

Abstract
Abstract Despite the best efforts of influenza scientists, companies and health officials to prepare for the next pandemic, most of the world’s people will not have access to affordable supplies of vaccines and antiviral agents. They will have to rely on 19th century public health ‘technologies’ to see them through. In the 21st century, science ought to be able to provide something better. Influenza scientists study the molecular characteristics of influenza viruses and their signaling effects in cell culture and animal models of infection. While these studies have been enormously informative, they have been unable to explain the system‐wide effects of influenza on the host, the increased mortality of younger adults in the 1918 influenza pandemic and the much lower mortality rates in children who were more commonly infected with the 1918 virus. Experiments by non‐influenza scientists have defined common cell signaling pathways for acute lung injury caused by different agents, including inactivated H5N1 influenza virus. These pathways include several molecular targets that are up‐regulated in acute lung injury and down‐regulated by anti‐inflammatory and immunomodulatory agents, including statins, fibrates, and glitazones. These agents also help reverse the mitochondrial dysfunction that accompanies multi‐organ failure, something often seen in fatal Influenza. Observational studies suggest that statins are beneficial in treating patients with pneumonia (there are no such studies for fibrates and glitazones). Other studies suggest that these agents might be able to ‘roll back’ the self‐damaging host response of young adults to the less damaging response of children and thus save lives. Research is urgently needed to determine whether these and other agents that modify the host response might be useful in managing H5N1 influenza and the next pandemic.
Keywords: Host response, influenza, pandemic, peroxisome proliferator activator receptor agonists, statins
‘It is the information carried by germs that we cannot abide … macromolecules are read by our tissues as the very worst of bad news … we are likely to turn on every defense at our disposal; we will bomb, defoliate, blockade, seal off, and destroy all the tissues in the area … It is a shambles. 1 ’
Vaccines and antiviral agents will fail to meet global needs for the next pandemic
Avian influenza A (H5N1) presents a serious and possibly imminent pandemic threat. 2 , 3 , 4 , 5 , 6 , 7 Vaccine companies have worked hard to develop more efficient H5N1 vaccines, 8 and the promise of using antigen sparing, adjuvanted inactivated vaccines that are cross‐protective is now well established. 9 In a few developed countries, stockpiling pre‐pandemic vaccines is being considered. 10 Nonetheless, if a pandemic virus should emerge within the next few years, the world would have to depend on a limited supply of egg‐based inactivated vaccines.
Two years ago it was estimated that all of the world’s influenza vaccine companies could produce in a 6‐month period (i.e., approximately 9 months after the emergence of the pandemic virus) enough doses of a new pandemic vaccine to vaccinate with two doses approximately 750 million people. 11 , 12 More recently, a report sponsored by the World Health Organization in collaboration with virtually all influenza vaccine companies estimated that 6 months after the emergence of a new pandemic virus, the companies could produce 860 million doses of vaccine. 13 These numbers are similar to the number of people living in the nine countries that produce almost all of the world’s seasonal influenza vaccines. During these early months, the pandemic virus will probably have already spread throughout the world.
Currently, there is no logistical plan for distributing supplies of pandemic vaccines to the ‘have not’ countries that will not be able to produce them. 11 , 12 These countries are home to approximately 88% of the world’s population. In all likelihood, people in these countries will not be able to obtain meaningful supplies of pandemic vaccines or they will get them too late.
Many health officials have placed their hopes on stockpiles of antiviral agents. 11 , 14 , 15 Recently, however, resistance to the most widely stockpiled agent – oseltamivir (Tamiflu®)– has emerged in seasonal influenza viruses, and there is concern now that similar resistance could develop in a future pandemic virus. More important, current government stockpiles of Tamiflu in ‘have not’ countries would be sufficient to treat approximately 1% of the people who live in these countries (DS Fedson, unpublished data). At a scientific meeting held in Singapore in early 2008, investigators reported that among people in Indonesia who had been infected with the H5N1 virus, 33/33 (100%) of those who received no antiviral treatment died. 15 This observation is terrifying: if this virus develops efficient human‐to‐human transmissibility and very few people have access to an effective antiviral agent, we could see a global population collapse. The possibility that this could occur was shown unequivocally in an experimental study published 35 years ago. 15 , 16
Our current ‘top down’ approach to confronting a newly emergent pandemic virus is based solely on developing and producing vaccines and antiviral agents. It relies on the decisions of relatively small groups of elite influenza scientists, national and international health officials and company executives. 14 , 15 As such, this process has been slow and difficult to manage. Despite best efforts, most of the world’s people will not have timely access to effective and affordable pandemic vaccines and antiviral agents. Instead, they will have to rely on 19th century public health ‘technologies’ to see them through. In the 21st century, scientists ought to be able to provide something better, especially as the consequences of not doing so could be disastrous.
This review will suggest ways in which 21st century science could be harnessed to provide new ways of confronting the next influenza pandemic. It will not cover the extraordinary developments in our understanding of the influenza virus itself, the cell signaling events that govern its replication and determine its virulence, or the intricacies of its effects on the humoral and cell‐mediated immunity; all issues covered at length in recent reviews. 2 , 3 , 4 , 5 , 6 , 7 Instead, it will draw on lessons that can be learned from the 1918 pandemic and review selectively new information on the host response to infectious and non‐infectious challenges that often result in multi‐organ failure and death. It will discuss recent studies of experimental influenza and the host response and suggest ways in which currently available agents might be used to manage the next global pandemic.
Understanding the virulence of 1918 and H5N1 influenza viruses alone will not help in managing the next global pandemic
In attempting to explain the high mortality of 1918 and H5N1 influenza, virologists have focused their attention on molecular characteristics of the virus that are associated with receptor specificity, efficient replication, virulence and transmissibility and on the molecular effects of virus replication in cell culture and experimental animals. 2 , 3 , 4 , 5 , 6 , 7 Experimental 1918‐like and H5N1 influenza virus infections are associated with elevated levels of pro‐inflammatory cytokines, often called a ‘cytokine storm’. 2 , 3 , 4 , 5 , 6 , 7 , 17 Similar findings characterize human H5N1 influenza. 18 A broad consensus has emerged that cytokine dysregulation can be controlled and clinical improvement achieved only by early administration of an effective antiviral agent. 19
The increased virulence of the 1918 and H5N1 influenza viruses compared with seasonal influenza viruses is beyond question. Yet laboratory studies have often shown a lack of correlation between levels of virus replication and survival [reviewed in Ref. (14)]. Furthermore, experience with H5N1 influenza has shown that early diagnosis is often not possible, 19 and for a global pandemic adequate supplies of antiviral agents and vaccines simply will not be available.
Bacterial pneumonia cannot account for the majority of deaths in the 1918 pandemic
An added level of complexity has been introduced recently by the suggestion that secondary bacterial pneumonia was the primary cause of mortality in the 1918 pandemic. 20 Support for this notion comes from recent experiments showing that antecedent influenza virus infection greatly increases the risk of death following subsequent pneumococcal pneumonia. 21 In a recent report, Morens et al. reviewed almost 8400 bacterial cultures of specimens taken from patients who died during the 1918 pandemic. 20 After considering their findings and related epidemiological evidence, they found little to implicate the influenza virus as the sole cause of mortality. Instead, they concluded ‘the vast majority of influenza deaths resulted from secondary bacterial pneumonia’. This conclusion has led to recommendations for pre‐pandemic stockpiling of antimicrobial agents as well as antivirals and vaccines.
Morens et al. have written that the ‘copathogenic properties of the 1918 influenza virus may have been generic and not specific to the 1918 virus or to influenza viruses in general. 22 They have found support for their view in the measles epidemics that affected US Army training camps in 1917–1918. Like the influenza pandemic 1 year later, most measles deaths were associated with bacterial pneumonia. The measles outbreaks were ‘the result of an unfortunate ‘natural experiment’ in which young men from remote rural areas, many of whom had escaped measles virus infection in childhood, were brought together in crowded barracks during the winter/spring season of measles circulation. 22 This explanation may be sufficient to explain why trainees became infected, but it does not explain why they died.
Clinicians have known since the time of Osler that not all people who die with pneumonia necessarily die because of pneumonia. 23 In patients with H5N1 influenza, virtually all who have come to medical attention have received antibiotic treatment, yet the mean duration of illness before death has been 10 days, 18 as it was in 1918, 24 For patients with H5N1 influenza who have presented late in the course of illness, bacterial pneumonia should have been evident, yet it has rarely been mentioned. Instead, most have gone on to develop acute respiratory distress syndrome (ARDS) and multi‐organ failure.
In the studies of bacterial pneumonia in the 1918 pandemic, one important epidemiologic feature has received little attention: children had higher influenza attack rates than young adults, yet they seldom developed bacterial pneumonia and their case fatality rates were much lower (Figure 1). 20 , 23 , 24 There is nothing to suggest that the experience of children with measles in 1917–1918 was any different from that of children in earlier years. 22 Virulence factors inherent in the 1918 influenza virus and the cytokine dysregulation caused by infection might help explain the increased mortality of younger adults, but there is no good reason to place the burden of ‘pathogenic responsibility’ entirely on the virus. Doing so overlooks a more important question: why did children live?
Figure 1.
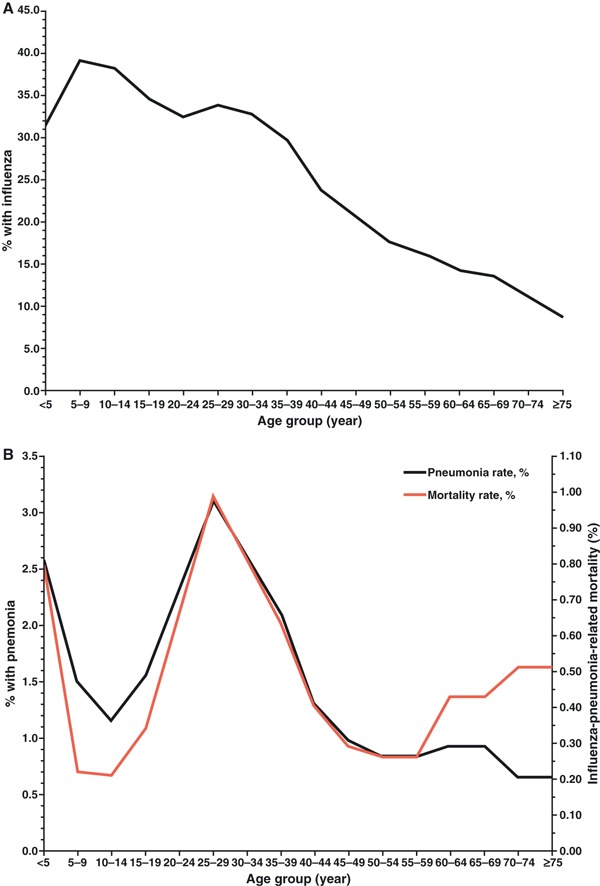
Panel (A) shows the estimated age group‐specific influenza case rates and panel (B) shows the estimated age group‐specific pneumonia rates and mortality rates based on household surveys conducted in 10 US communities [from figure 5 in Ref. (24)].
In attempting to understand the high mortality in the 1918 pandemic, influenza scientists have focused their attention on young adults. 25 Previous exposure to 1918‐like viruses in the late 19th century seems to have given older adults a degree of protection that spared them the mortality experienced by younger adults. For children, however, there has been no satisfactory explanation for why the same virus failed to cause most of them serious harm.
Acute lung injury is associated with many conditions and it is different in children and adults
It is worth considering the host responses of children and adults with sickle cell disease, and malaria and those who experience severe trauma. Persons with any one of these conditions are at risk of developing acute lung injury, yet the disease is mild in children whereas in adults it is more severe and there is greater risk of dying [reviewed in Ref. (23)]. In none of these conditions do bacterial infections contribute significantly to the course of illness. Likewise, children with malaria and schistosomiasis have less vigorous immune responses to their infections than do young adults. 26 , 27 The switch from one host response to the other seems to occur at the time of puberty/menarche/adrenarche. 23 The differences between the two are poorly understood and rarely studied.
In many bacterial, viral and parasitic diseases, there is an appreciable risk of developing acute lung injury. 28 , 29 A similar risk is seen in non‐infectious disorders such as extensive trauma or severe ischemia. In each of these conditions, the cell signaling pathways that respond to pathogen‐associated molecular patterns associated with infection and danger‐associated molecular patterns seen in non‐infectious disorders are much the same. 30 The severe acute lung injury and ARDS that develop in patients with H5N1 and other severe influenza virus infections are probably due to similar mechanisms. 31
Recent studies suggest the host response determines the outcome to severe influenza virus infection
Given the overwhelming need for alternatives to vaccination and antiviral treatment, agents that improve the host response to influenza virus infection must be considered. 11 , 14 , 15 , 31 Although most influenza scientists doubt this approach will work, several studies suggest it might be effective.
In a study of mice massively infected with H5N1 influenza virus (1000 LD50), treatment with zanamivir begun 48 hours after infection reduced lung virus titers but led to little improvement in survival. 32 However, when two immunomodulatory agents (celecoxib and mesalazine) were added, virus titers remained much the same but survival improved significantly. Unfortunately, the investigators failed to include a group of mice that were treated with celecoxib and mesalazine alone. If they had measured survival rates and virus titers in the two groups (two immunomodulators with and without an antiviral agent), they could have determined whether the antiviral agent was necessary for improving survival.
A commentary that accompanied this study emphasized that co‐administration of the two anti‐inflammatory and immunomodulatory agents along with an antiviral agent was essential. 33 Improved survival was ascribed to inhibition of cyclooxygenase (COX)‐2, but it is unlikely that it was due to COX‐2 inhibition alone. The cell signaling pathways involved in the regulation of COX‐2 expression are complex. 34 In other models of acute lung injury, COX‐2 inhibition actually impairs resolution of pulmonary inflammation, probably because it prevents the up‐regulation of pro‐resolution factors such as lipoxin A4. 35 Moreover, mesalamine is not simply a COX‐2 inhibitor; it is primarily a peroxisome proliferator activator receptor (PPAR)γ agonist 36 (see below).
Another report has compared survival rates of mice sequentially infected with influenza virus and S. pneumoniae who were treated with either a cell wall‐active antibiotic (ampicillin) or one of two macrolides known to inhibit bacterial protein synthesis (clindamycin or azithromycin). 37 Ampicillin‐treated mice had lower survival rates, presumably because of increased inflammation caused by the lysis of bacterial cell walls. The better survival of macrolide‐treated animals ‘appeared to be mediated by decreased inflammation as manifested by lower levels of inflammatory cells and pro‐inflammatory cytokines. 37 However, the greater survival of macrolide‐treated mice was probably due to more than inhibition of pneumococcal protein synthesis. Macrolides have well‐documented anti‐inflammatory and immunomodulatory effects that improve the host response to a wide variety of non‐infectious as well as infectious conditions. 38 , 39 , 40 , 41
A masterly study by Imai et al. has defined common set of major cell signaling events in acute lung injury due to different causes. 42 It is well known that in mice, intra‐tracheal instillation of acid or lipopolysaccharide (LPS) reliably induces severe acute lung injury. Imai et al. showed that both of these insults activated pulmonary macrophages. This led to oxidative stress and the formation of reactive oxygen species (ROS). ROS in turn generated large amounts of oxidized phospholipids (OxPLs) derived mostly from cellular debris. OxPLs then triggered the production of pro‐inflammatory cytokines [e.g., interleukin (IL)‐6] via a TLR4/TRIF/TRAF6/nuclear factor (NF)‐kappaB signaling cascade (Figure 2).
Figure 2.

Schematic diagram of the signaling cascade that leads from Toll‐like receptor 4 (TLR4) through TIR‐domain‐containing adaptor‐inducing IFN‐β (TRIF), tumor necrosis factor‐receptor associated factor (TRAF6), and NF‐kappaB to the up‐regulation of pro‐inflammatory cytokines and resultant acute lung injury [from figure 2(J) in Ref. (42)].
What is important about this study is that the same degree of acute lung injury and the same cell signaling cascade was observed following intra‐tracheal instillation of inactivated (not live) H5N1 virus. 42 The same pattern was seen in human peripheral blood mononuclear cells when they were exposed to inactivated H5N1 virus. The pulmonary lesions in mice were histologically identical to those seen in fatal cases of H5N1 influenza. None of these changes was seen with inactivated H1N1 virus.
Among the many factors contributing to pulmonary defenses, several investigators have shown that heme‐oxygenase (HO)‐1 directly affects the initiation of the TLR4 signaling cascade described above. 43 , 44 , 45 , 46 , 47 HO‐1 is a stress responsive enzyme that degrades heme to carbon monoxide, biliverdin and iron. The location of TLRs within cells determines their signaling effects. 46 Carbon monoxide derived from endogenous HO‐1 activity limits ROS‐induced TLR4 signaling by inhibiting the relocalization of TLR4 from the cytoplasm to lipid rafts on macrophage cell membranes. 43 This is one of several ways in which HO‐1 contributes to pulmonary host defenses and improves survival in a wide range of pathologic disorders. 47
The host response to acute lung injury due to any cause is far too complex to be captured in a single study. 48 , 49 The same can be said about the host response in sepsis. 50 , 51 , 52 , 53 Early and late (e.g. high molecular group box 1 (HMGB1)) mediators of inflammation, the balance between inflammatory and anti‐inflammatory (e.g., lipoxin A4) factors, the contributions of innate and adaptive immunity (especially late immunosuppression 50 , 53 ) (see below), disorders of the complement and coagulation systems (and their interactions), autonomic system involvement (cholinergic anti‐inflammatory pathway), endocrine and metabolic dysfunction and disturbances in energy homeostasis all affect outcome in these conditions. Maintaining or restoring a balanced host response seems to be the key to recovery.
Little is known about the molecular events that characterize the overall response of influenza patients who progress to multi‐organ failure and death. Nonetheless, no virus replication occurred in the study of Imai et al., 42 so under these experimental conditions, antiviral treatment would not have affected the outcome.* These findings call into question the claim that antiviral agents are essential if treatment of H5N1 or any other severe influenza virus infection is to be effective. 19 They also suggest that agents capable of interrupting one or more of the steps in the cell signaling cascade demonstrated by Imai et al. might reduce the severity of the acute lung injury seen in H5N1 and pandemic influenza, and in doing so prevent or reverse multi‐organ failure and improve survival.
Anti‐inflammatory and immunomodulatory agents might be effective for treatment and prophylaxis of H5N1 and pandemic influenza
Several lines of experimental and clinical evidence suggest that three classes of drugs – statins, PPARα agonists (fibrates), and PPARγ agonists (glitazones) might individually or in combination prevent H5N1‐associated acute lung injury [reviewed in Refs (11, 14, 15, 31)]. Each of these groups of agents (as well as several others) has been shown to inhibit the cell signaling pathways set in motion by inactivated H5N1 virus (Table 1; DS Fedson, unpublished data). 42
Table 1.
Cell signaling in acute lung injury and the opposing effects of statins, fibrates and glitazones*
| Cell signaling molecules | Acute lung injury | Statins | PPARα fibrates | PPARγ glitazones |
|---|---|---|---|---|
| ROS | ↑ | ↓ | ↓ | ↓ |
| TLR4 | ↑ | ↓ | ↓ | ↓ |
| NFκB ↑ | ↑ | ↓ | ↓ | ↓ |
| IL‐6 | ↑ | ↓ | ↓ | ↓ |
| HO‐1 | – | ↑ | ↑ | ↑ |
*The cell signaling molecules and their activities in acute lung injury are based on findings reported in Ref. (42). The activities of statins, fibrates, and glitazones are based on the author’s unpublished data.
ROS, reactive oxygen species; TLR4, Toll‐like receptor 4; NFκB, nuclear factor‐kappaB; IL‐6, interleukin‐6; HO‐1, heme‐oxygenase‐1.
Statins [hydroxymethyl glutaryl ‐ coenzyme A (HMG‐ CoA) reductase inhibitors] are taken every day by millions of patients with cardiovascular diseases to lower their low density lipoprotein (LDL) cholesterol levels. These agents also have anti‐inflammatory (pleiotropic) effects, 54 and investigators are studying their use in treating patients with sepsis 55 and pneumonia. Several retrospective studies have suggested that prescriptions for statins are associated with an approximately 50% reduction in pneumonia hospitalizations and deaths [reviewed in Refs (14, 15)]. A preliminary report of a randomized controlled trial of statin treatment in 67 ICU pneumonia patients showed that hospital mortality was reduced by 51%. 56 Thus far, no reports have been published showing beneficial effects of statins in cell culture or animal influenza virus infections, although lack of benefit has been mentioned in one report. 5
Investigators often observe a lack of correlation between the results of treatments in experimental animals and in humans. 49 , 57 The evidence for statin benefit in humans with sepsis and pneumonia justifies further studies, including randomized controlled trials. These studies must pay close attention to the pharmacokinetics of each agent; early evidence indicates that acute blood levels of atorvastatin might be much higher in patients with severe acute illness than they are in normal subjects. 58 If statins prove to be effective against pneumonia, they might be similarly effective against H5N1 and pandemic influenza.
Many investigators believe that fibrates – PPARα agonists that lower cholesterol levels – and glitazones – PPARγ agonists used to increase insulin sensitivity in diabetic patients – could also be used to treat acute lung injury. 14 , 15 , 59 Like statins, these agents have anti‐inflammatory and immunomodulatory activities. 59 , 60 Moreover, there is considerable molecular cross‐talk between statins, fibrates, and glitazones, and the pleiotropic effects of statins are achieved because of their interactions with PPARs. 61 , 62 In experimental studies, the cell signaling effects of statins and PPAR agonists (both α and γ) can be additive. 63 , 64 Likewise, in patients with cardiovascular diseases, the effects of therapy on biomarkers of disease are greater with combination than with single agent treatment. Given many years of use in clinical practice, the safety profile for each group of agents is well established. 14
An important study published in 2007 showed that in H2N2 influenza virus‐infected mice, treatment with a common PPARα agonist (gemfibrozil) reduced mortality by 54%. 65 Some have criticized this study because pulmonary virus titers were not measured. Nonetheless, it was structured like a randomized controlled trial of an acute treatment; gemfibrozil was started 4 days following infection when mice were beginning to show signs of clinical illness. Moreover, the investigators used an unambiguous end‐point (death) and like a clinical trial they chose a sample size (96) that gave them statistically significant results.
More recently, Aldridge et al. studied the effects of treatment with pioglitazone (a PPARγ agonist) in influenza‐infected mice. 66 They found that a subset of dendritic cells (DCs) known as tumor necrosis factor (TNF)α/inducible nitric oxide synthase DCs (TipDCs) accumulated with high frequency in the lungs of mice infected with highly pathogenic PR8 virus. TipDCs are known to recruit CCR2‐positive mononuclear cells from the bone marrow and traffic them to sites of pulmonary infection. CCR2‐deficient mice are generally more susceptible to non‐viral infections, but CCR2‐positive monocyte‐derived cells have been shown to be a major cause of the immunopathology of influenza. 67 Aldridge et al. speculated that pioglitazone suppression of CCL2 (the pro‐inflammatory ligand for CCR2) would reduce the number of CCR2‐positive mononuclear cells and increase protection. The results showed that with 3 days of pre‐treatment, mortality fell from 92% to 50%. However, they also found that TipDCs increased the frequency of virus‐specific CD8+ T‐cells in the later stages of infection. As CD8+ T‐cells are critical for influenza virus clearance, TipDCs appeared to induce a protective response. Yet, protection was not reflected in pulmonary virus titers; they were the same in control and pioglitazone‐treated animals. Thus, although pioglitazone was able to ‘tip the balance’ in favor of protection, 68 it must have done so through mechanisms that were independent of its effects on virus replication and clearance.
The study by Aldridge et al. was not designed to test whether pioglitazone could be used to treat an already established infection, unlike the gemfibrozil study discussed above. 65 As such, the findings are similar to those obtained in the observational studies that have shown that patients already taking statins have reduced rates of pneumonia hospitalization and death (i.e., both act as prophylactic agents). 14 , 15 Interestingly, if reducing the number of CCR2‐positive mononuclear cells has any role to play in recovery from influenza, statins are known to suppress CCR2 gene expression and monocyte recruitment, 69 , 70 and might have effects similar to those seen with pioglitazone.
Several other agents with anti‐inflammatory and immunomodulatory or even antiviral activities should be considered for treatment and prophylaxis of H5N1 and pandemic influenza [reviewed in Ref. (14)]. For example, in cell culture chloroquine, a classic anti‐malaria drug, impairs lysosomal acidification, preventing the release into the cytoplasm of viral nucleic acid from H3N2 and H1N1 but not H5N1 influenza viruses [discussed in Ref. (14)]. The many effects of catechins (found in green tea) and curcumin (turmeric in curry) on inflammation and the host response suggest that they too might be beneficial against influenza.
A potentially important but overlooked compound is resveratrol, a commonly available polyphenol found naturally in dark grapes and red wine. In a study of influenza PR8‐infected mice, resveratrol treatment inhibited virus replication and reduced mortality by half. 71 Resveratrol has statin‐like effects on HMG‐CoA, 72 activates PPARα 73 and PPARγ, 74 and synergizes with statins in protecting against experimental myocardial infarction. 75 The effects of resveratrol on ROS, TLR4, NF‐kappaB, pro‐inflammatory cytokines (e.g., TNFα, IL‐6), and HO‐1 are the same as those of statins, fibrates, and glitazones (see Table 1). 76 , 77 , 78 In experimental Serratia marcescens pneumonia in rats, resveratrol has been shown to down‐regulate NF‐kappaB, TNFα, IL‐6, and IL‐1β, increase macrophage infiltration, decrease neutrophil infiltration, reduce the bacterial burden in the lung and improve survival. 78
The report on the efficacy of resveratrol treatment of influenza in mice was published in 2005 by investigators who work outside the influenza scientific community. 71 Remarkably, this important study has gone unnoticed by mainstream influenza scientists.
Other aspects of the host response might be affected by statins, fibrates and glitazones
The pathologic effects of influenza virus infection are mediated though several pathways, of which three might be targets of treatments that modify the host response.
Inflammasomes
Much attention has been given recently to the role of inflammasomes in the host response to influenza virus infection. Inflammasomes are multi‐protein complexes that are responsible for the activation of caspase‐1 that, in turn, generates two pro‐inflammatory cytokines – IL‐1β and IL‐18. 79 Among the three major groups of pattern recognition receptors – TLRs, retinoic acid inducible gene‐I‐like receptors and the Nod‐like receptors (NLRs) – inflammasomes are part of the NLR family of receptors, and they participate in the innate and adaptive immune response. For influenza virus infection, the NLRP3 inflammasome seems to be important.
Two recent studies by Allen et al. 80 and Thomas et al. 81 have examined the responses of PR8‐infected knockout mice deficient in caspase‐1 or NLRP3. Compared with wild‐type mice, mice with either deficiency had lower survival rates and reduced numbers of mononuclear cells and neutrophils in their lungs. Although the histological findings in the lungs of knockout mice in the two studies differed, it was clear from both studies that NLRP3 was protective. Both macrophages and epithelial cells were involved in early NLRP3 signaling, but compared with wild‐type mice, much lower levels of IL‐1β and IL‐18 were found in the bronchoalveolar fluid of mice deficient in caspase‐1 and NLRP3. Neither deficiency, however, had an appreciable effect on the adaptive immune response. 80 , 81 , 82
These studies demonstrate the importance of NLRP3 signaling pathways in mounting a controlled inflammatory response to influenza virus infection. 83 Moreover, the study by Thomas et al. 81 showed that the NLRP3 inflammasome response could be triggered by intra‐peritoneal administration of influenza viral RNA alone. In other words, virus replication was not required to trigger a protective inflammatory response.
These findings might be relevant to those obtained in a study of patients with sepsis. Reductions in caspase‐1 signaling were found in those with septic shock compared with other critically ill patients who were not in shock. 84 Down regulation of caspase‐1 signaling suggested that mononuclear cell dysfunction appeared in patients with more severe illness. Importantly, an experimental study of mitogen‐activated mononuclear cells has shown that statins activate caspase‐1 and increase IL‐18 secretion, thus reversing mononuclear cell dysfunction. 85 Whether statins and other agents (e.g., fibrates and glitazones) would produce the same response in influenza virus infections remains to be determined.
Apoptosis and autophagy
Almost all patients with seasonal and pandemic influenza survive, but for those who die there is little understanding of the factors responsible for their deaths. The emergence of H5N1 influenza and its high case fatality rate has focused attention on the ‘cytokine storm’ that accompanies infection. 2 , 3 , 4 , 5 , 6 , 7 This is surely not the only factor and perhaps not even the main factor responsible. A better understanding of the probable pathogenesis of fatal influenza can gained from studies of fatal sepsis. 50 , 53 . Among sepsis patients who die, few die within the first few days. Most develop a sustained ‘immunoparalysis’ and die much later. Apoptosis (programmed cell death) is the central feature of this late stage of disease. With apoptosis, there is a profound decrease in the numbers of lymphocytes – B‐cells and CD4+ T‐cells, the critical effector cells of the adaptive immune response. DCs are also lost, compromising antigen presentation. Uptake of apoptotic cells by macrophages and DCs stimulates the release of anti‐inflammatory cytokines (e.g., IL‐10 and TGF‐β) and induces immune suppression. Apoptotic cell death can follow extrinsic (caspase‐8‐mediated) or mitochondria‐initiated (caspase‐9‐mediated) pathways, and both are involved in sepsis‐induced lymphocyte depletion. Experimental studies show that caspase inhibition improves survival, but it has been difficult to develop caspase inhibitors suitable for clinical use. 50 , 86
Autophagy is a cellular pathway that is central to cell preservation and turnover. 87 It involves the self‐digestion of proteins and cell organelles that are part of normal homeostatic cell function, but it also involves the response to stress (e.g., starvation, infection). The molecular interactions between autophagy and apoptosis are not well understood, 87 , 88 but ‘coordinated regulation of ‘self‐digestion’ by autophagy and ‘self‐killing’ by apoptosis may underlie diverse aspects of … disease pathogenesis’. 87
Autophagy and apoptosis are features of influenza virus replication. That autophagy is involved is not surprising, 89 as the virus must use the building blocks at hand to form new virus particles. Apoptosis also accompanies influenza virus infection. In cell cultures of human blood macrophages, the onset of apoptosis induced by H5N1 viruses is delayed compared with that for H1N1 viruses, 90 suggesting that intracellular persistence of the H5N1 virus might have something to do with its pathologic effects. In other studies, H5N1 virus (but not H5N2 or H5N3 viruses) was shown to induce caspase‐dependent apoptosis in porcine alveolar epithelial cells, although levels of virus replication for all three viruses were the same. 91 The H5N1 NS1 protein has also been shown to cause caspase‐dependent apoptosis in human lung epithelial cells. 92
In a splendid study of murine influenza, the PB1‐F2 protein of the 1918 influenza virus was shown to cause severe viral and secondary pneumococcal pneumonia. 93 PBI‐F2 is known to have no major effect on virus replication. Instead, by localizing to the inner and outer mitochondrial membranes, it disrupts mitochondrial morphology and dissipates mitochondrial energy potential, causing apoptosis and cell death. It is thought that apoptosis of immune cells prevents efficient maturation of the adaptive immune response, and that this explains its pathologic effects. Remarkably, the effects of the 1918 PB1‐F2 protein can be produced by intranasal administration of only the C‐terminal portion of the protein. 93
In a limited study of two patients who died of H5N1 influenza, apoptosis was seen in alveolar epithelial cells and pulmonary leukocytes. 94 Apoptotic lymphocytes were also found in the spleen but there was no evidence of virus replication, suggesting that unidentified host factors were responsible. Considered together, the findings from the studies discussed above indicate that apoptosis is not directly related to high levels of influenza virus replication. Instead, it is caused by poorly defined host factors that respond to the molecular features of the virus such as the PB1‐F2 and NS‐1 proteins and perhaps viral RNA 81 and those of the host cells damaged by infection.
There have been no studies that report the effects of statins, fibrates, or glitazones on autophagy or apoptosis in acute lung injury due to any cause. However, the apoptosis observed in septic cardiomyopathy is reduced with statin treatment, with a corresponding improvement in cardiac function. 95 Furthermore, in a rat model of hepatic ischemia/reperfusion injury, simvastatin pre‐treatment reduced the amount of apoptosis, with an associated improvement in liver function. 96
Mitochondria
Studies of experimental and human sepsis 97 , 98 have shown that mitochondrial dysfunction and the disruption of energy homeostasis could be responsible for much of the loss of pulmonary integrity and the multi‐organ failure seen in acute lung injury. Thus, mitochondrial dysfunction could play a fundamental role in determining the outcome of H5N1 and pandemic influenza virus infection.
Mitochondrial dysfunction is responsible for oxidant‐induced acute lung injury 99 , 100 in a process that is regulated by peroxisome proliferator activator receptorγ co‐activator (PGC)‐1a. 101 , 102 In mice with experimental bacterial sepsis 103 and in critically ill patients, 104 restoration of mitochondrial function clearly separates those who recover from those who die. Mitochondrial biogenesis can be restored by up‐regulating HO‐1 105 and by glitazones. 106 In a model of LPS‐induced mitochondrial dysfunction in murine neutrophils, inhibition of mitochondrial respiratory complex 1 with metformin led to decreased activity of NF‐kappaB and lower levels of pro‐inflammatory cytokines. 107 In whole animal studies, LPS‐exposed mice treated with metformin showed an inhibition of mitochondrial respiratory complex 1 in the lungs and a reduction in the severity of acute lung injury. 107 Glitazones and fibrates have the same down regulating effect on mitochondrial respiratory complex 1 as metformin. 108 , 109
Mitochondrial dysfunction with diminished cardiac function is a late occurring event in the myocardial depression seen in sepsis, but these changes might actually be protective. 110 By reducing energy expenditure when mitochondrial energy generation is compromised, a state analogous to hibernation is induced that maintains myocardial integrity until recovery sets in.
Unfortunately, there is little information on the effects of statins, fibrates, and glitazones on mitochondrial functioning in acutely ill patients, and it must be remembered that the toxic effects of each group of drugs are thought to be due to their effects on mitochondria. 111 Nonetheless, in a clinical trial conducted in children with severe burn injury and mitochondrial dysfunction, mitochondrial biogenesis was restored by treatment with fenofibrate. 112
Research on the host response should determine whether anti‐inflammatory and immunomodulatory agents could be used to manage the next pandemic
In focusing on the structural characteristics of influenza viruses that are associated with receptor specificity, replication efficiency, virulence and transmissibility and on factors that affect virus‐induced cell signaling and cytokine dysregulation, 2 , 3 , 4 , 5 , 6 , 7 influenza scientists have largely ignored the system‐wide effects of the disease (multi‐organ failure) and have left unexplored differences in system‐wide molecular pathophysiology that might explain the remarkably different mortality rates in children and young adults seen in the 1918 pandemic. The well‐known ability of some species (e.g., guinea pigs 113 ) to support high levels of influenza virus replication without developing illness must reflect intrinsic host factors that differ from those of other animals that develop severe disease. The molecular consequences of influenza’s effects on cardiac function and on organs other than the lung are hardly known. The pulmonary infiltrates seen in patients with H5N1 influenza that have been attributed to local cytokine dysregulation, could well be due to the influx of pro‐inflammatory factors generated in the liver 114 or perhaps other organs.
The similarities in the clinical course of patients with 1918 and H5N1 influenza and that of patients with sepsis are striking. The median duration of illness from onset until death in 1918 and H5N1 influenza has been similar to that seen in sepsis. 18 , 24 The time courses for the development of lymphocyte depletion and multi‐organ failure are generally the same. Bacterial super‐infection is often associated with late immunosuppression seen in patients with sepsis and in those with acute lung injury due to non‐infectious conditions like severe trauma. Thus, it is reasonable to assume that many of the pneumonia deaths seen in the 1918 pandemic had a similar cause. It is also reasonable to assume that agents shown to be effective in treating one condition might also be effective in treating the other, as has already been suggested for statins in both sepsis 55 and pneumonia. 14 , 15 As noted recently by Hotchkiss et al., ‘reengagement or preserving host immune function will be the next major advance in the management of patients with sepsis. 53 The same could be said for the management of patients with severe and pandemic influenza.
Surgeons have provided clues on how pandemic influenza could be managed
Surgeons who care for patients with multiple trauma have noted that secondary acute lung injury is less common and less severe in children than it is in young adults, and that it also has a lower mortality rate. 115 The cytokine profiles of children and adults with severe burns differ greatly. 116 Surgeons have also shown that the inflammatory responses of peritoneal macrophages harvested from children and young adults differ. When challenged in vitro with LPS 117 or IL‐1β, 118 macrophages from children showed a greater predominance of anti‐inflammatory activity compared with those from adults.
A model of severe acute inflammation in mice has provided a clue to how anti‐inflammatory and immunomodulatory treatment might improve outcomes in severe influenza. Shin and his surgical colleagues showed that following ischemia/reperfusion injury in mice, inflammation was much more severe in ‘young adults’ (10–12 week old mice) than it was in ‘children’ (4‐ to 5‐weeks old) (Table 2). 119 The liver cells of ‘children’ showed up‐regulation of PPARγ that was more pronounced and lasted longer that what was seen in ‘young adults.’ In ‘children,’ PPARγ activity was retained in the nucleus, whereas in ‘young adults’ it leaked into the cytoplasm. The liver cells of ‘children’ also showed greater evidence of autophagy than did those of ‘young adults.’ Importantly, when ‘young adults’ were treated with a PPARγ agonist (rosiglitazone), PPARγ activity was up‐regulated in liver cells to levels similar to that seen in untreated ‘children’.
Table 2.
Treatment of age‐related hepatic ischemia/reperfusion injury in mice
| Findings in liver cells | Children (4–5 weeks) | Young adults (10–12 weeks) | Older adults (9–12 months) |
|---|---|---|---|
| PPARγ activity | ++ | + | ± |
| PPARγ– nuclear | ++ | – | – |
| PPARγ– cytoplasm | – | + | ++ |
| Autophagy | ++ | + | – |
| Rosiglitazone effect on autophagy | nd | ++ | nd |
Adapted from Ref. (119).
PPAR, peroxisome proliferator activator receptor; nd, not done.
Knowledge such as this could help us prepare for the next influenza pandemic. If PPARγ agonists are able to control acute lung injury, as has been suggested, 120 the results of the study by Shin et al. suggest that treatment of young adults might, in effect, ‘roll back’ the host response of someone who is sexually mature and a poor PPARγ responder (and who might die) to that of a sexually immature child who is a better PPARγ responder (and more likely to live). Whether treatment with fibrates or statins would have a similar effect is unknown and should be studied.
If effective agents can be found, treatment in a pandemic might need to be given only to patients who are on the verge of developing severe, life‐threatening illness and continued only until they had recovered. Since most patients infected with a pandemic influenza virus might be expected to have a balanced host response and recover uneventfully (perhaps H5N1 excepted), it would not be necessary to make treatment available for entire populations. Instead, smaller supplies could be stockpiled and reserved for treating only those individuals (perhaps only 2–10% of a population) who might truly benefit.
Speculation such as this is based largely on observations in settings other than experimental and human influenza. These ideas might eventually be shown to be incorrect. However, they serve to illustrate the narrowness of our current knowledge of the host response to severe influenza. They also emphasize how this narrowness continues to limit our search for practical ways to manage the next pandemic.
It should be evident that any treatment shown to be effective in modifying the host response and reducing mortality in patients with pandemic or seasonal influenza should also be useful in treating patients with sepsis and other conditions characterized by a dysregulated inflammatory response and late immunosuppression. One such treatment (rosiglitazone) has already been shown to be effective in treating experimental cerebral malaria. 121
The global relevance of research on the host response to influenza
What is overwhelmingly important about the anti‐inflammatory and immunomodulatory agents discussed here – statins, fibrates, glitazones, and several others – is that they are now being produced as inexpensive generic medications and are widely available in ‘have not’ countries. 11 , 14 , 15 , 31 For example, in the US a 5‐day course of treatment with a generic statin would cost $3·20 and a course of resveratrol would cost $1·80 (DS Fedson, unpublished data). In developing countries they would cost even less. They could be stockpiled and made available in each country on the first pandemic day. It is unlikely that this will ever be said for pandemic vaccines and antiviral agents.
More research is needed to determine whether agents that modify the host response will be useful for the treatment and prophylaxis of H5N1 and pandemic influenza. A five‐point research agenda was recently proposed, and an adaptation is shown in Table 3. 15 The first two points address studies to be carried out by laboratory‐based investigators. If these studies are focused on identifying promising candidates for patient treatment, they should not require a great deal of time or money, but they would need to be sponsored. As pointed out recently, 33 studies of generic agents for pandemic treatment are of no commercial interest to pharmaceutical companies and there is no identifiable patient group to advocate they be undertaken. Thus, the studies can only be undertaken if there is strong support from governments or foundations.
Table 3.
A research agenda to establish whether anti‐inflammatory and immunomodulatory agents could be used for treatment and prophylaxis of an H5N1‐like influenza pandemic*
| Test promising treatment regimens in mice, ferrets, and non‐human primates to identify specific agents that might be effective in managing an H5N1‐like pandemic |
| Later study promising treatments in cell culture and animals to define the molecular mechanisms that explain their beneficial effects against H5N1 and 1918‐like viruses |
| Conduct a global analysis to identify developing countries where these agents are produced and determine quantities produced, surge capacities, patterns of distribution and costs to public programmes |
| Establish an international process to coordinate or manage the stockpiling of these agents and/or their distribution once a pandemic virus has emerged |
| Plan to conduct randomized controlled trials of promising treatments immediately after the emergence of a new pandemic virus |
*Adapted from table 3 in Ref. (15).
Randomized controlled trials of promising treatments will be more difficult than the laboratory studies. 122 The results of sepsis trials of these agents might not be available for many years and there might not be sufficient time or opportunity to organize and conduct large‐scale clinical trials for treating seasonal influenza. Thus, it will be essential to undertake detailed planning so that clinical trials can be conducted in the first few weeks of the next pandemic when large numbers of patients will be immediately available and can be easily enrolled. 15
The research agenda outlined in Table 3 is much too important to be left to influenza scientists alone. It must involve other investigators with expertise in the cellular and molecular biology of inflammation, immunity, sepsis, cardiopulmonary diseases, critical care, endocrinology and metabolism and mitochondrial function. Influenza scientists must realize that they cannot do this work by themselves; they need the help of others. Likewise, scientists in other disciplines must be persuaded that their expertise will be essential if we are to adequately prepare ourselves for the next pandemic.
Conclusions
There is no guarantee that treatment with one or more anti‐inflammatory and immunomodulatory agents alone will mitigate the effects of an H5N1 or pandemic influenza virus infection. Nonetheless, scientists and the health officials who support their work must choose whether to undertake the necessary laboratory and clinical research before the pandemic arrives, and perhaps discover that generic agents will not be useful, or undertake it after the pandemic has passed, only to discover that millions could have been saved. This choice can no longer be avoided.
Most of the world’s people lack realistic alternatives for confronting the next pandemic. Without the availability of treatments that will prevent multi‐organ failure and death, millions are expected to die, and with an H5N1 pandemic the global death toll could reach hundreds of millions. For this reason alone, it is essential that the research outlined here be undertaken immediately.
Footnotes
References
- 1. Thomas L. Lives of a Cell. Notes of a Biology Watcher. New York: Penguin Books, 1974, pp 78–79. [Google Scholar]
- 2. Peiris JS, De Jong MD, Guan Y. Avian influenza A (H5N1): a threat to human health. Clin Microbiol Rev 2007; 20:243–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gambotto A, Barratt‐Boyes SM, De Jong MD, Neumann G, Kawaoka Y. Human infection with highly pathogenic H5N1 influenza virus. Lancet 2008; 371:1464–1475. [DOI] [PubMed] [Google Scholar]
- 4. Kortweg C, Gu J. Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans. Am J Pathol 2008; 172:1155–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maines TR, Szretter KJ, Perrone L et al. Pathogenesis of emerging avian influenza viruses in mammals and the host innate immune response. Immunol Rev 2008; 225:68–84. [DOI] [PubMed] [Google Scholar]
- 6. Basler CF, Aguilar PV. Progress in identifying virulence determinants of the 1918 H1N1 and Southeast Asian H5N1 influenza A viruses. Antiviral Res 2008; 79:166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salomon R, Webster RG. The influenza virus enigma. Cell 2009; 136:402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palache B, Krause R. Progress with human H5N1 vaccines: a perspective from industry. Exp Rev Vaccines 2009; 8:375–400. [DOI] [PubMed] [Google Scholar]
- 9. Leroux‐Roels I, Leroux‐Roels G. Current status and progress of pre‐pandemic and pandemic influenza vaccine development. Exp Rev Vaccines 2009; 8:401–423. [DOI] [PubMed] [Google Scholar]
- 10. Jennings L, Monto AS, Chan PKS, Szucs TD, Nicholson KG. Stockpiling prepandemic influenza vaccines: a new cornerstone of pandemic preparedness plans. Lancet Infect Dis 2008; 8:650–658. [DOI] [PubMed] [Google Scholar]
- 11. Fedson DS, Dunnill P. From scarcity to abundance: pandemic vaccines and other agents for ‘have not’ countries. J Public Health Policy 2007; 28:322–340. [DOI] [PubMed] [Google Scholar]
- 12. Fedson DS. New technologies for meeting the global demand for pandemic influenza vaccines. Biologicals 2008; 36:346–349. [DOI] [PubMed] [Google Scholar]
- 13. News Release . International Federation of Pharmaceutical Manufacturers Associations. Authoritative New Study Reveals Global Pandemic Influenza Vaccine Capacity. Available on: http://www.ifpma.org (Accessed 24 February 2009).
- 14. Fedson DS. Confronting an influenza pandemic with inexpensive generic agents: can it be done? Lancet Infect Dis 2008; 8:571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fedson DS. Meeting the challenge of influenza pandemic preparedness in developing countries. Emerg Infect Dis 2009; 15:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Webster RG, Campbell CH. Studies on the origin of pandemic influenza. IV. Selection and transmission of ‘new’ influenza viruses in vivo. Virology 1974; 62:404–413. [DOI] [PubMed] [Google Scholar]
- 17. Baskin CR, Bielefeldt H, Tumpey TM et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci USA 2009; 106:3455–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Writing Committee of the Second World Health Organization Consultation on clinical aspects of human infection with avian influenza A (H5N1) virus: update on avian influenza A (H5N1) virus infection in humans. New Engl J Med 2008; 358:261–273. [DOI] [PubMed] [Google Scholar]
- 19. De Jong MD, Simmons CP, Thanh TT et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 2006; 12:1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morens DM, Taubenberger JF, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 2008; 198:962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 2006; 19:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morens DM, Taubenberger JK, Fauci AS. Commentary on fatalities in the 1918–1919 influenza pandemic. J Infect Dis 2009; 199:913–914. [DOI] [PubMed] [Google Scholar]
- 23. Fedson DS. Was bacterial pneumonia the predominant cause of death in the 1918–1919 influenza pandemic? J Infect Dis 2009; 199:1408–1409. [DOI] [PubMed] [Google Scholar]
- 24. Brundage JF, Shanks GD. Death from bacterial pneumonia during 1918–19 influenza pandemic. Emerg Infect Dis 2008; 8:1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahmed R, Oldstone MBA, Palese P. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat Immunol 2006; 11:1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leenstra T, Ter Kuile FO, Kariuki SK et al. Dehydroepiandrosterone sulfate levels associated with decreased malaria parasite density and increased hemoglobin concentration in pubertal girls from western Kenya. J Infect Dis 2003; 188:297–304. [DOI] [PubMed] [Google Scholar]
- 27. Kurtis JD, Friedman JF, Leenstra T et al. Pubertal development predicts resistance to infection and reinfection with Schistosoma japonicum . Clin Infect Dis 2006; 42:1092–1098. [DOI] [PubMed] [Google Scholar]
- 28. Clark IA, Alleva LM, Budd AC, Cowden WB. Understanding the role of inflammatory cytokines in malaria and related diseases. Travel Med Infect Dis. 2008; 6:67–81. [DOI] [PubMed] [Google Scholar]
- 29. Clark IA, Budd AC, Alleva LM. Sickness behaviour pushed too far – the basis for the syndrome seen in severe protozoal, bacterial and viral diseases and post‐trauma. Malaria J 2008; 7:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007; 81:1–5. [DOI] [PubMed] [Google Scholar]
- 31. Fedson DS. Pandemic influenza: a potential role for statins in treatment and prophylaxis. Clin Infect Dis 2006; 43:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng BJ, Chan KW, Lin YP et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci USA 2008; 105:8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simmons C, Farrar JT. Insights into inflammation. N Engl J Med 2008; 359:1621–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsatanis C, Androulidaki A, Venihaki M, Margioris AN. Signalling networks regulating cyclooxygenase‐2. Int J Biochem Cell Biol 2006; 38:1654–1661. [DOI] [PubMed] [Google Scholar]
- 35. Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol 2005; 174:5033–5039. [DOI] [PubMed] [Google Scholar]
- 36. Rousseaux C, Lefebvre B, Dubuquoy L et al. Intestinal anti‐inflammatory effect of 5‐aminosalicylic acid is dependent on peroxisome proliferator‐activated receptor‐gamma. J Exp Med 2005; 201:1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karlstrom A, Boyd KL, English BK, McCullers JA. Treatment with protein synthesis inhibitors improves outcomes of secondary bacterial pneumonia after influenza. J Infect Dis 2009; 199:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tamaoki J, Kadota J, Takizawa H. Clinical implications of the immunomodulatory effects of macrolides. Am J Med 2004; 117 (Suppl. 9A):5S–11S. [DOI] [PubMed] [Google Scholar]
- 39. Hodge S, Hodge G, Brozyna S, Jersmann H, Holmes M, Reynolds PN. Azithromycin increases phagocytosis of apoptotic bronchial epithelial cells by alveolar macrophages. Eur Respir J 2006; 28:486–495. [DOI] [PubMed] [Google Scholar]
- 40. Sugiyama K, Shirai R, Mukae H et al. Differing effects of clarithromycin and azithromycin on cyokine production in murine dendritic cells. Clin Exp Immunol 2007; 147:540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hodge S, Hodge G, Jersmann H et al. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 178:139–148. [DOI] [PubMed] [Google Scholar]
- 42. Imai Y, Kuba K, Neely GG et al. Identification of oxidative stress and Toll‐like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008; 133:235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nakahira K, Kim HP, Geng XH et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS‐induced trafficking of TLRs to lipid rafts. J Exp Med 2006; 203:2377–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ryter SW, Kim HP, Nakahira K, Zuckerbraun BS, Morse D, Choi AM. Protective functions of heme oxygenase‐1 and carbon monoxide in the respiratory system. Antioxid Redox Signal 2007; 9:2157–2173. [DOI] [PubMed] [Google Scholar]
- 45. Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme‐oxygenase‐1‐derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest 2008; 118:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chaturvedi A, Pierce SK. How location governs Toll‐like receptor signaling. Traffic 2009; 10:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Soares MP, Bach FH. Heme oxygenase‐1: from biology to therapeutic potential. Trends Mol Med 2009; 15:50–57. [DOI] [PubMed] [Google Scholar]
- 48. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med 2008; 358:716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mizgerd JP, Skerrett SJ. Animal models of human lung disease. Am J Physiol Lung Cell Mol Physiol 2008; 294:L387–L398. [DOI] [PubMed] [Google Scholar]
- 50. Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol 2006; 6:813–822. [DOI] [PubMed] [Google Scholar]
- 51. Van Der Poll T, Opal SM. Host‐pathogen interactions in sepsis. Lancet Infect Dis 2008; 8:32–43. [DOI] [PubMed] [Google Scholar]
- 52. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol 2008; 8:776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. Tilting toward immunosuppression. Nat Med 2009; 15:496–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med 2007; 14:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Terblanche M, Almog Y, Rosenson RS, Smith TS, Hackam DG. Statins and sepsis: multiple modifications at multiple levels. Lancet Infect Dis 2007; 7:358–368. [DOI] [PubMed] [Google Scholar]
- 56. Choi HS, Park MJ, Kang HM, Lim IH, Choi CW, You JH. Statin use and mortality in sepsis due to pneumonia (abstract). Crit Care Med 2007; 35 (Suppl. 12):1362. 17414732 [Google Scholar]
- 57. Dyson A, Singer M. Animal models of sepsis: why does preclinical efficacy fail to translate to the clinical setting? Crit Care Med 2009; 37 (Suppl. 1):S30–S37. [DOI] [PubMed] [Google Scholar]
- 58. Kruger PS, Freir NM, Venkatesh B, Robertson TA, Roberts MS, Jones MA. Preliminary study of atorvastatin plasma concentrations in critically ill patients with sepsis. Intensive Care Med 2009; 35:717–721. [DOI] [PubMed] [Google Scholar]
- 59. Straus DS, Glass CK. Anti‐inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol 2007; 28:551–558. [DOI] [PubMed] [Google Scholar]
- 60. Smith MR, Standiford TJ, Reddy RC. PPARs in alveolar macrophage biology. PPAR Res 2007; 2007:23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yano MY, Matsumura T, Senokuchi T et al. Statins activate peroxisome proliferator‐activated receptorγ through extracellular signal‐regulated kinase 1/2 and p38 mitogen‐activated protein kinase‐dependent cyclooygenase‐2 expression in macrophages. Circ Res 2007; 100:1442–1451. [DOI] [PubMed] [Google Scholar]
- 62. Paulmelle R, Staels B. Cross‐talk between statins and PPAR in cardiovascular diseases: clinical evidence and basic mechanisms. Trends Cardiovasc Med 2008; 18:73–78. [DOI] [PubMed] [Google Scholar]
- 63. Inoue I, Itoh F, Aoyagi S et al. Fibrate and statin synergistically increase the transcriptional activities of PPARalpha/RXRalpha and decrease the transactivation of NFkappaB. Biochem Biophys Res Commun 2002; 290:131–139. [DOI] [PubMed] [Google Scholar]
- 64. Birnbaum Y, Ye Y, Lin Y et al. Augmentation of myocardial production of 15‐epi‐lipoxin‐A4 by pioglitazone and atorvastatin in the rat. Circulation 2006; 114:929–935. [DOI] [PubMed] [Google Scholar]
- 65. Budd A, Alleva L, Alsharifi M et al. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob Agents Chemother 2007; 51:2965–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Aldridge JR, Moseley CE, Boltz DA et al. TNF/iNOS‐producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci USA 2009; 106:5306–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte‐derived dendritic cells and exudate macrophages produce influenza‐induced immune pathology and mortality. J Immunol 2008; 180:2562–2572. [DOI] [PubMed] [Google Scholar]
- 68. Pamer EG. Tipping the balance in favor of protective immunity during influenza virus infection. Proc Natl Acad Sci USA 2009; 106:4961–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Han KH, Ryu J, Hong KH et al. HMG‐CoA reductase inhibition reduces monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein‐1‐mediated monocyte recruitment in vivo. Circulation 2005; 111:1439–1447. [DOI] [PubMed] [Google Scholar]
- 70. Veillard NR, Braunersreuther V, Arnaud C et al. Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis 2006; 188:51–58. [DOI] [PubMed] [Google Scholar]
- 71. Palamara AT, Nencioni L, Aquilano K et al. Inhibition of influenza A virus replication by resveratrol. J Infect Dis 2005; 191:1719–1729. [DOI] [PubMed] [Google Scholar]
- 72. Cho IJ, Ahn JY, Kim S, Choi MS, Ha TY. Resveratrol attenuates the expression of HMG‐CoA reductase mRNA in hamsters. Biochem Biophys Res Comm 2008; 367:190–194. [DOI] [PubMed] [Google Scholar]
- 73. Inoue H, Jiang XF, Katayama T, Osada S, Umesono K, Namura S. Brain protection by resveratrol and fenofibrate against stroke requires peroxisome proliferator‐activated receptor alpha in mice. Neurosci Lett 2003; 352:203–206. [DOI] [PubMed] [Google Scholar]
- 74. Ge H, Zhang JF, Guo BS et al. Resveratrol inhibits macrophage expression of EMMPRIN by activating PPARgamma. Vascul Pharmacol 2007; 46:114–121. [DOI] [PubMed] [Google Scholar]
- 75. Penumathsa SV, Thirunavukkarasu M, Koneru S et al. Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J Mol Cell Cardiol 2007; 42:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Donnelly LE, Newton R, Kennedy GE et al. Anti‐inflammatory effects of resveratrol in lung epithelial cells: molecular mechanisms. Am J Physiol Lung Cell Mol Physiol 2004; 287:L774–L783. [DOI] [PubMed] [Google Scholar]
- 77. Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. Specific inhibition of My88‐independent signaling pathways of TLR3 and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J Immunol 2005; 175:3339–3346. [DOI] [PubMed] [Google Scholar]
- 78. Lu CC, Lai HC, Hsieh SC, Chen JK. Resveratrol ameliorates Serratia marcescens‐induced acute pneumonia in rats. J Leukoc Biol 2008; 83:1028–1037. [DOI] [PubMed] [Google Scholar]
- 79. Franchi L, Eigenbrod T, Muñoz‐Planillo R, Nunez G. The inflammasome: a caspase‐1‐activation platform that regulates immune response and disease pathogenesis. Nat Immunol 2009; 3:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Allen IC, Scull MA, Moore CB et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009; 30:556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thomas PG, Dash P, Aldridge JR Jr et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via regulation of caspase‐1. Immunity 2009; 30:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 2009; 206:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Owen DM, Gale M. Fighting flu with inflammasomes signalling. Immunity 2009; 30:476–478. [DOI] [PubMed] [Google Scholar]
- 84. Frahy RJ, Exline MC, Gavrillin MA et al. Inflammasome mRNA expression in human monocytes during early septic shock. Am J Respir Crit Care Med 2008; 177:983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Coward WR, Marei A, Yang AL, Vasa‐Nicotera MM, Chow SC. Statin‐induced proinflammatory response in mitogen‐activated peripheral blood mononuclear cells through the activation of caspase‐1 and IL‐18 secretion in monocytes. J Immunol 2006; 176:5284–5292. [DOI] [PubMed] [Google Scholar]
- 86. Hallett JM, Leitch AE, Riley NA, Duffin R, Haslett C, Rossi AG. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol Sci 2008; 29:250–257. [DOI] [PubMed] [Google Scholar]
- 87. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature 2008; 451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hsieh YC, Athar M, Chaudry IH. When apoptosis meets autophagy: deciding cell fate after trauma and sepsis. Trends Mol Med 2009; 15:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhou Z, Jiang X, Liu D et al. Autophagy is involved in influenza A virus replication. Autophagy 2009; 5:321–328. [DOI] [PubMed] [Google Scholar]
- 90. Mok CKP, Lee DCW, Cheung CY, Peiris M, Lau ASY. Differential onset of apoptosis in influenza A virus H5N1‐ and H1N1‐infected human blood macrophages. J Gen Virol 2007; 88:1275–1280. [DOI] [PubMed] [Google Scholar]
- 91. Daidoji T, Koma T, Du A et al. H5N1 avian influenza virus induces apoptotic cell death in mammalian airway epithelial cells. J Virol 2008; 82:11295–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lam WY, Tang JW, Yeung AC, Chiu LC, Sung JJ, Chan PK. Avian influenza virus A/HK/483/97 (H5N1) NS1 protein induces apoptosis in human airway epithelial cells. J Virol 2008; 82:2741–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. McAuley JL, Hornung F, Boyd KL et al. Expression of the 1918 influenza A virus PB1‐F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007; 2:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Uiprasertkul M, Kitphati R, Puthavanthana P et al. Apoptosis and pathogenesis of avian influenza A (H5N1) virus in humans. Emerg Infect Dis 2007; 13:708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Buerke U, Carter JM, Schlitt A et al. Apoptosis contributes to septic cardiomyopathy and is improved by simvastatin therapy. Shock 2008; 29:497–503. [DOI] [PubMed] [Google Scholar]
- 96. Lai IR, Chang KJ, Tsai HW, Chen CF. Pharmacological preconditioning with simvastatin protects liver from ischemia‐reperfusion injury by heme‐oxygenase‐1 induction. Transplantation 2008; 85:732–738. [DOI] [PubMed] [Google Scholar]
- 97. Carre JE, Singer M. Cellular energetic metabolism in sepsis: the need for a systems approach. Biochim Biophys Acta 2008; 1777:763–771. [DOI] [PubMed] [Google Scholar]
- 98. Exline MC, Crouser ED. Mitochondrial mechanisms for sepsis‐induced organ failure. Front Biosci 2008; 13:5031–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant lung injury. J Biol Chem 2007; 282:2355–2362. [DOI] [PubMed] [Google Scholar]
- 100. Carraway MS, Suliman HB, Kliment C et al. Mitochondrial biogenesis in the pulmonary vasculature during inhalational lung injury and fibrosis. Antioxid Redox Signal 2008; 10:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rohas LM, St.‐Pierre J, Uldry M, Jager S, Handschin C, Spiegelman BM. A fundamental system of cellular energy homeostasis regulated by PGC‐1. Proc Natl Acad Sci USA 2007; 104:7933–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC‐1α pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab 2008; 8:249–256. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103. Haden DW, Suliman HB, Carraway MS et al. Mitochondrial biogenesis restores oxidative metabolism during Staphyloccus aureus sepsis. Am J Respir Crit Care Med 2007; 176:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cote HCF, Day AG, Heyland DK. Longitudinal increases in mitochondrial DNA levels in blood cells are associated with survival in critically ill patients. Crit Care 2007; 11:R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci 2007; 120:299–308. [DOI] [PubMed] [Google Scholar]
- 106. Wang YL, Frauwirth KA, Rangwala SM, Lazar MA, Thompson CB. Thiazolidinedione activation of peroxisome proliferator‐activated receptorγ can enhance mitochondrial potential and promote cell survival. J Biol Chem 2002; 277:31781–31788. [DOI] [PubMed] [Google Scholar]
- 107. Zmiljewski JW, Lorne E, Zhao X et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med 2008; 178:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Brunmair B, Staniek K, Gras F et al. Thiozolidinediones, like metformin, inhibit respiratory complex I. A common mechanism contributing to their antidiabetic actions? Diabetes 2004; 53:1052–1059. [DOI] [PubMed] [Google Scholar]
- 109. Brunmair B, Lest A, Staniek K et al. Fenofibrate impairs mitochondrial function by inhibition of respiratory complex I. J Pharmacol Exp Ther 2004; 311:109–114. [DOI] [PubMed] [Google Scholar]
- 110. Rudger A, Singer M. Mechanisms of sepsis‐induced cardiac dysfunction. Crit Care Med 2007; 35:1599–1608. [DOI] [PubMed] [Google Scholar]
- 111. Hanai J, Cao P, Tanksale P et al. The muscle‐specific ubiquitin ligase atrogin‐1/MAFbx mediates statin‐induced muscle toxicity. J Clin Invest 2007; 117:3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cree MG, Zwetsloot JJ, Herndon DN et al. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann Surg 2007; 245:214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lowen AC, Mubareka S, Tumpey TM, Garcia‐Sastre A, Palese P. The guinea pig as a transmission model for human influenza viruses. Proc Natl Acad Sci USA 2006; 103:9988–9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Siore AM, Parker RE, Stencenko AA et al. Endotoxin‐induced acute lung injury requires interaction with the liver. Am J Physiol Lung Cell Mol Physiol 2005; 289:L769–L776. [DOI] [PubMed] [Google Scholar]
- 115. Calkins CM, Bensard DD, Moore EE et al. The injured child is resistant to multiple organ failure: a different inflammatory response? J Trauma 2002; 53:1058–1063. [DOI] [PubMed] [Google Scholar]
- 116. Finnerty CC, Jeschke MG, Herndon DN et al. Temporal cytokine profiles in severely burned patients: a comparison of adults and children. Mol Med 2008; 14:553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Barsness KA, Bensard DD, Partrick DA et al. IL‐1β induces an exaggerated pro‐ and anti‐inflammatory response in peritoneal macrophages of children compared with adults. Pediatr Surg Int 2004; 20:238–242. [DOI] [PubMed] [Google Scholar]
- 118. Barsness KA, Bensard DD, Partrick DA, Calkins CM, Hendrickson RJ, McIntryre RC Jr. Endotoxin induces an exaggerated interleukin‐10 response in peritoneal macrophages of children compared with adults. J Pediatr Surg 2004; 39:912–915. [DOI] [PubMed] [Google Scholar]
- 119. Shin T, Kuboki S, Huber N et al. Activation of perioxisome proliferator‐activated receptor‐gamma during hepatic ischemia is age‐dependent. J Surg Res 2008; 147:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Di Paola R, Cuzzocrea S. Peroxisome proliferator‐activated receptors and acute lung injury. PPAR Res 2007; 2007:63745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Serghides L, Patel SN, Ayi K et al. Rosiglitazone modulates the innate immune response to Plasmodium falciparum infection and improves outcome in experimental cerebral malaria. J Infect Dis 2009; 199:1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Opal SM, Patrozou E. Translational research in the development of novel sepsis therapeutics: logical deductive reasoning or mission impossible? Crit Care Med 2009; 37 (Suppl.):S10–S15. [DOI] [PubMed] [Google Scholar]