling Basics.
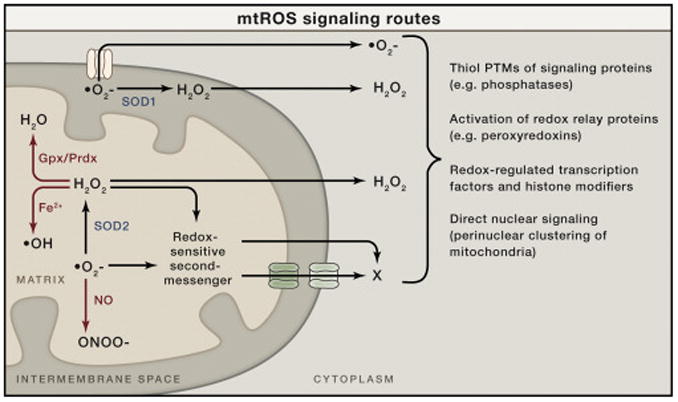
Superoxide (O2–) is generated on both sides of the inner mitochondrial membrane and hence arises in the matrix or the intermembrane space (IMS). Superoxide can be converted to hydrogen peroxide (H2O2) by superoxide dismutase enzymes (SOD1 in the IMS or SOD2 in the matrix). The resulting hydrogen peroxide can cross membranes and enter the cytoplasm to promote redox signaling. Superoxide is not readily membrane permeable but may be released into the cytoplasm through specific outer membrane channels, as shown (see main text). In addition to signaling in the cytoplasm directly, both superoxide and hydrogen peroxide could, in principle, oxidize or modify other molecules in mitochondria that can be released into the cytoplasm to signal (redox-sensitive second messenger; X). These mitochondrial ROS (mtROS) can generate signaling responses and changes in nuclear gene expression in multiple ways (shown to the right). There are other fates of mtROS that would prevent signaling (or potentially enact other signaling and damage responses). For example, superoxide can react with nitric oxide (NO) to form peroxinitrite (ONOO–). This would prevent its conversion to hydrogen peroxide, could cause damage by the highly reactive peroxynitrite, and could potentially limit NO availability for its own type of signaling. Hydrogen peroxide can be eliminated enzymatically by glutathione peroxidase (Gpx) in the matrix or peroxiredoxins (Prdx) in the matrix and elsewhere in the cell. Peroxyredoxins can also promote redox signaling by promoting disulfide bond formation in target proteins. Finally, in the presence of transition metals, hydrogen peroxide can generate damaging hydroxyl radicals (OH).
That hydrogen peroxide has signaling roles in cells was initially elucidated through studies of receptor tyrosine kinase, growth-factor signaling that showed bursts of ROS production by NADPH oxidase (NOX) enzymes. A major mechanism at play in this scenario is the inactivation of redox-sensitive protein tyrosine phosphatases that normally downregulate these receptors (via dephosphorylation) by localized NOX-dependent production of hydrogen peroxide. Several paradigms emerge from these studies that are relevant to mitochondrial hydrogen peroxide acting as a signal (Finkel, 2012). First, many NOX enzymes produce extracellular superoxide that dismutates to hydrogen peroxide that is then is transported across the plasma membrane, perhaps in a regulated manner through aquaporin channels, to effect localized redox signaling. In a similar manner, superoxide produced in the mitochondrial inner-membrane space or matrix can be converted to hydrogen peroxide by SOD1 or SOD2, respectively, allowing it to diffuse into the cytoplasm to signal (Figure 1). Whether this involves free diffusion or facilitated diffusion through specific channels in mitochondrial membranes remains unclear. Second, the inactivation of phosphatases by hydrogen peroxide occurs through the modification of specific reactive thiol side chains (e.g., cysteine). It is now recognized that cysteine residues on many proteins can undergo a variety of redox-dependent modifications, including sequential oxidation (to sulfenic, sulfinic, and sulfonic acid), glutathiolation, and S-nitrosation (Go et al., 2015). Like phosphorylation, ubiquitination, and other post-translational modifications, these redox modifications can alter protein structure and function and be regulatory. Therefore, selective oxidation or modification of redox-dependent thiols in regulatory proteins allows for intricate cellular redox-switch control mechanisms (Finkel, 2012; Go et al., 2015). Redox regulatory proteins that associate with or are otherwise selectively tuned to readout mitochondrial hydrogen peroxide production would provide a mechanism for mtROS signaling (Figure 1). Perinuclear clustering of mitochondria has also been postulated to be a mechanism for direct mitochondrial-nuclear signaling via mtROS (Al-Mehdi et al., 2012).
Superoxide is often summarily dismissed as a relevant signaling molecule because of its chemical properties. For example, unlike hydrogen peroxide, it is a negatively charged molecule and hence not able to easily diffuse across cell membranes, and it does not engage in protein cysteine oxidation reactions conducive to known redox-switch mechanisms of signaling (Winterbourn, 2008). However, as we will discuss, physiologically relevant, superoxide-mediated signaling does appear to exist that is distinct from hydrogen-peroxide-mediated signaling pathways. Although not “freely” diffusible, mitochondrial superoxide can be released from the intermembrane space into the cytoplasm through the voltage-dependent anion channel that spans the outer mitochondrial membrane (Han et al., 2003) (Figure 1). This includes superoxide that is generated in the intermembrane space by complex III, as well as that generated in the matrix by complex I (and potentially by other enzymes) (Lustgarten et al., 2012). However, the latter may require superoxide levels to cross a critical threshold (e.g., when antioxidant defenses are limiting). In the budding yeast, S. cerevisiae, mitochondrial superoxide is released into the cytoplasm by a specific isoform of the voltage-dependent anion channel (Por1p) or, in the absence of Por1p, through the TOM protein import complex (Budzińska et al., 2009). Thus, superoxide released from mitochondria can, in principle, participate directly in cytoplasmic signaling processes (Figure 1). Finally, it is possible that mitochondrial matrix superoxide signals to the cytoplasm via second-messenger systems that involved released oxidized components (e.g., heme or FeS clusters) or other redox sensors have yet to be defined (Figure 1).
Mitochondrial ROS Signaling in Organismal Physiology: Lessons from Model Systems
There is now extensive evidence from the study of model organisms supporting an active role for mtROS signaling in organismal physiology and adaptive responses (Hamanaka and Chandel, 2010; Ristow and Zarse, 2010; Yun and Finkel, 2014). The tractability of these genetic model systems has led to elucidation of new molecular details and signaling pathways underlying these responses. In this regard, much has been learned in the context of aging and longevity studies, which will be highlighted here.
In the nematode worm, C. elegans, several pathways that extend lifespan involve increased mitochondrial ROS production and signaling. These studies have called into question the “mitochondrial” and “free radical” theories of aging (at least as originally formulated) by implicating ROS as pro-longevity signals as opposed to damaging, pro-aging agents, as they've long been viewed. Ristow and colleagues broke initial ground in this area by showing that reduced glucose availability leads to increased mitochondrial respiration and mtROS production that delays worm aging (Schulz et al., 2007). They and others have subsequently found that increased mtROS is a common downstream event in many conserved longevity-promoting interventions, which has led to the concept of “mitohormesis” (Ristow and Zarse, 2010; Yun and Finkel, 2014). Recent work in this area includes mtROS signaling in the anti-aging effects of reduced insulin/IGF signaling and D-glucosamine supplementation (Weimer et al., 2014; Zarse et al., 2012). Inhibition of the mitochondrial ETC by certain mutations or inactivation of mitochondrial SOD2 increases worm lifespan and has been causally linked to increased mtROS production (Dancy et al., 2014). Hekimi and colleagues have recently shown that this involves a unique form of activation of apoptotic signaling cascades to promote protective stress responses rather than apoptosis (Yee et al., 2014). Longevity-extending effects of mtROS in worms are also mediated by HIF1 and AMP kinase signaling and are linked to some degree to enhanced immunity (Hwang et al., 2014; Lee et al., 2010). Like in worms, mtROS signaling extends chronological lifespan in S. cerevisiae, which, in part, is how reduced TORC1 signaling mediates longevity in this organism (Bonawitz et al., 2007; Pan et al., 2011; Schroeder et al., 2013). Here, the mtROS signal activates the DNA-damage-sensing kinases, Tel1p and Rad35p (yeast orthologs of ATM and Chk2), leading to enhanced subtelomeric silencing via inactivation of the jumonji-family, H3K36 demethylase, Rph1p (Schroeder et al., 2013). This response, vis-à-vis the mtROS-mediated, apoptotic-signaling response in worms discussed above (Yee et al., 2014), suggests that a new paradigm is emerging whereby canonical stress-response pathways (e.g., DNA repair and apoptosis) are utilized differentially to sense mtROS to elicit adaptive, homeostatic responses instead of the emergency and cell death responses for which they were defined originally.
While the above discussion was limited largely to examples from worms and yeast, it is important to note that similar mtROS longevity pathways have been shown to operate in other invertebrates and in mice (Hekimi et al., 2011; Ristow and Schmeisser, 2011). Furthermore, these pathways are not limited to anti-aging responses. For example, mtROS signaling has also been implicated in other homeostatic pathways and processes, including wound healing (Xu and Chisholm, 2014), survival under hypoxia (Schieber and Chandel, 2014), intracellular pH homeostasis (Johnson et al., 2012), cell differentiation (Hamanaka and Chandel, 2010; Hamanaka et al., 2013; Tormos et al., 2011), and innate immunity (West et al., 2011). Accordingly, the remainder of this Review will be devoted to the role of mtROS in whole-body physiology, with the main focus on neuroendocrine control of systemic metabolism in mammals.
ROS Generation and Central Control of Whole-Body Metabolism
The amount of mtROS generated in metabolic processes depends on the fuel load and type (lipid, carbohydrate, protein), as well as the amount, composition, activity, and dynamics of mitochondria in the cell or tissue involved. Because ROS are de facto by-products of mitochondrial oxidative metabolism, it may not be surprising that studies have connected mtROS to neuroendocrine control of metabolism, including feeding behavior, energy expenditure, and glucose homeostasis (Andrews et al., 2008; Benani et al., 2007; Diano et al., 2011; Horvath et al., 2009; Leloup et al., 2006; Long et al., 2014).
Modulation of mtROS Production by Uncoupling Protein 2 in the Brain
The discovery of new members of the uncoupling protein (UCP) family in 1997 (Fleury et al., 1997) and the localization of UCP2 to specific brain areas (Horvath et al., 1999; Richard et al., 1998) initiated studies by several groups to unmask what these proteins might do in neurons and in other brain cells. Regardless of the wealth of information gained in these studies, there remains a great ambiguity about the precise role of these UCPs in cellular functions (Brand and Esteves, 2005).
There is little debate regarding the functional relevance of UCP1 in brown adipose tissue, where it promotes mitochondrial fatty acid oxidation by uncoupling mitochondrial electron transport from ATP generation under adrenergic and thyroid control (Ricquier, 1998). Under this scenario, the gained energy is dissipated in the form of heat, which is the hallmark of non-shivering thermogenesis (Ricquier, 1998). UCP2 is clearly not an uncoupler, as UCP1 and its precise mode of action remain unclear. Furthermore, none of the cells in the brain that express UCP2 display the aforementioned features of brown adipocytes. At baseline, UCP2 in rodents and primates is expressed predominantly in neurons of basal structures of the brain (Diano et al., 2000; Horvath et al., 1999; Richard et al., 1998). However, UCP2 is induced in many brain sites in response to cellular injury inflicted by physical insults (Bechmann et al., 2002), epileptic seizures (Diano et al., 2003), or ischemia (Deierborg et al., 2008). These seminal observations, together with studies unmasking the bidirectional regulatory relationship between UCP2 and ROS (Arsenijevic et al., 2000; Echtay et al., 2002; Negre-Salvayre et al., 1997), underscore the potential importance of this mitochondrial protein in mtROS control during cellular stress. Pursuit of the physiological functions of UCP2 in normal brain gave further support for this notion.
Initial studies in normal brain implicated UCP2 protein expression in specific subpopulations of neurons that control hunger, energy expenditure, glucose metabolism, and circadian rhythms (Horvath et al., 1999). Simultaneously, but independently, the sites of action of the hunger-promoting peripheral hormone, ghrelin (Cowley et al., 2003), revealed virtually complete overlap of ghrelin action in the brain and UCP2 expression. Eventually it became clear that most cells in the brain that express ghrelin receptors also express UCP2 (Andrews et al., 2008). These observations spurred the interrogation of whether the influence of ghrelin on appetite and food intake is mediated by UCP2 and, if so, via what cellular and intercellular mechanisms (Andrews et al., 2008; Diano and Horvath, 2012; Horvath et al., 2009). Through these investigations, the following chain of events was uncovered regarding UCP2, mtROS, and neuronal activity (Figure 2): (1) ghrelin induces NPY/AgRP neuronal firing via activation of its receptor, GHSR (growth hormone secretogouge receptor), which in turn activates AMP kinase (AMPK); (2) AMPK activation suppresses acetyl CoA carboxylase (ACC) activity, eliminating the inhibitory effect of malonyl-CoA on carnitine palmitoyl transferase 1 (CPT1) activity; (3) CPT1 activation enhances long-chain fatty acid oxidation by mitochondria and the generation of mtROS; (4) ROS, together with fatty acids, promotes UCP2 gene transcription and activity; (5) UCP2, via enhancing proton leak, tempers mtROS production, allowing continuous fatty acid oxidation without oxidative stress burden and transcription of genes that promote mitochondrial biogenesis and activity (e.g., NRF1), enabling continuous support of the bioenergetic needs of sustained firing of NPY/AgRP cells; and (6) activity of NPY/AgRP neurons results in activity-dependent synaptic plasticity and inhibition of POMC neurons. The intracellular signaling of AgRP neurons is supportive of neuronal activation and decreases vulnerability of these neurons to cellular stress. It was also suggested that the long-chain fatty acyl CoAs utilized under these conditions arise from the periphery under the control of the hypothalamic NPY/AgRP neurons (Andrews et al., 2008). Knocking out UCP2 diminished the ability of AgRP neurons to inhibit mtROS production and maintain low levels of cellular ROS, which, in turn, impairs their neuronal functions (Andrews et al., 2008). Consistent with this model, cellular and electric activity of AgRP neurons is restored, together with reversal of impaired feeding behavior, when a ROS-scavenging cocktail containing L-cysteine is infused into the parenchyma of the hypothalamus (Andrews et al., 2008). These results strongly indicate that behaviors associated with low-energy availability hinge significantly on mtROS signaling associated with lipid metabolism in key neurons that drive these organismal adaptations. However, we recognize that UCP2 likely controls mtROS indirectly, via alteration of mitochondrial fuel utilization, and that other consequences of UCP2 activity in regulating metabolism may also be important (Andrews et al., 2008; Diano and Horvath, 2012; Horvath et al., 2009; Pecqueur et al., 2009; Vozza et al., 2014).
Figure 2. Schematic Illustration of Hypothalamic Control of Negative Energy Metabolism with Low ROS.
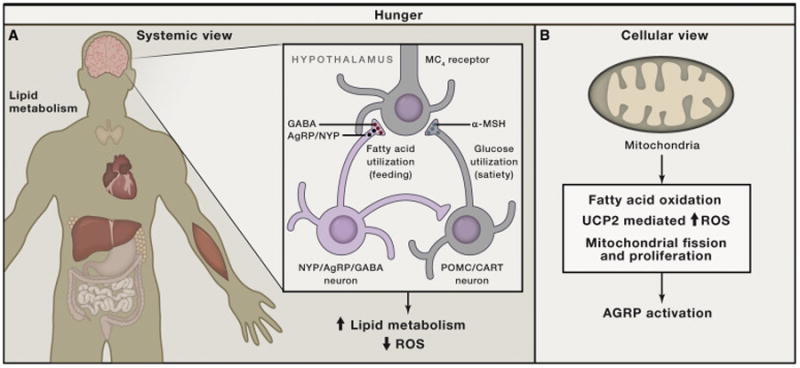
(A) In the brain, the hypothalamus contains neuronal populations that control hunger (negative energy balance) and satiety (positive energy balance). Hunger state is promoted by neurons (green) that produce Agouti-related peptide (AgRP) and neuropeptide Y (NPY), as well as GABA. When these neurons are active (hunger, calorie restriction, starvation), systemic metabolism is shifting to lipid metabolism with an overall lower level of mtROS production in all tissues.
(B) The activation of AgRP neurons during negative energy balance is promoted by pathways enabling long-chain fatty acid oxidation in the mitochondria, which is enabled by maintenance of low mtROS generation by engagement of UCP2 and mechanisms that propagate fission and/or proliferation of mitochondria (NRF1, Sirt1, and PGC1α).
(C) UCP2 is believed to function as a conditional mitochondrial uncoupler in the presence of long-chain fatty acids and, as such, reduces mtROS production and overall cellular ROS levels.
The studies outlined above also suggested the exact opposite scenario for those neurons that support cessation of eating once enough food is consumed (satiety). The hypothalamic neurons that produce pro-opiomelanocortin (POMC) and related peptides are located in the same area as the hunger-promoting AgRP neurons that, when active, suppress POMC neuronal activity. POMC neurons appear to have elevated ROS, as indicated by intracellular dihydroethidium (DHE) staining (Andrews et al., 2008; Diano et al., 2011), when they are active to promote satiety and increased energy expenditure after food consumption (Figure 3). Elevated mtROS production is a logical POMC neuronal activator, since it would likely correlate with mitochondrial activation during full oxidation of glucose, the main fuel of these neurons (Parton et al., 2007). When ROS levels are suppressed chemically, POMC neurons are hyperpolarized and their firing rate declines (Diano et al., 2011). Conversely, when in-slice preparations of POMC neurons are exposed to hydrogen peroxide, they become depolarized and their firing rate is elevated (Diano et al., 2011). These results indicate that it is actually mtROS, rather than glucose itself, that instigate POMC neuronal firing (Diano et al., 2011; Long et al., 2014). Because hypothalamic POMC neurons are involved in both behavioral and autonomic control of energy and glucose metabolism, it is not surprising that hypothalamic ROS control has been tied to all of these processes (Figure 3).
Figure 3. Schematic Illustration of Hypothalamic Control of Positive Energy Metabolism with Elevated ROS.
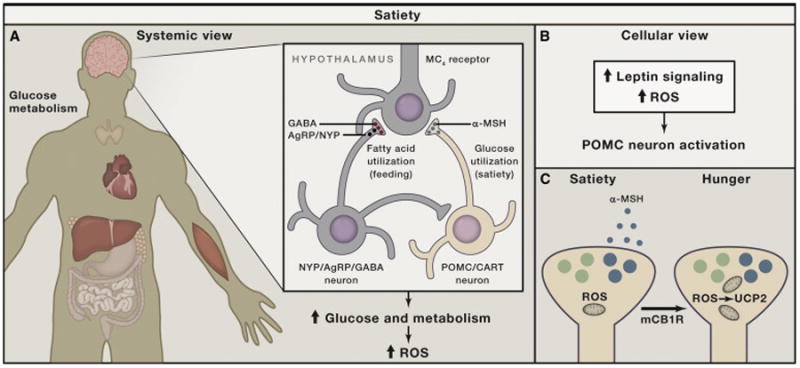
(A) Satiety (feeling full) is promoted by hypothalamic neurons (blue) that produce pro-opiomelanocortin (POMC)-derived peptides, such as α-MSH, which, in turn, act on melanocortin-4-receptor-containing neurons. When these neurons are active, systemic metabolism is shifting toward glucose utilization, with enhanced mtROS production contributing to increased cellular ROS in various tissues.
(B) The activation of POMC neurons after a meal is accomplished by ROS, in part driven by increased mtROS production, and is supported by intracellular leptin (Jak/Stat) and insulin (PI-3K/PTEN) signaling, involving altered K-ATP channel activity.
(C) Recent studies indicate that, under unique circumstances (e.g., activation of cannabinoid receptors), POMC neurons, while still driven by ROS, will become promoters of hunger rather than satiety because they shift to releasing appetite-stimulating opiates (β-endorphin) though UCP2-dependent mitochondrial adaptations.
ROS are Satiety Signals
From a behavioral and systemic perspective, under normal conditions, elevated hypothalamic ROS levels are permissive of suppression of eating, increased energy expenditure, and glucose utilization by peripheral tissues (Andrews et al., 2008; Benani et al., 2007; Diano et al., 2011; Leloup et al., 2006; Long et al., 2014). In fact, it is reasonable to conclude that ROS signaling in the brain, as well as in the periphery, is fundamental for appropriate behavioral and autonomic adaptations to energy surplus, for example, after the consumption of a meal. If one interferes with ROS in these physiological processes, both behavioral and autonomic correlates of proper fuel management of the body will be impaired (Andrews et al., 2008; Diano et al., 2011; Horvath et al., 2009; Long et al., 2014). Since mitochondrial perturbations via UCP2 modulate these responses, we conclude that mtROS signaling plays a fundamental and crucial role in physiological regulation of systemic metabolism.
The relentless pursuit of available energy sources in the environment is mandatory for organismal survival, and hence, hunger and hunger-controlled circuits motivate this critical behavior. However, advanced animal species, including humans, also developed the capacity to maintain energy reserves, for example, in the form of fat or glycogen, so they do not have to continuously feed to survive. This evolutionary advantage, by default, demands that “gauges” and “switches” are in place to shut off feeding when storage capacity is sufficient. The role of the hypothalamic melanocortin system appears to represent both the gauge and the switch, with ROS, likely driven by mtROS production, being the sensor. When cellular ROS reach a threshold, they activate POMC neurons, enabling the cessation of feeding and initiation of storage and utilization of fuels via processes controlled by insulin and leptin (Varela and Horvath, 2012). The control of insulin release itself is regulated by redox events (Bashan et al., 2009). At the same time, elevating ROS in the hypothalamus reduces activity of AgRP neurons that propagate hunger (Andrews et al., 2008) (Figure 2). While it remains unknown what underlies the differential effect of ROS on POMC and AgRP neurons, cell-specific expression of plasma membrane channels could be involved. For example, superoxide can directly alter the activity of potassium channels (Avshalumov and Rice, 2003), which play important roles in glucose sensing by hypothalamic neurons and under regulatory control by UCP2 (Parton et al., 2007). Likewise, it remains unclear how mtROS signals reach neuronal perikarya. In this regard, it is noteworthy that mitochondria in both AgRP and POMC neurons dynamically change their morphology and localization within a short period of time (Andrews et al., 2008; Coppola et al., 2007; Dietrich et al., 2013; Schneeberger et al., 2013). Mitochondrial fission and fusion capacity and the physical interaction between mitochondria and the endoplasmic reticulum may be involved in mtROS production or associated signaling events (Dietrich et al., 2013; Nasrallah and Horvath, 2014; Schneeberger et al., 2013).
It is important to note that ROS are likely not the satiety signal in the hypothalamus under all circumstances. For example, the known effect of cannabinoids on promoting ferocious appetite, regardless of metabolic satiety, is actually mediated by mtROS-driven POMC neurons (Koch et al., 2015). However, under cannabinoid influence, POMC neurons reverse their function and promote appetite because they switch from the release of its satiety-promoting neuropeptide, α-melanocyte-stimulating hormone (α-MSH), to β-endorphin (Koch et al., 2015). This switch is enabled by a mitochondrial adaptive response controlled by UCP2 (Koch et al., 2015) (Figure 3C). Whether this response is specific for this pharmacological situation or relevant to regulation of metabolism under certain physiological circumstances is unknown.
Mitochondrial ROS and Exercise
A key feature of animal species is their need to physically relocate in a rapid and predictable fashion to find food, reproduce, or escape danger. This is accomplished, in grosso modo, through behavioral adaptations, which are the sum of coordinating sensory and effector functions via the communication between the nervous system (the sensory component) and the musculoskeletal system (the effector component). Movement and exercise, in general, have long been recognized as key to supporting not only the aforementioned fundamental biological needs, but also tissue health and longevity. Ristow and colleagues showed that suppressing ROS generation during aerobic exercise (likely, in large part, mtROS) diminishes beneficial outcomes on many exercise-related parameters (Ristow et al., 2009). They also argue for the critical relevance of mtROS signaling transients as important contributors to longevity, as well as mediators of other signaling responses that promote healthspan and longevity (Schmeisser et al., 2013; Zarse et al., 2012). In support of the notion that mtROS transients (and not sustained elevated ROS levels) mediate the benefits of exercise on integrative physiology, UCP2 was found crucial to support exercise-induced synaptogenesis in the dentate gyrus of the hippocampal formation, a key site of spatial learning (Dietrich et al., 2008). This same mechanism is also relevant to hippocampal development (Simon-Areces et al., 2012) and lifespan determination (Andrews and Horvath, 2009).
Short- and Long-Term Effects of ROS
The distinction between physiologically beneficial short ROS bursts and pathological, sustained high ROS levels on cellular and circuit integrity is best illustrated in the response of the hypothalamus to exposure to calorie-dense diets containing high levels of fats and carbohydrates (Diano et al., 2011; Parton et al., 2007). On regular chow diet, which contains <20% fat, ROS levels fluctuate between hunger and satiety states and mice maintain a positive correlation between hypothalamic ROS levels, circulating leptin levels (the adipose hormone that signals to the hypothalamus when sufficient amount of food is consumed), and activity of POMC neurons (Diano et al., 2011). However, when animals are placed on calorie-dense diets (>40% fat), homeostatic control of energy metabolism is gradually deregulated. When this occurs, animals steadily increase their fat stores, which results in steady elevation of circulating leptin. Under homeostatic conditions, this elevated leptin would decrease feeding and enable maintenance of fat stores. This inability of elevated leptin levels to avoid or reverse weight gain is called leptin resistance, for which many cellular and tissue mechanisms have been proposed to explain. One of these mechanisms relates to the aforementioned mtROS control in POMC and AgRP neurons. On a high-fat diet, the positive correlation between hypothalamic ROS, circulating leptin levels, and POMC neuronal activity in mice deteriorates (Diano et al., 2011). Under these conditions, hypothalamic ROS levels plateau and do not follow the robust and steady elevation in circulating leptin concentrations. At the same time, POMC neuronal activity is diminished (Diano et al., 2011). The underlying cause for this dysregulation is tied to PPARγ-related proliferation of peroxisomes and increased ROS elimination by catalase (Diano et al., 2011; Long et al., 2014). We suggest that leptin affects this process by enabling increased glucose uptake in POMC and AgRP neurons, which is accompanied by increased lipid load. Initially, this will lead to increased mtROS production and crossing cellular ROS thresholds that promote satiety by activating POMC neurons. However, rising leptin levels on high-fat diet will continue to promote glucose uptake by these neurons, which, together with increasing lipid load, will overburden these postmitotic cells. The scenario in which lipid and carbohydrate load is increasing in cells provides a perfect energetic basis for growth. Carbohydrate oxidation will dominate mitochondrial OXPHOS and ATP generation, while long-chain fatty acids are diverted from mitochondria via the malonyl-CoA shuttle for biogenesis of membranes and cell growth. However, in cells whose growth is strictly limited, such as neurons of the adult central nervous system, lipids cannot be continuously utilized for membrane biogenesis and they will accumulate in various intracellular compartments, including the endoplasmic reticulum. The activation of peroxisome proliferation under these circumstances provides an alternative mechanism through which excess fat within cells can be eliminated via mitochondrial β oxidation that is less coupled to ATP generation. While this is a beneficial process to prevent lipotoxicity, peroxisomal catalase activity will limit ROS generation needed for proper signaling from the hypothalamus to diminish feeding and increase energy expenditure (Diano et al., 2011). These alterations may have multiple negative effects on cellular signaling mechanisms and organelle integrity and function.
A “Fuel Hypothesis” of Cellular Function
While the above details on mtROS-related mechanisms were described in relation to a hypothalamic circuit that controls feeding behavior and peripheral fuel partitioning and utilization, these processes are likely relevant to the functionality and impairment of neurons in various parts of the brain. For example, UCP2-dependent control of mtROS was also found in dopamine neurons in the midbrain substantia nigra, where both normal functioning of these cells and their protection under cellular stress were attributed to this mechanism (Andrews et al., 2005, 2006; Conti et al., 2005). Dopamine cells in this area of the brain are connected to control of fine motor functions and complex motivated behaviors. A role for the peripheral metabolic hormone, ghrelin, was identified to modulate the activity of these neurons and to prevent their impairment and death in models of Parkinson's disease (Abizaid et al., 2006; Andrews et al., 2009). The intracellular signaling pathway that enabled these beneficial effects of ghrelin was related to ROS control by the same machinery as described above in relation to the control of feeding (Andrews et al., 2008). Similar ghrelin action was also found in the hippocampus to promote learning and memory and to ameliorate deficits of animals in a model of Alzheimer's disease (Diano et al., 2006). Because the changing metabolic state (hunger ↔ satiety) is closely tied to predictable changes in complex behaviors, it is reasonable to suggest that fluctuating ROS levels (mediated by alterations in mtROS output) in all or part of the brain play a critical regulatory role in the synchronization of neuronal circuit activity in support of continuous and appropriate behavioral adaptations. Furthermore, mtROS-controlled neuronal activity in the hypothalamus is sufficient to affect complex behaviors beyond feeding. For example, actute activation of hypothalamic AgRP neurons rapidly alters stereotypic behaviors, locomotion, and anxiety (Dietrich et al., 2015). Finally, we speculate that purposeful alterations in mtROS production to effect cellular redox signaling pathways (Figure 1) will regulate homeostasis in other tissues. In simple terms, it is reasonable to assume that cellular functions in any tissue are determined by fuel availability, uptake, and utilization and that these “fuel” principles drive and orchestrate signaling modalities, including mtROS signaling, to control homeostatic and adaptive responses. For example, intracellular metabolic pathways have distinct and dominant impacts on various immune cell types (Caro-Maldonado et al., 2012; Procaccini et al., 2010), and UCP2-dependent mtROS regulation is connected to both adaptive and innate immune cell functions (Arsenijevic et al., 2000; Horvath et al., 2003; Krauss et al., 2002). How other cells and tissues respond to such signals and the intersection between cell-intrinsic signaling and control by the CNS is an exciting area of future research. In this regard, determining whether the aforementioned CNS processes are mediated by cell-non-autonomous mtROS-mediated signals similar to those documented in C. elegans downstream of ETC disruption and mtROS (Durieux et al., 2011; Schieber and Chandel, 2014) will be important to consider.
Challenges in ROS Research and Ramifications of ROS as Central Controllers of Organismal Homeostasis
In this Review, we have summarized how changes in mtROS production can impact cellular ROS thresholds and redox signaling events that control basal physiological functions and adaptive responses. As such, we argue that these pathways are critical for organismal homeostasis, stress responsiveness, health, and longevity. However, these pathways are far from understood and are in need of more intensive study. Some current impediments and other relevant considerations as the field moves forward in this area are covered below.
At present, it remains very difficult to effectively measure ROS in cells and in vivo. The use of commercially fluorescent ROS probes is widespread but often without the knowledge that these do not always readout specific ROS species faithfully and are prone to other confounding artifacts (Kalyanaraman et al., 2012). That is not to say that these are not useful to a degree, but they should not be used as the only line of evidence to implicate ROS in a response. Development of better ROS assays and probes is ongoing (Ezeriņa et al., 2014; Logan et al., 2014; Woolley et al., 2013), yet there remains a great need for additional forward progress in this important area.
There are important implications for ROS as physiological signaling molecules that impact therapeutic strategies. Two that immediately come to mind are the use of antioxidants and anti-obesity strategies that target the CNS.
Antioxidants have long been considered potential therapeutics for a number of conditions involving oxidative stress. In general, trials using these have failed, likely, in part, because of unintentional inhibition of important basal and adaptive ROS signaling pathways. It has even been argued that taking antioxidants as daily dietary supplements might also perturb these ROS pathways in ways that are not beneficial or even harmful (Ristow, 2014). Similarly, strategies that target activation of POMC neurons (to promote satiety) as an anti-obesity/anti-diabetic strategy might also be confounded due to perturbation of ROS signaling circuits that we have described herein (Dietrich and Horvath, 2012). That is, we assert that many compounds that activate POMC neurons will, by default, upregulate ROS production and signaling that governs their activity (Diano et al., 2011). If this scenario is maintained for a prolonged period of time (hours, days, months), weight loss may be accomplished, but sustained ROS levels could have a multitude of unintended detrimental consequences. As these examples point out, previously held views of ROS as just damaging agents that need to be eliminated are out of date, and a new appreciation of their signaling roles is important to consider going forward. The fact that mitochondria are major producers of ROS also highlights the importance of better understanding what controls their activity and rate of mtROS production, both in terms of redox signaling and oxidative stress. In this regard, the greater recent appreciation of mitochondria as important signaling hubs (Chandel, 2014; West et al., 2011) has begun to transcend older, over-simplified views of these organelles as just sites of intermediary metabolism and ATP production.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5:ra47. doi: 10.1126/scisignal.2002712. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Erion D, Beiler R, Liu ZW, Abizaid A, Zigman J, Elsworth JD, Savitt JM, DiMarchi R, Tschoep M, et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci. 2009;29:14057–14065. doi: 10.1523/JNEUROSCI.3890-09.2009. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elsworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci. 2005;25:184–191. doi: 10.1523/JNEUROSCI.4269-04.2005. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Horvath TL. Uncoupling protein-2 regulates lifespan in mice. Am J Physiol Endocrinol Metab. 2009;296:E621–E627. doi: 10.1152/ajpendo.90903.2008. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Rivera A, Elsworth JD, Roth RH, Agnati L, Gago B, Abizaid A, Schwartz M, Fuxe K, Horvath TL. Uncoupling protein-2 promotes nigrostriatal dopamine neuronal function. Eur J Neurosci. 2006;24:32–36. doi: 10.1111/j.1460-9568.2006.04906.x. PubMed. [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. PubMed. [DOI] [PubMed] [Google Scholar]
- Avshalumov MV, Rice ME. Activation of ATP-sensitive K+ (K(ATP)) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc Natl Acad Sci USA. 2003;100:11729–11734. doi: 10.1073/pnas.1834314100. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev. 2009;89:27–71. doi: 10.1152/physrev.00014.2008. PubMed. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Diano S, Warden CH, Bartfai T, Nitsch R, Horvath TL. Brain mitochondrial uncoupling protein 2 (UCP2): a protective stress signal in neuronal injury. Biochem Pharmacol. 2002;64:363–367. doi: 10.1016/s0006-2952(02)01166-8. PubMed. [DOI] [PubMed] [Google Scholar]
- Benani A, Troy S, Carmona MC, Fioramonti X, Lorsignol A, Leloup C, Casteilla L, Pénicaud L. Role for mitochondrial reactive oxygen species in brain lipid sensing: redox regulation of food intake. Diabetes. 2007;56:152–160. doi: 10.2337/db06-0440. PubMed. [DOI] [PubMed] [Google Scholar]
- Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007;5:265–277. doi: 10.1016/j.cmet.2007.02.009. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. PubMed. [DOI] [PubMed] [Google Scholar]
- Budzińska M, Gałgańska H, Karachitos A, Wojtkowska M, Kmita H. The TOM complex is involved in the release of superoxide anion from mitochondria. J Bioenerg Biomembr. 2009;41:361–367. doi: 10.1007/s10863-009-9231-9. PubMed. [DOI] [PubMed] [Google Scholar]
- Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. PubMed. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25–44. doi: 10.1146/annurev-genom-082509-141720. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Maldonado A, Gerriets VA, Rathmell JC. Matched and mismatched metabolic fuels in lymphocyte function. Semin Immunol. 2012;24:405–413. doi: 10.1016/j.smim.2012.12.002. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34. doi: 10.1186/1741-7007-12-34. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti B, Sugama S, Lucero J, Winsky-Sommerer R, Wirz SA, Maher P, Andrews Z, Barr AM, Morale MC, Paneda C, et al. Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. J Neurochem. 2005;93:493–501. doi: 10.1111/j.1471-4159.2005.03052.x. PubMed. [DOI] [PubMed] [Google Scholar]
- Coppola A, Liu ZW, Andrews ZB, Paradis E, Roy MC, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao XB, Diano S. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5:21–33. doi: 10.1016/j.cmet.2006.12.002. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschöp M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661. doi: 10.1016/s0896-6273(03)00063-1. PubMed. [DOI] [PubMed] [Google Scholar]
- Dancy BM, Sedensky MM, Morgan PG. Effects of the mitochondrial respiratory chain on longevity in C. elegans. Exp Gerontol. 2014;56:245–255. doi: 10.1016/j.exger.2014.03.028. PubMed. [DOI] [PubMed] [Google Scholar]
- Deierborg T, Wieloch T, Diano S, Warden CH, Horvath TL, Mattiasson G. Overexpression of UCP2 protects thalamic neurons following global ischemia in the mouse. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1186–1195. doi: 10.1038/jcbfm.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S, Horvath TL. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol Med. 2012;18:52–58. doi: 10.1016/j.molmed.2011.08.003. PubMed. [DOI] [PubMed] [Google Scholar]
- Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med. 2011;17:1121–1127. doi: 10.1038/nm.2421. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S, Matthews RT, Patrylo P, Yang L, Beal MF, Barnstable CJ, Horvath TL. Uncoupling protein 2 prevents neuronal death including that occurring during seizures: a mechanism for preconditioning. Endocrinology. 2003;144:5014–5021. doi: 10.1210/en.2003-0667. PubMed. [DOI] [PubMed] [Google Scholar]
- Diano S, Urbanski HF, Horvath B, Bechmann I, Kagiya A, Nemeth G, Naftolin F, Warden CH, Horvath TL. Mitochondrial uncoupling protein 2 (UCP2) in the nonhuman primate brain and pituitary. Endocrinology. 2000;141:4226–4238. doi: 10.1210/endo.141.11.7740. PubMed. [DOI] [PubMed] [Google Scholar]
- Dietrich MO, Andrews ZB, Horvath TL. Exercise-induced synaptogenesis in the hippocampus is dependent on UCP2-regulated mitochondrial adaptation. J Neurosci. 2008;28:10766–10771. doi: 10.1523/JNEUROSCI.2744-08.2008. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich MO, Horvath TL. Limitations in anti-obesity drug development: the critical role of hunger-promoting neurons. Nat Rev Drug Discov. 2012;11:675–691. doi: 10.1038/nrd3739. PubMed. [DOI] [PubMed] [Google Scholar]
- Dietrich MO, Liu ZW, Horvath TL. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell. 2013;155:188–199. doi: 10.1016/j.cell.2013.09.004. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich MO, Zimmer MR, Bober J, Horvath TL. Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell. 2015;160:1222–1232. doi: 10.1016/j.cell.2015.02.024. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem. 2002;277:47129–47135. doi: 10.1074/jbc.M208262200. PubMed. [DOI] [PubMed] [Google Scholar]
- Ezeriņa D, Morgan B, Dick TP. Imaging dynamic redox processes with genetically encoded probes. J Mol Cell Cardiol. 2014;73:43–49. doi: 10.1016/j.yjmcc.2013.12.023. PubMed. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, Bouillaud F, Seldin MF, Surwit RS, Ricquier D, Warden CH. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet. 1997;15:269–272. doi: 10.1038/ng0397-269. PubMed. [DOI] [PubMed] [Google Scholar]
- Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic Biol Med. 2015;84:227–245. doi: 10.1016/j.freeradbiomed.2015.03.022. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Glasauer A, Hoover P, Yang S, Blatt H, Mullen AR, Getsios S, Gottardi CJ, DeBerardinis RJ, Lavker RM, Chandel NS. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development Sci Signal. 2013;6:ra8. doi: 10.1126/scisignal.2003638. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. PubMed. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013;23:311–318. doi: 10.1016/j.tcb.2013.02.002. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21:569–576. doi: 10.1016/j.tcb.2011.06.008. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Andrews ZB, Diano S. Fuel utilization by hypothalamic neurons: roles for ROS. Trends Endocrinol Metab. 2009;20:78–87. doi: 10.1016/j.tem.2008.10.003. PubMed. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, Miyamoto S, Barry S, Gatti S, Alberati D, Livak F, Lombardi A, Moreno M, Goglia F, et al. Uncoupling proteins-2 and 3 influence obesity and inflammation in transgenic mice. International journal of obesity and related metabolic disorders: journal of the International Association for the Study of Obesity. 2003;27:433–442. doi: 10.1038/sj.ijo.0802257. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Warden CH, Hajos M, Lombardi A, Goglia F, Diano S. Brain uncoupling protein 2: uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J Neurosci. 1999;19:10417–10427. doi: 10.1523/JNEUROSCI.19-23-10417.1999. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang AB, Ryu EA, Artan M, Chang HW, Kabir MH, Nam HJ, Lee D, Yang JS, Kim S, Mair WB, et al. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2014;111:E4458–E4467. doi: 10.1073/pnas.1411199111. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, Allman E, Nehrke K. Regulation of acid-base transporters by reactive oxygen species following mitochondrial fragmentation. Am J Physiol Cell Physiol. 2012;302:C1045–C1054. doi: 10.1152/ajpcell.00411.2011. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519:45–50. doi: 10.1038/nature14260. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Lowell BB. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci USA. 2002;99:118–122. doi: 10.1073/pnas.012410699. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 2014;30:357–391. doi: 10.1146/annurev-cellbio-101011-155756. PubMed. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup C, Magnan C, Benani A, Bonnet E, Alquier T, Offer G, Carriere A, Périquet A, Fernandez Y, Ktorza A, et al. Mitochondrial reactive oxygen species are required for hypothalamic glucose sensing. Diabetes. 2006;55:2084–2090. doi: 10.2337/db06-0086. PubMed. [DOI] [PubMed] [Google Scholar]
- Logan A, Cochemé HM, Li Pun PB, Apostolova N, Smith RA, Larsen L, Larsen DS, James AM, Fearnley IM, Rogatti S, et al. Using exomarkers to assess mitochondrial reactive species in vivo. Biochim Biophys Acta. 2014;1840:923–930. doi: 10.1016/j.bbagen.2013.05.026. PubMed. [DOI] [PubMed] [Google Scholar]
- Long L, Toda C, Jeong JK, Horvath TL, Diano S. PPARγ ablation sensitizes proopiomelanocortin neurons to leptin during high-fat feeding. J Clin Invest. 2014;124:4017–4027. doi: 10.1172/JCI76220. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustgarten MS, Bhattacharya A, Muller FL, Jang YC, Shimizu T, Shirasawa T, Richardson A, Van Remmen H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem Biophys Res Commun. 2012;422:515–521. doi: 10.1016/j.bbrc.2012.05.055. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15:634–646. doi: 10.1038/nrm3877. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah CM, Horvath TL. Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol. 2014;10:650–658. doi: 10.1038/nrendo.2014.160. PubMed. [DOI] [PubMed] [Google Scholar]
- Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1997;11:809–815. [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Schroeder EA, Ocampo A, Barrientos A, Shadel GS. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011;13:668–678. doi: 10.1016/j.cmet.2011.03.018. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. PubMed. [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Alves-Guerra C, Ricquier D, Bouillaud F. UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life. 2009;61:762–767. doi: 10.1002/iub.188. PubMed. [DOI] [PubMed] [Google Scholar]
- Procaccini C, De Rosa V, Galgani M, Abanni L, Calì G, Porcellini A, Carbone F, Fontana S, Horvath TL, La Cava A, Matarese G. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–941. doi: 10.1016/j.immuni.2010.11.024. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard D, Rivest R, Huang Q, Bouillaud F, Sanchis D, Champigny O, Ricquier D. Distribution of the uncoupling protein 2 mRNA in the mouse brain. J Comp Neurol. 1998;397:549–560. PubMed. [PubMed] [Google Scholar]
- Ricquier D. Neonatal brown adipose tissue, UCP1 and the novel uncoupling proteins. Biochem Soc Trans. 1998;26:120–123. doi: 10.1042/bst0260120. PubMed. [DOI] [PubMed] [Google Scholar]
- Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med. 2014;20:709–711. doi: 10.1038/nm.3624. PubMed. [DOI] [PubMed] [Google Scholar]
- Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011;51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. PubMed. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45:410–418. doi: 10.1016/j.exger.2010.03.014. PubMed. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Blüher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. PubMed. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM, 3rd, Bohr VA. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015;25:158–170. doi: 10.1016/j.tcb.2014.11.002. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M, Chandel NS. TOR signaling couples oxygen sensing to lifespan in C. elegans. Cell Rep. 2014;9:9–15. doi: 10.1016/j.celrep.2014.08.075. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S, Priebe S, Groth M, Monajembashi S, Hemmerich P, Guthke R, Platzer M, Ristow M. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol Metab. 2013;2:92–102. doi: 10.1016/j.molmet.2013.02.002. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, Garcia A, Esteban Y, Gonzalez-Franquesa A, Rodríguez IC, Bortolozzi A, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder EA, Raimundo N, Shadel GS. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013;17:954–964. doi: 10.1016/j.cmet.2013.04.003. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. PubMed. [DOI] [PubMed] [Google Scholar]
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. PubMed. [DOI] [PubMed] [Google Scholar]
- Simon-Areces J, Dietrich MO, Hermes G, Garcia-Segura LM, Arevalo MA, Horvath TL. UCP2 induced by natural birth regulates neuronal differentiation of the hippocampus and related adult behavior. PLoS ONE. 2012;7:e42911. doi: 10.1371/journal.pone.0042911. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, Chandel NS. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011;14:537–544. doi: 10.1016/j.cmet.2011.08.007. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela L, Horvath TL. AgRP neurons: a switch between peripheral carbohydrate and lipid utilization. EMBO J. 2012;31:4252–4254. doi: 10.1038/emboj.2012.287. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D, et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci USA. 2014;111:960–965. doi: 10.1073/pnas.1317400111. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer S, Priebs J, Kuhlow D, Groth M, Priebe S, Mansfeld J, Merry TL, Dubuis S, Laube B, Pfeiffer AF, et al. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat Commun. 2014;5:3563. doi: 10.1038/ncomms4563. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. PubMed. [DOI] [PubMed] [Google Scholar]
- Woolley JF, Stanicka J, Cotter TG. Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem Sci. 2013;38:556–565. doi: 10.1016/j.tibs.2013.08.009. PubMed. [DOI] [PubMed] [Google Scholar]
- Xu S, Chisholm AD. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev Cell. 2014;31:48–60. doi: 10.1016/j.devcel.2014.08.002. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell. 2014;157:897–909. doi: 10.1016/j.cell.2014.02.055. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Finkel T. Mitohormesis. Cell Metab. 2014;19:757–766. doi: 10.1016/j.cmet.2014.01.011. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–465. doi: 10.1016/j.cmet.2012.02.013. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]