

Abstract
Background In recent years, several avian influenza subtypes (H5, H7 and H9) have transmitted directly from birds to man, posing a pandemic threat.
Objectives We have investigated the immunogenicity and protective efficacy of a cell based candidate pandemic influenza H7 vaccine in pre‐clinical animal models.
Methods Mice and ferrets were immunised with two doses of the split virus vaccine (12–24 μg haemagglutinin) with or without aluminium hydroxide adjuvant and challenged 3 weeks after second dose with the highly pathogenic A/chicken/Italy/13474/99 (H7N1) virus. The H7N1‐specific serum antibody response was also measured. After challenge, viral shedding, weight loss, disease signs and death (only mice) were recorded.
Results Low‐to‐modest serum antibody titres were detected after vaccination. Nevertheless, the vaccine induced significant protection from disease after challenge with the wild‐type virus. In the murine lethal challenge model, vaccination effectively prevented death and, furthermore, formulation with adjuvant reduced excessive weight loss and viral shedding. In ferrets, vaccination reduced viral shedding and protected against systemic spread of the virus.
Conclusions We have extended to the H7 subtype the finding that protective efficacy may not be directly correlated with the pre‐challenge levels of serum antibodies, a finding which could be of great importance in assessing the potential effectiveness of pandemic influenza vaccines.
Keywords: Challenge, ferret, highly pathogenic, influenza H7, mouse, protection
Introduction
In recent years, several avian influenza subtypes have transmitted directly from birds to humans, causing disease and death. The H5 virus has been responsible for the majority of these zoonoses, resulting in over 60% mortality in infected individuals. 1 In Europe and North America, however, human cases of avian influenza have almost exclusively been of the H7 subtype and caused by direct transmission from poultry infected with highly pathogenic avian H7 viruses. 2 , 3 , 4 , 5 The major symptom of H7 infection in humans has been conjunctivitis, sometimes with influenza‐like‐illness (reviewed in Ref. 6). Nevertheless, during an H7N7 outbreak in the Netherlands in 2003, this virus also led to acute respiratory distress syndrome and the subsequent death of a veterinarian. 2 , 3 Recently, it has been reported that some H7 isolates of the North American lineage have acquired human α2,6 sialic acid receptor binding properties. 7 It is thus clear that in addition to avian H5, the H7 avian subtype also constitute a pandemic threat, as it is a novel virus that has replicated and caused both respiratory and systemic disease in humans. However, a pandemic virus must also exhibit efficient human‐to‐human transmission, a property not yet acquired by viruses of the H5 or H7 subtypes.
It is safe to assume that a timely and widespread use of an effective vaccine will limit the burden of disease in an influenza pandemic. Pandemic modelling also suggests that even a pandemic vaccine of relatively low efficacy will be useful during a pandemic. 8 , 9 One critical issue in responding to an influenza pandemic is the time lag between the emergence of a pandemic virus and the availability of an appropriate seed virus approved for vaccine production. The use of reverse genetics (RG) to attenuate a highly pathogenic pandemic influenza strain allows rapid construction of a safe high yielding seed virus. 10 , 11 Furthermore, this allows vaccine production in embryonated hens’ eggs, the substrate used by most influenza vaccine manufacturers. However, the supply of eggs may be a limiting factor in a pandemic scenario. A cell culture based influenza vaccine is an attractive alternative to supplement current egg based vaccine production, as the production output may be more readily scaled up. Furthermore, studies of seasonal strains have shown that cell‐based seed viruses may be more antigenically relevant than egg‐derived seed viruses. 12 , 13 To date, three different cell lines have been used in clinical trials of influenza vaccines; the Madin Darby canine kidney (MDCK) cells, 14 the African green monkey kidney (Vero) cell line 15 , 16 and the PER.C6® cell line derived from a human retina; the latter being used in this study.
Avian influenza pandemic vaccine candidates have been shown to be weakly immunogenic in naïve adults. 17 Therefore, strategies to increase the immunogenicity of these vaccines are urgently needed. The ability of several different adjuvants to augment the immune response after vaccination has been investigated in pre‐clinical 18 , 19 , 20 and clinical studies. 21 , 22 , 23 Of these adjuvants, aluminium salt adjuvants have been the most frequently used, and have enhanced the immune response after vaccination in most studies, as well as reduced the morbidity after challenge in H5 and H7 animal studies. 18 , 24 , 25 , 26 , 27 The proprietary oil‐in‐water emulsion systems such as MF59 and AS, have effectively enhanced antibody responses after H5 vaccination in man. 22 , 23 , 28 , 29 , 30 , 31
In this study, we have evaluated a novel cell‐based influenza H7 vaccine, based on the Eurasian highly pathogenic avian influenza strain, A/chicken/Italy/13474/99 (H7N1) that was isolated from poultry in northern Italy in 1999. The attenuated vaccine seed (RD3) was generated by RG, using the neuraminidase and modified haemagglutinin (HA) from A/chicken/Italy/13474/99 (H7N1) and the internal protein genes from the vaccine donor strain A/Puerto Rico/8/34 (PR8) (H1N1). 32 RD3 was propagated in the PER.C6® cell line and subsequently used to produce a split virion vaccine for use in the pre‐clinical studies reported here using the human doses used in a phase I clinical trial. 33 We evaluated the protective efficacy of this vaccine to reduce disease and death in a lethal murine challenge model. In mice, low levels of antibodies were detected after vaccination, but nevertheless vaccination reduced disease and protected animals from death after challenge with the parent highly pathogenic virus, A/chicken/Italy/13474/99 (H7N1). Furthermore, the protective efficacy of the aluminium hydroxide adjuvanted vaccine was evaluated in a pilot study in ferrets and found to effectively reduce viral shedding and prevent systemic spread of the highly pathogenic parent virus.
Materials and methods
Virus strains and vaccine preparation
The highly pathogenic A/chicken/Italy/13474/99 (H7N1) virus was isolated during an outbreak in Italian poultry in 1999. 34 Due to its lethality in both mice and embryonated eggs, it was chosen as the basis for the pandemic vaccine candidate used in this study. The vaccine virus, called RD3, was generated by RG using the surface antigens HA and neuraminidase from the A/chicken/Italy/13474/99 (H7N1) virus and the internal protein genes from the vaccine donor strain A/Puerto Rico/8/34 (PR8) (H1N1). 32 The recovered virus was only propagated on two different mammalian cell lines (Vero and PER.C6®). The multibasic cleavage site of HA was genetically modified to contain only one basic amino acid residue, which has previously been demonstrated to reduce virulence of highly pathogenic avian influenza viruses. 35 The RD3 virus was subsequently propagated in PER.C6® cells under Biological Safety Level (BSL) 2 enhanced (pandemic influenza vaccine) and an inactivated split virion vaccine was produced (sanofi pasteur, Marcy l’Etoile, France). The HA content of the vaccine was measured by single radial diffusion. The aluminium hydroxide adjuvant (AlOOH) was provided in 0·5 ml pre‐filled syringes containing 600 μg Al.
Immunisation of animals
All procedures were carried out according to the UK Home Office Licence regulations and by individuals with appropriate personal Home Office Licenses. The study was approved by the National Institute for Biological Standards and Control Ethical Review Process. The vaccine was formulated at 47 μg/ml of HA in phosphate‐buffered saline (PBS) for a phase I clinical trial. Animals were immunised with 12–24 μg HA reflecting the dose used in this phase I clinical trial. Ferrets were immunised intramuscularly with 24 μg HA, whereas mice were immunised subcutaneously with 12 and 20 μg HA due to limits of the injectable volume. These vaccine doses were determined based on data from the preliminary immunogenicity studies in mice and toxicology in rabbits and to allow the vaccine to fulfil the regulatory requirements to enter human clinical trial. All the challenge studies were conducted under BSL‐3 conditions.
Murine protective efficacy study
Six‐ to 8 weeks‐old female BALB/c mice were purchased from Charles River Laboratories (Kent, UK) and housed according to the appropriate national regulations. Groups of 20 mice were vaccinated subcutaneously with two doses (18 days apart) of the split virion vaccine, 12 or 20 μg HA with or without aluminium hydroxide adjuvant (AlOOH) (60 μg Al). The aluminium hydroxide adjuvant was mixed with the vaccine antigen immediately before injection, as described earlier. 21 The injected volume was adjusted correspondingly to administer the correct dose with mice receiving 12 μg HA (0·25 ml), adjuvanted 12 μg HA (0·3 ml), 20 μg HA (0·43 ml) or adjuvanted 20 μg HA (0·48 ml). A control group consisted of unimmunised mice. Serum samples were collected at 18 days after the first and second dose of vaccine from a tail vein or by cardiac puncture post‐challenge (at day 14 or earlier if moribund).
Three weeks after the second immunisation, animals were anaesthetised with ketamine (Vetalar; Pfizer Limited, Kent, UK) and challenged intranasally with 100MID50/10LD50 (20 μl) of egg grown A/chicken/Italy/13474/99 (H7N1) virus. Following challenge, animals were weighed and observed for clinical signs of disease (ruffled fur, neurological signs, respiratory and ocular symptoms) for 14 days. Nasal wash samples were collected for the initial 6 days after challenge to monitor viral shedding. Awake mice (non‐anaesthetised) were restrained by scruffing, and 500 μl PBS/0·14% bovine serum albumin (BSA) (PBS/BSA) administered drop wise to the nasal cavity. 36 The exhaled PBS/BSA was collected and aliquots screened for infectious virus by inoculation onto MDCK. The remainder of the washings were stored at −70°C for subsequent quantitative assay of infectious virus in MDCK cells.
Pilot ferret protective efficacy study
The adjuvanted highest human dose of vaccine used in the phase I clinical trial 33 was evaluated in a pilot study in ferrets. Groups of four adult male ferrets (accredited supplier, UK) 6‐ to 12‐months old were vaccinated intramuscularly with two doses (21 days apart) of either 24 μg HA of the split virion vaccine adjuvanted with AlOOH (600 μg Al) (1 ml) or saline (control group). The adjuvant was mixed with the vaccine immediately before injection, as described above. All ferrets were seronegative by haemagglutination inhibition assays (HI titres <8) to the H7N1 virus and currently circulating H3, H1 and B strains. Ferrets were housed in open pens after vaccination and then transferred to individual cages prior to challenge. Animals were fed twice daily with tinned cat food/dry cat biscuit mix and following challenge the diet was supplemented with cat milk.
Twenty‐one days after the second immunisation, animals were sedated with 0·2 ml/kg of ketamine/xylazine and challenged by intranasally administering A/chicken/Italy/13474/99 (H7N1) virus [108·5 egg infectious dose (EID50) in 0·4 ml PBS/BSA]. Animals were bled from the tail vein prior to each vaccination and 21 days after the second immunisation and by cardiac puncture when killed. Serum samples were stored at −70°C until they were tested in the modified HI and microneutralisation (MN) assays.
After challenge, body weights, temperatures and clinical symptoms were recorded at the same time daily for six consecutive days. Animals were sedated as described previously on days 1, 3 and 5 post‐challenge and nasal washings collected using drop‐wise administration of 2 ml PBS/BSA to the nostrils. Exhaled nasal washings were collected and immediately frozen at −70°C. 37
On day 6, two animals from each group were killed and nasal washings, eye swabs (placed in 1 ml PBS/BSA) and samples of lung, spleen and the whole brain including the olfactory bulbs collected from each animal. Lung samples comprised tissue collected from each lobe except the cardiac lobe. The remaining animals were observed for clinical signs of disease until 14 days post‐challenge, when nasal washings and eye swabs were collected and the animals were killed. All eye swabs, spleen and brain tissues were frozen immediately at −70°C.
Virus recovery from the nasal washes of mice
The presence of replicative virus in the nasal washes was quantified in confluent monolayers of MDCK cells for mice as previously described. 38 On the day of collection, nasal washings were inoculated onto MDCK cells in flat bottom (FB‐96) plates, two wells/wash, 50 μl/well, adsorbed for 30 minutes at room temperature and then replaced with 100 μl infection medium, Dulbecco’s modified Eagles Medium with 40 mm l‐glutamine, 2·5 μg/ml TPCK trypsin, 100 IU/ml penicillin, 100 μg/ml streptomycin, 2·5 μg/ml amphotericin and 0·14% BSA. Unused aliquots of nasal washings were stored at −70°C. Cells were incubated at 35°C for 3 days, when 50 μl aliquots of the medium were transferred to U‐bottom plates and 50 μl of 0·7% turkey erythrocyte suspension added to each well to detect virus. Positive nasal washings were thawed and diluted 10−1 to 10−4 in infection medium and the dilutions added to confluent monolayers of MDCK cells in FB‐96 plates, 100 μl/well, and 10 wells/dilution. Plates were incubated and processed as before and the number of virus positive wells for each dilution recorded. Negative nasal washings were inoculated at 10−1 dilution only. Each set of assays included a control titration of A/chicken/Italy/13474/99 (H7N1) virus. Titres were calculated by the method of Spearman‐Karber. 39
Virus recovery from nasal washings, eye swabs and tissue samples from ferrets
Tissue samples were thawed, weighed and homogenized in 2 ml of PBS/BSA containing 4000 IU penicillin for each gram of tissue. The homogenates were clarified by micro‐centrifugation at 15 kg for 3 minutes. Nasal washings, eye swab collection fluids and clarified tissue homogenates were inoculated undiluted or diluted (10−1 to 10−6) into 10‐day embryonated hens’ eggs (0·1 ml per egg, three eggs for each dilution). The undiluted samples were similarly inoculated except for the addition of 2000 IU/ml of penicillin. Eggs were incubated at 35°C for a minimum of 48 hours and then chilled at +4°C for a minimum of 4 hours before harvesting allantoic fluid. The presence of replicative virus was detected by using an HA assay as follows 50 μl samples of allantoic fluid from each egg were transferred to wells of U‐well micro‐titre plates and 50 μl of 0·7% turkey erythrocytes added to each well to detect virus growth by 100% haemagglutination. The 50% EID50 was calculated by the method of Spearman‐Karber. 39
Serological analyses
Pre‐ and post‐challenge serum samples from each individual were tested at the same time in all serological assays using the PER.C6® cell grown RD3 virus. A post‐infection ferret serum against RD3 was used as a positive control in the HI and MN assays. Cross‐reactive HI assays were also conducted with egg grown A/mallard/Netherlands/12/2000 (H7N3) and A/New York/107/03 (H7N2) strains.
Haemagglutination inhibition assay
Sera were treated to remove non‐specific inhibitors by 1/4 dilution in receptor destroying enzyme and incubation for 18 hours at 37°C followed by 45 minutes incubation at 56°C and tested by a modified HI assay, using 1% horse erythrocytes. 21 , 40 HI assay for antibody against cell grown RD3 virus was performed by standard micro‐titre method using twofold dilutions of sera in 25 μl volumes, four haemagglutinating units of RD3 and 1% horse erythrocytes. Positive control sera included a post‐RD3 infection ferret serum and a goat hyper‐immune serum raised against the A/equine/Prague/56 (H7N7) purified HA. Each serum sample was tested at least twice and the results reported as the geometric mean of the readings. The serum HI titre was expressed as the reciprocal of the highest dilution at which haemagglutination was inhibited and titres <8 were assigned a value of four for calculation purposes.
Microneutralisation assay
Neutralising antibody titres were determined in ferret serum using a previously described MN assay 41 with MDCK cells. Positive control sera 32 were included in the assay. Serum samples were tested in duplicate on at least two occasions from initial dilutions of 1:20. Sera with titres of 20 or greater were considered positive. Antibody titres <20 were assigned a value of 10 for calculation purposes.
Enzyme‐linked immunosorbent assay
The murine influenza‐specific serum IgG, IgG1 and IgG2a antibodies were quantified using the enzyme‐linked immunosorbent assay (ELISA) assay, as previously described. 42 , 43 Briefly, ELISA plates were coated with 0·5 μg/well of whole influenza RD3 virus and capture goat anti‐mouse IgG, IgG1 and IgG2a antibodies overnight at 4°C. Serially diluted sera and immunoglobulin standards were then incubated for 2 hours at room temperature, followed by 1‐hour incubation with biotinylated goat anti‐mouse IgG class or subclass antibodies and 1‐hour incubation with extravidin peroxidase. Serum samples from each of the vaccination groups were included on each ELISA plate. The antibody concentrations (μg/ml) were calculated using the IgG, IgG1 and IgG2a standards and linear regression of the log‐transformed readings.
Statistical analyses
Statistical analyses were performed using spss for Windows (version 14.0.2, SPSS Inc., Chicago, IL, USA). The clinical score data up to 14 days post‐challenge from the murine protective efficacy study was analysed using a non‐parametric analysis of variance and the Wilcoxon rank test was used for performing pair‐wise comparisons. The murine serum IgG, IgG1 and IgG2a response was analysed using the two sample or the paired Student’s t‐test. A multiple comparisons approach with the Bonferroni corrections was used to analyse the groups of mice for virus recovery or weight loss. The only statistical analysis performed on the ferret study was a mixed linear regression model which was used to test the viral recovery from nasal washes. P‐values ≤ 0·05 were considered significant.
Results
Murine protective efficacy study
The protective efficacy of the vaccine (12 or 20 μg HA) with or without aluminium adjuvant was investigated using a lethal virus challenge model. Unimmunised mice served as a control group. Mice were challenged with the highly pathogenic A/chicken/Italy/13474/99 (H7N1) strain 3 weeks after the second dose. Pre‐ and post‐challenge H7N1 antibody titres were measured by HI, MN and ELISA assays. Viral shedding in the nasal washes was quantified and weight loss, clinical signs of disease and death were recorded after challenge.
Viral challenge enhanced the post‐vaccination HI response
No or little HI antibody was detected 21 days after the first vaccination (data not shown). All groups had detectable antibodies after the second immunisation although HI titres were low ranging from not detectable to 64, with the highest HI titres observed in the animals vaccinated with 20 μg HA with or without AlOOH (Fig. 1). The adjuvanted vaccines generally elicited higher HI titres than the non‐adjuvanted formulations after the second vaccination. Antibody titres increased significantly after challenge in all groups.
Figure 1.
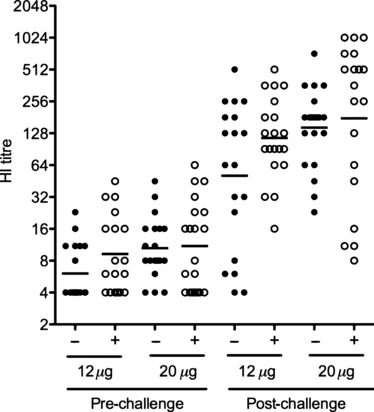
The haemagglutination inhibition (HI) antibody response induced after vaccination and challenge. Groups of 20 mice were vaccinated with two doses of vaccine (12 or 20 μg HA), alone or with aluminium hydroxide adjuvant (60 μg Al). Unimmunised mice served as a control group. Mice were challenged with the highly pathogenic A/chicken/Italy/13474/99 (H7N1) strain 3 weeks after the second dose. Serum HI titres were measured by a modified HI assay using cell grown H7N1 virus and horse red blood cells at 18 days after the second vaccination (pre) and 14 days after challenge (post). The horizontal lines represent the geometric mean titre (GMT), and each symbol represents one animal from the adjuvanted (+) or non‐adjuvanted (−) groups. The limit of detection of the HI assay was eight, and negative titres were assigned a value of four.
Vaccination induced a Th2 skewed response
Low antibody levels of IgG consisting mainly of IgG1 were measured 21 days after the second vaccine dose. A dose response was observed with the lowest concentrations of IgG detected in 12 μg HA group and the highest response observed in the 12 or 20 μg HA adjuvanted groups. Virus challenge significantly boosted these antibody responses in all groups (P < 0·05), with the highest antibody concentrations observed in the adjuvanted groups. The IgG response was entirely dominated by the IgG1 subclass in all vaccinated mice, whereas little or no IgG2a was detected in these animals either after vaccination or following challenge (Figure 2). In contrast, IgG2a was detected in higher concentrations than IgG1 following challenge, albeit at very low antibody concentrations, 900 and 400 ng/ml respectively.
Figure 2.
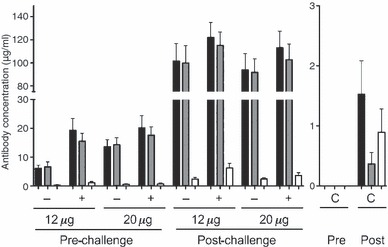
The IgG subclass distribution in mice after vaccination and challenge. Groups of 20 mice were unimmunised (controls) or vaccinated with two doses of 12 or 20 μg HA vaccine with (60 μg Al) or without aluminium hydroxide adjuvant. Mice were challenged with the highly pathogenic A/chicken/Italy/13474/99 (H7N1) strain 3 weeks after the second dose. An ELISA assay was used to measure the RD3 influenza specific IgG (black), IgG1 (grey) and IgG2a (open) concentrations (μg/ml) ± SEM at 18 days after the second vaccination in (pre) and 14 days after challenge (post) samples. The different groups adjuvanted (+), non‐adjuvanted (−) and unvaccinated controls (C) are shown below the axis. The ELISA results were analysed using the two sample or the paired Student’s t‐test. The limit of detection of the ELISA is 10–15 ng/ml.
Immunisation reduced viral shedding after viral challenge
Vaccination significantly reduced viral shedding in the nasal washes compared to the unvaccinated controls (P < 0·0001). However, no significant differences were found in viral shedding between the different vaccine groups. Peak viral titres were observed at 2 or 3 days post‐challenge in the vaccinated groups, and later on day 4 in the control animals (Figure 3A). By day 6, and with the exception of one animal in the 20 μg HA adjuvanted group, no detectable virus was found in any of the vaccinated mice, whereas virus was still recovered on day 6 from all control mice. Nasal wash virus was recovered from fewer challenged mice in the adjuvanted than in the non‐adjuvanted vaccine groups.
Figure 3.
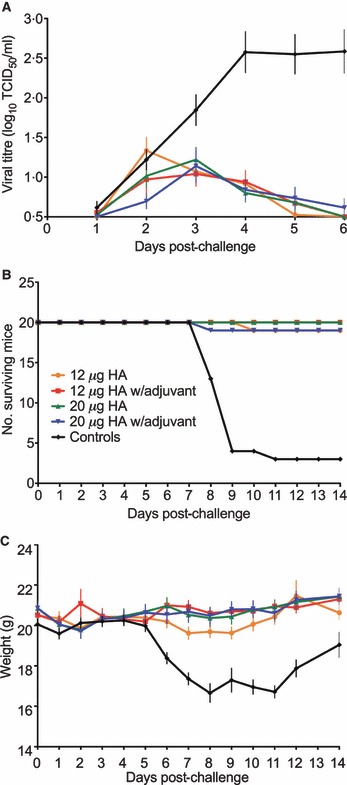
Protective efficacy of the H7N1 split virus vaccine in mice. (A) The viral shedding in the nasal washes. The mean viral titres (TCID50/ml ± SEM) are presented for each group. The lower limit of detection was 100·5 TCID50/ml. (B) The number of mice surviving viral challenge from each group of 20 animals. (C) Weight loss in challenged animals. The data is presented as mean weight loss (grams) ± SEM. Mice were examined for day 6 (viral shedding) or 14 days (death and weight loss) after challenge and the data is presented for each vaccine group as follows: 12 μg (orange), adjuvanted 12 μg (red), 20 μg (green), adjuvanted 20 μg (blue) and unvaccinated controls (black). Statistical comparisons between the groups for virus recovery were based on the mean virus shedding over 6 days or for weight loss on the mean weight over 14 days for each animal and an analysis of variance was carried out using a multiple comparisons approach, with a Bonferroni corrections.
A cell‐derived H7N1 vaccine protected mice from disease and weight loss
Vaccination induced a highly significant reduction in the signs of infection (P < 0·0001) (Table 1, Figure 3B). All unvaccinated control animals exhibited multiple signs of influenza disease after challenge, typically starting 5 days post‐infection with severe symptoms developing several days later. Early signs of disease consisted of severe ruffled fur and reduced activity, whereas rapid and/or laboured breathing and excessive weight loss (>10% of pre‐infection weight) were observed later (Figure 3C). Almost half of the unvaccinated control mice developed ocular symptoms probably conjunctivitis and/or neurological signs (tremor and/or ataxia). Seventeen of the unvaccinated mice died or were killed for humane reasons before the end of the study period. Signs of infection were significantly reduced in all groups of vaccinated mice compared to the controls (P < 0·0001). The adjuvanted vaccine groups also had higher numbers of mice without any signs of infection than the non‐adjuvanted groups. Two mice in each of the 12 μg HA and adjuvanted 20 μg HA groups developed clinical symptoms, and of these, one mouse in each group died or was killed due to excessive weight loss. Thus, formulation of both vaccine strengths with AlOOH reduced the number of mice exhibiting weight loss and viral shedding.
Table 1.
Signs of infection in mice challenged with A/chicken/Italy/13474/99 (H7N1) virus
| Vaccine | No. of mice (n = 20) | ||||
|---|---|---|---|---|---|
| Died | Weight loss >10% | Virus recovery | Clinical symptoms | Disease free | |
| 12 μg | 1** | 9 | 15 | 2 | 4 |
| 12 μg + AlOOH | 0** | 4 | 12 | 0 | 7 |
| 20 μg | 0** | 8 | 16 | 0 | 3 |
| 20 μg + AlOOH | 1** | 3 | 12 | 2 | 8 |
| None | 17 | 20 | 20 | 20 | 0 |
Following viral challenge, animals were weighed and observed for death and clinical symptoms (ruffled fur, neurological signs, respiratory and ocular symptoms) for 14 days and nasal washings were collected for 6 days to monitor viral shedding. The table shows the number of mice exhibiting signs of infection on any of the observation days. Disease‐free animals are defined as mice that did not show any of the following: weight loss, virus recovery and clinical symptoms. The statistical analysis for the clinical scores was based on the data collected from 1 to 14 days post‐challenge. As the clinical score data were not normally distributed, a non‐parametric analysis of variance and the Wilcoxon rank test was used for performing pair‐wise comparisons.
**Significantly lower than the control (none) group (P < 0·001).
A cell‐derived H7N1 vaccine protected mice from death
Vaccination induced a highly significant reduction in the signs of infection (P < 0·0001) and marked protection from death after viral challenge (Table 1, Figure 3B). Only two vaccinated animals died (one out of 20 mice in each of the 12 μg HA and adjuvanted 20 μg HA groups). In contrast, four control mice died and 13 were killed. The three remaining control animals survived and appeared disease free at the end of the study. One mouse in the 12 μg HA group died 10 days post‐infection and one mouse in the adjuvanted 20 μg HA group was killed due to excessive weight loss.
Protective efficacy in ferrets
The protective efficacy of this vaccine was further investigated in a pilot study in ferrets by vaccinating with two doses of adjuvanted 24 μg HA or saline (control group) and challenging 21 days later with the highly pathogenic H7N1 parent virus. Previously we have not detected systemic spread of virus at 3 days post‐infection 32 so in this study two ferrets from each group were killed at 6 days post‐vaccination when the occular symptoms first occurred.
Vaccination induced modest antibody responses
Low HI antibody responses were detected after the first vaccination [geometric mean titre (GMT) 10] and the antibody response was boosted after the second dose of vaccine (GMT 76), whereas no HI antibody was detected in the control group (Figure 4A). No MN antibody was detected until after the second dose in vaccinated ferrets (MN titres 42–200) and no antibody was detected in control animals at any time point. Six days following challenge low HI responses were detectable in control animals (GMT 8), whereas HI (GMT 64) and MN responses (MN titres 51–206) remained at the pre‐challenge level in vaccinated animals. Viral challenge further increased HI (GMT 256) and MN (MN titres 264–313) antibody titres by two‐ to fourfold by day 14 post‐challenge.
Figure 4.
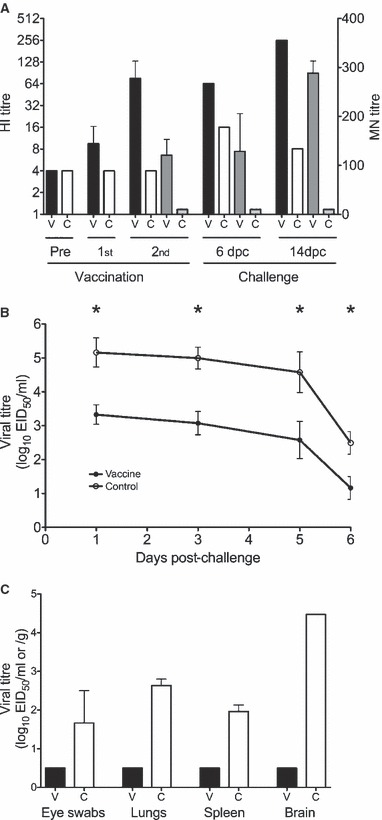
The protective efficacy of the vaccine in ferrets. Groups of four ferrets were vaccinated with two doses of 24 μg HA aluminium adjuvanted H7N1 vaccine or saline (controls) and challenged 3 weeks after the second dose with the highly pathogenic H7N1 virus. Two animals from each group were killed at 6 days post‐challenge and the remaining two at 14 days post‐challenge. (A) The serum HI and MN antibody responses measured pre‐ and 21 days after the first (1st) and second (2nd) doses of vaccine (n = 4 per group) and at 6 and 14 days post‐challenge (dpc) (2 ferrets per group). The HI data is presented as the geometric mean titre and 95% confidence interval of vaccinated (filled black bars) or control (open white bars) animals. The MN titres are presented as the mean ± SEM of vaccinated (dark grey bars) or control (light grey bars) animals after the second vaccination and at 6 and 14 days post‐challenge. For calculation purposes, HI and MN titres <8 and 20, respectively, were assigned values of 4 and 10. (B) The viral shedding in the nasal wash measured from four animals per group at 1, 3 and 5 days post‐challenge, and two animals per group at day 6. The data is presented as mean are presented as the mean ± SEM by filled circles and the control ferrets by open circles. A mixed linear regression model was used to test the viral recovery from nasal washes and the vaccinated ferrets had significantly lower viral shedding than control animals (*P < 0·05). (C) Viral recovery from the eye, spleen, lung and brain detected 6 days after viral challenge of vaccinated (filled black bars) and controls (open bars). The log viral titres were determined in the nasal wash (log EID50/ml) or in the tissues (log EID50 per ml eye swab fluid or per gram of tissue) and are presented as the mean ± SEM. The lower limit of detection was 100·5 EID50/ml for virus recovery from nasal washes, tissues and eye swabs.
The ability of the vaccine to elicit cross‐reactive HI antibody response 18 days after the second vaccination was examined to a contemporary Eurasian egg grown virus [A/mallard/Netherlands/12/2000 (H7N3)] and a North American H7 subtype human isolate [A/New York/107/03 (H7N2)]. The vaccine elicited low but detectable titres to a prototype Eurasian strain (all animals HI titre 8) and North American (range in HI titres 8–16) strains.
A cell‐based H7N1 vaccine reduced viral shedding and prevents systemic spread of virus in ferrets
Vaccination reduced clinical signs of infection
No febrile responses were detected in any of the animals after viral challenge. All vaccinated animals continued to gain weight throughout the study, whereas the weight of control animals stabilized from 9 days post‐challenge (results not shown). One control animal developed occular symptoms, with excess watery and coloured tearing, between 6 and 8 days post‐challenge.
A cell‐based H7N1 vaccine reduced viral shedding
There were significantly higher (P < 0·05) levels of virus recovery in the nasal washes of the controls (20‐ to 100‐fold higher) than the H7N1 vaccinated animals on each of the sampling days (Figure 4B). Individual viral titres were lower in the vaccinated than the control ferrets on all days except on day 5, when one immunised animal had a twofold higher titre than the highest titre recorded for the controls. Nasal wash virus was recovered from fewer challenged mice in the adjuvanted than in the non‐adjuvanted vaccine groups.
Vaccination prevented lower respiratory tract and systemic spread of virus
No virus was recovered from eye swab, lung, spleen and brain from vaccinated animals at 6 days post‐challenge, whereas all of these samples were positive for virus in the control animals (Figure 4C). The titres for lung, spleen and brain homogenates for the two control animals were very similar (≤2‐fold difference for each organ), but a 50‐fold difference was observed for the eye swabs. The viral recovery from the brain (log 4·5) was much higher than from the lungs (79‐fold), spleen (316‐fold) and eye swabs (631‐fold) in the control animals. The whole brain was collected and included the olfactory bulbs which may have increased the level of virus detected.
Discussion
In 2003, an avian influenza strain of the H7 subtype caused one human death in the Netherlands, as well as 86 cases of conjunctivitis and respiratory symptoms in people who handled infected poultry. 2 , 3 Avian H7 viruses have also caused zoonoses in Italy, the USA (2003), Canada (2004) and Great Britain (2007). 4 , 44 , 45 The H7 viruses can be divided into two geographical and phylogenetic groups, termed the Eurasian and North American lineages. 46 Interestingly, the Eurasian strains have been found to elicit cross‐reactive antibodies to both the Eurasian and North American viruses. 47 To increase pandemic preparedness against the H7 subtype, we have prepared a PER.C6 cell‐derived vaccine based on a highly pathogenic Eurasian H7N1 strain. The vaccine was formulated with or without aluminium hydroxide adjuvant to investigate the immunogenicity and protective efficacy in pre‐clinical animal models. Recently, this candidate H7N1 vaccine has also been evaluated in a human phase I clinical trial 33 and found to elicit only low HI and MN titres, although the addition of aluminium adjuvant significantly augmented the antibody response.
Candidate pandemic inactivated avian influenza virus vaccines have been shown to be weakly immunogenic compared to seasonal strains and two doses of adjuvanted vaccine are generally necessary to induce protective antibody levels (reviewed in Ref. 48). Most pandemic vaccination studies have concentrated on the H5N1 subtype and there are only a limited number of candidate H7 vaccine strains available. 26 , 32 , 49 , 50 In this study, we have shown that cell‐based inactivated split virion H7N1 vaccine elicited low antibody levels in mice after two doses of either 12 or 20 μg HA whether it was formulated with or without AlOOH. Our murine data suggests that there is also a need for more effective adjuvants other than aluminium salts with candidate H7 influenza vaccines, such as e.g. the promising proprietary oil‐in‐water emulsion systems MF59 and AS. Furthermore, the vaccine also stimulated low levels of cross‐reactive HI antibodies in mice (results not shown) and ferrets (Table 2) to a North American H7 subtype human isolate and the A/mallard/Netherlands/12/2000 (H7N3) virus, which was the likely ancestor of the highly pathogenic virus that caused the Netherlands outbreak in 2003.
Table 2.
The cross‐reactivity of the post‐vaccination HI antibody response in ferrets to influenza A H7 strains
| Ferret | Virus strain | ||
|---|---|---|---|
| RD‐3 | A/mallard/ Netherlands/ 12/2000 (H7N7) | A/New York/ 107/03 (H7N2) | |
| Vaccinated | 76 | 8 | 11 |
| Control | –* | –* | –* |
Groups of four ferrets were vaccinated with two doses of 24 μg HA aluminium adjuvanted H7N1 vaccine or saline (controls) and blood collected 18 days post‐vaccination. Serum HI titres 8 days after the second vaccination were measured by a modified HI assay using cell grown RD‐3 (H7N1) or egg grown A/mallard/Netherlands/12/2000 (H7N3) and A/New York/107/03 (H7N2) viruses and horse red blood cells. The geometric mean titre (GMT) of each strain is given for the vaccinated and control groups. The limit of detection of the HI assay was 8 and HI antibodies were detected in all immunised ferrets to all strains.
*Negative HI titres (<8).
Importantly, this H7 vaccine induced significant protection from disease and death in mice, despite low pre‐challenge serum HI and MN antibody titres. In contrast, 17 of 20 unvaccinated control mice died or had to be killed due to severe illness. Similarly, other studies have found that whole virus H7 vaccine alone (10 μg HA) or formulated with aluminium salt adjuvant protected mice against challenge with both homologous and heterologous H7 strains. 26 , 49 In contrast, a H7N7 subunit virus vaccine did not elicit protection against homologous challenge, unless formulated with ISCOM adjuvant, 19 suggesting that type of vaccine (whole, split or subunit) may play a role in generation of protective immunity. In our study, all vaccine formulations tested provided protection from disease and death. The adjuvanted vaccine groups also had higher numbers of mice without any signs of infection and lower numbers of mice suffering from weight loss and viral shedding than the non‐adjuvanted vaccine groups, although not statistically significant. These findings suggest that the formulation of the vaccine with aluminium hydroxide adjuvant increased priming against subsequent infection, reducing disease manifestations. Others have also found that adjuvanting with aluminium salts reduced viral shedding and prevented systemic spread of the H5 or H7 virus after challenge. 26 , 27 , 49 In human clinical trials of candidate pandemic vaccines, aluminium adjuvant has ranged from augmenting to suppressing the antibody response (reviewed in Ref. 51) and the oil‐in‐water emulsions have proved most effective at enhancing the antibody response to pandemic candidate vaccines.
The intrinsic ability of a vaccine formulation to elicit an immune response is influenced by activation of the innate immune response via pathogen‐associated molecular patterns. The ability to induce T‐helper response can be assessed by using the IgG2a antibody response as an indicator of a Th1 phenotype and IgG1 as a marker of a Th2‐biased response. 52 We have previously observed that whole virus vaccine elicits high levels of IgG2a antibodies particularly after the first dose of vaccine. In mice, inactivated seasonal split influenza virus vaccines have been shown to elicit a mixed Th1/Th2 response, 42 , 53 , 54 whereas formulation of vaccines with AlOOH is known to promote a Th2‐skewed immune response (reviewed in Ref. 55). Surprisingly, in this study IgG1 completely dominated the antibody response in all vaccine groups and almost no IgG2a was detected either after vaccination or challenge. We have previously produced a whole virus vaccine from the highly pathogenic strain used in these studies and found that this vaccine elicited IgG2a antibody and a mixed IgG2a/IgG1 profile 56 . Similarly, despite only low antibody concentrations in the unimmunised control mice after challenge, a mixed IgG2a/IgG1 profile was observed and this may be associated with the viral ssRNA activating toll‐like receptor 7. This suggests that the formulation of a vaccine used for priming influences the subsequent IgG2a/IgG1 profile after viral challenge, as found previously. 38 IgG1 antibodies have been found to be associated with virus neutralization, whilst IgG2a has been shown to correlate with clearance of virus and increased protection against lethal viral challenge. 57 In our study, protection of mice from disease and death after a Th2 skewed response, highlights that priming by vaccination may have induced memory B and T cells which could be reactivated upon viral challenge and play an important role in protection.
Ferrets are the most relevant animal model for influenza vaccine studies. Our particular highly pathogenic H7N1 virus induced a relatively mild disease in infected ferrets (e.g. no fever and no weight loss) yet the virus caused conjunctivitis and spread to the lungs and beyond, which contrasts with the severe disease and death observed in mice. However, in both these animal models, we observed ocular symptoms and conjunctivitis which is similar to H7 infection of man where the majority of infected patients present with conjunctivitis, and some cases are also associated with influenza‐like illness. We demonstrated in ferrets that whilst 24 μg HA of this H7 vaccine formulated with AlOOH evoked modest HI and MN antibody titres, it effectively prevented spread of the highly pathogenic virus to the eye, the lower respiratory tract (lungs) and systemically to the brain and spleen at 6 days post‐challenge. Furthermore, vaccination also reduced nasal viral shedding, thus potentially reducing transmission of the virus and preventing spread of the virus to the lower respiratory tract. Indeed, protective efficacy studies using pandemic candidate H5 vaccines in animal models have shown that correlates of resistance to serious illness and death to avian viruses may not be solely reflected by levels of circulating serum antibodies. 18 , 50 , 58 , 59 Furthermore, vaccination may still protect against disease and death after challenge in the absence of detectable antibodies, as mice were protected against lethal H5N1 challenge despite no or very low pre‐challenge HI and neutralisation antibody titres. 18 , 50 , 57 Other factors, such as the intrinsic ability of the vaccine to stimulate the innate response, effective priming of the adaptive immune system particularly the cellular immune response and its associated cytokine production may also play a role. These findings may be highly relevant to the development of human candidate pandemic or pre‐pandemic influenza vaccines, as there is widespread concern about the low serum antibody response detected in humans after immunisation with avian influenza vaccine candidates.
In this study, we have investigated the protective efficacy of a PER.C6® cell grown H7N1 split influenza virus vaccine in pre‐clinical animal models. Importantly, whilst this vaccine induced modest antibody titres in these animals it effectively prevented systemic spread, reduced viral shedding and reduced illness and/or death after challenge with the highly pathogenic parent avian H7 virus. The ability of this vaccine to provide cross‐protection against Eurasian and North American strains which have caused human illness is important and would require further investigation. In a pandemic scenario, the ability of a vaccine to reduce transmission and protect people from disease and death is paramount. Thus, our results confirm and extend the findings with H5 vaccine candidates that HI and MN antibody titres alone are not necessarily the optimal correlates of protection and that more research is needed to define these factors.
Conflict of interest
John Wood and Diane Major are employed by an organisation that receives funding from the International Federation of Pharmaceutical Manufacturers and Associations for a member of staff and research into improved influenza vaccine viruses. Jon Smith and Frederick Vogel are both employed by Sanofi Pasteur.
Acknowledgements
We thank all partners in the European Union Flupan (QLK2‐CT‐2001‐01786) project, Pauline Lloyd, Rachel Landon and Rose Curran at NIBSC for excellent technical assistance. The virus strain was provided by Isabella Donatelli, Istituto Superiore di Sanita, Rome, Italy.
References
- 1. World Health Organization . Cumulative Number of Confirmed Human Cases of Avian Influenza A/(H5N1) Reported to WHO. 2008. [updated 2008; cited 2007]; Available from: http://www.who.int/csr/don/2009_03_11/en/index.html.
- 2. Fouchier RA, Schneeberger PM, Rozendaal FW et al. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc Natl Acad Sci USA 2004; 3:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koopmans M, Wilbrink B, Conyn M et al. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet 2004; 363:587–593. [DOI] [PubMed] [Google Scholar]
- 4. Skowronski DM, Li Y, Tweed SA et al. Protective measures and human antibody response during an avian influenza H7N3 outbreak in poultry in British Columbia, Canada. CMAJ 2007; 176:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. HPA Press Statement. Confirmation of Avian Influenza H7N2 Infection. 2007. [updated 2007; cited 2007, 25 May]; Available from: http://www.hpa.org.uk/hpa/news/articles/press_releases/2007/070525_avian_flu_H7N2.htm.
- 6. Wong SS, Yuen KY. Avian influenza virus infections in humans. Chest 2006; 129:156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belser JA, Blixt O, Chen LM et al. Contemporary North American influenza H7 viruses possess human receptor specificity: implications for virus transmissibility. Proc Natl Acad Sci USA 2008; 27:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Longini IM Jr, Nizam A, Xu S et al. Containing pandemic influenza at the source. Science 2005; 309:1083–1087. [DOI] [PubMed] [Google Scholar]
- 9. Ferguson NM, Cummings DA, Fraser C, Cajka JC, Cooley PC, Burke DS. Strategies for mitigating an influenza pandemic. Nature 2006; 442:448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoffmann E, Krauss S, Perez D, Webby R, Webster RG. Eight‐plasmid system for rapid generation of influenza virus vaccines. Vaccine 2002; 20:3165–3170. [DOI] [PubMed] [Google Scholar]
- 11. Webby RJ, Perez DR, Coleman JS et al. Responsiveness to a pandemic alert: use of reverse genetics for rapid development of influenza vaccines. Lancet 2004; 363:1099–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schild GC, Oxford JS, De Jong JC, Webster RG. Evidence for host‐cell selection of influenza virus antigenic variants. Nature 1983; 303:706–709. [DOI] [PubMed] [Google Scholar]
- 13. Katz JM, Naeve CW, Webster RG. Host cell‐mediated variation in H3N2 influenza viruses. Virology 1987; 156:386–395. [DOI] [PubMed] [Google Scholar]
- 14. Palache AM, Scheepers HS, De Regt V et al. Safety, reactogenicity and immunogenicity of Madin Darby Canine Kidney cell‐derived inactivated influenza subunit vaccine. A meta‐analysis of clinical studies. Dev Biol Stand 1999; 98:115–125. Discussion 33–4. [PubMed] [Google Scholar]
- 15. Ehrlich HJ, Muller M, Oh HM et al. A clinical trial of a whole‐virus H5N1 vaccine derived from cell culture. N Engl J Med 2008; 358:2573–2584. [DOI] [PubMed] [Google Scholar]
- 16. Kistner O, Barrett PN, Mundt W, Reiter M, Schober‐Bendixen S, Dorner F. Development of a mammalian cell (Vero) derived candidate influenza virus vaccine. Vaccine 1998; 16:960–968. [DOI] [PubMed] [Google Scholar]
- 17. Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M. Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N Engl J Med 2006; 354:1343–1351. [DOI] [PubMed] [Google Scholar]
- 18. Ninomiya A, Imai M, Tashiro M, Odagiri T. Inactivated influenza H5N1 whole‐virus vaccine with aluminum adjuvant induces homologous and heterologous protective immunities against lethal challenge with highly pathogenic H5N1 avian influenza viruses in a mouse model. Vaccine 2007; 25:3554–3560. [DOI] [PubMed] [Google Scholar]
- 19. De Wit E, Munster VJ, Spronken MI et al. Protection of mice against lethal infection with highly pathogenic H7N7 influenza A virus by using a recombinant low‐pathogenicity vaccine strain. J Virol 2005; 79:12401–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pushko P, Tumpey TM, Van Hoeven N et al. Evaluation of influenza virus‐like particles and Novasome adjuvant as candidate vaccine for avian influenza. Vaccine 2007; 25:4283–4290. [DOI] [PubMed] [Google Scholar]
- 21. Bresson JL, Perronne C, Launay O et al. Safety and immunogenicity of an inactivated split‐virion influenza A/Vietnam/1194/2004 (H5N1) vaccine: phase I randomised trial. Lancet 2006; 367:1657–1664. [DOI] [PubMed] [Google Scholar]
- 22. Nicholson KG, Colegate AE, Podda A et al. Safety and antigenicity of non‐adjuvanted and MF59‐adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a randomised trial of two potential vaccines against H5N1 influenza. Lancet 2001; 357:1937–1943. [DOI] [PubMed] [Google Scholar]
- 23. Leroux‐Roels I, Borkowski A, Vanwolleghem T et al. Antigen sparing and cross‐reactive immunity with an adjuvanted rH5N1 prototype pandemic influenza vaccine: a randomised controlled trial. Lancet 2007; 370:580–589. [DOI] [PubMed] [Google Scholar]
- 24. Lu X, Tumpey TM, Morken T, Zaki SR, Cox NJ, Katz JM. A mouse model for the evaluation of pathogenesis and immunity to influenza A (H5N1) viruses isolated from humans. J Virol 1999; 73:5903–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu X, Edwards LE, Desheva JA et al. Cross‐protective immunity in mice induced by live‐attenuated or inactivated vaccines against highly pathogenic influenza A (H5N1) viruses. Vaccine 2006; 24:6588–6593. [DOI] [PubMed] [Google Scholar]
- 26. Pappas C, Matsuoka Y, Swayne DE, Donis RO. Development and evaluation of an influenza virus subtype H7N2 vaccine candidate for pandemic preparedness. Clin Vaccine Immunol 2007; 14:1425–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruat C, Caillet C, Bidaut A, Simon J, Osterhaus AD. Vaccination of macaques with adjuvanted formalin‐inactivated influenza A virus (H5N1) vaccines: protection against H5N1 challenge without disease enhancement. J Virol 2008; 82:2565–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stephenson I, Bugarini R, Nicholson KG et al. Cross‐reactivity to highly pathogenic avian influenza H5N1 viruses after vaccination with nonadjuvanted and MF59‐adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a potential priming strategy. J Infect Dis 2005; 191:1210–1215. [DOI] [PubMed] [Google Scholar]
- 29. Stephenson I, Nicholson KG, Colegate A et al. Boosting immunity to influenza H5N1 with MF59‐adjuvanted H5N3 A/Duck/Singapore/97 vaccine in a primed human population. Vaccine 2003; 21:1687–1693. [DOI] [PubMed] [Google Scholar]
- 30. Baras B, Stittelaar KJ, Simon JH et al. Cross‐protection against lethal H5N1 challenge in ferrets with an adjuvanted pandemic influenza vaccine. PLoS ONE 2008; 3:e1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bernstein DI, Edwards KM, Dekker CL et al. Effects of adjuvants on the safety and immunogenicity of an avian influenza H5N1 vaccine in adults. J Infect Dis 2008; 197:667–675. [DOI] [PubMed] [Google Scholar]
- 32. Whiteley A, Major D, Legastelois I et al. Generation of candidate human influenza vaccine strains in cell culture – rehearsing the European response to an H7N1 pandemic threat. Influenza Other Respir Viruses 2007; 1:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cox RJ, Madhun AS, Hauge S et al. A phase I clinical trial of the safety and immunogenicity of the first cell grown influenza H7 virus vaccine. Vaccine 2009; (doi:DOI: 10.1016/j.vaccine.2009.01.116). [DOI] [PubMed] [Google Scholar]
- 34. Banks J, Speidel ES, Moore E et al. Changes in the haemagglutinin and the neuraminidase genes prior to the emergence of highly pathogenic H7N1 avian influenza viruses in Italy. Arch Virol 2001; 146:963–973. [DOI] [PubMed] [Google Scholar]
- 35. Nicolson C, Major D, Wood JM, Robertson JS. Generation of influenza vaccine viruses on Vero cells by reverse genetics: an H5N1 candidate vaccine strain produced under a quality system. Vaccine 2005; 23:2943–2952. [DOI] [PubMed] [Google Scholar]
- 36. Cox RJ, Mykkeltvedt E, Robertson J, Haaheim LR. Non‐lethal viral challenge of influenza haemagglutinin and nucleoprotein DNA vaccinated mice results in reduced viral replication. Scand J Immunol 2002; 55:14–23. [DOI] [PubMed] [Google Scholar]
- 37. Govorkova EA, Rehg JE, Krauss S et al. Lethality to ferrets of H5N1 influenza viruses isolated from humans and poultry in 2004. J Virol 2005; 79:2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hovden AO, Cox RJ, Madhun A, Haaheim LR. Two doses of parenterally administered split influenza virus vaccine elicited high serum IgG concentrations which effectively limited viral shedding upon challenge in mice. Scand J Immunol 2005; 62:342–352. [DOI] [PubMed] [Google Scholar]
- 39. Mahy BWJ, Kangro HO. Virology Methods Manual. London: Academic Press, 1996, pp. 36–37. [Google Scholar]
- 40. Stephenson I, Wood JM, Nicholson KG, Charlett A, Zambon MC. Detection of anti‐H5 responses in human sera by HI using horse erythrocytes following MF59‐adjuvanted influenza A/Duck/Singapore/97 vaccine. Virus Res 2004; 103:91–95. [DOI] [PubMed] [Google Scholar]
- 41. Rowe T, Abernathy RA, Hu‐Primmer J et al. Detection of antibody to avian influenza A (H5N1) virus in human serum by using a combination of serologic assays. J Clin Microbiol 1999; 37:937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hovden AO, Cox RJ, Haaheim LR. Whole influenza virus vaccine is more immunogenic than split influenza virus vaccine and induces primarily an IgG2a response in BALB/c mice. Scand J Immunol 2005; 62:36–44. [DOI] [PubMed] [Google Scholar]
- 43. Hauge S, Madhun A, Cox RJ, Haaheim LR. Quality and kinetics of the antibody response in mice after three different low‐dose influenza virus vaccination strategies. Clin Vaccine Immunol 2007; 14:978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Puzelli S, Di Trani L, Fabiani C et al. Serological analysis of serum samples from humans exposed to avian H7 influenza viruses in Italy between 1999 and 2003. J Infect Dis 2005; 192:1318–1322. [DOI] [PubMed] [Google Scholar]
- 45. Tweed SA, Skowronski DM, David ST et al. Human illness from avian influenza H7N3, British Columbia. Emerg Infect Dis 2004; 10:2196–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Banks J, Speidel EC, McCauley JW, Alexander DJ. Phylogenetic analysis of H7 haemagglutinin subtype influenza A viruses. Arch Virol 2000; 145:1047–1058. [DOI] [PubMed] [Google Scholar]
- 47. Joseph T, McAuliffe J, Lu B, Jin H, Kemble G, Subbarao K. Evaluation of replication and pathogenicity of avian influenza a H7 subtype viruses in a mouse model. J Virol 2007; 81:10558–10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. European Centre of Disease Prevention and Control (ECDC) . Technical Report. Expert Advisory Groups on Human H5N1 Vaccines. Scientific Questions. 2007. [updated 2007; cited August 2007]; Available from: http://ecdc.europa.eu/pdf/Sci%20Questions%20final.pdf.
- 49. Jadhao SJ, Achenbach J, Swayne DE, Donis R, Cox N, Matsuoka Y. Development of Eurasian H7N7/PR8 high growth reassortant virus for clinical evaluation as an inactivated pandemic influenza vaccine. Vaccine 2008; 26:1742–1750. [DOI] [PubMed] [Google Scholar]
- 50. Lipatov AS, Hoffmann E, Salomon R, Yen HL, Webster RG. Cross‐protectiveness and immunogenicity of influenza A/Duck/Singapore/3/97(H5) vaccines against infection with A/Vietnam/1203/04(H5N1) virus in ferrets. J Infect Dis 2006; 194:1040–1043. [DOI] [PubMed] [Google Scholar]
- 51. Keitel WA, Atmar RL. Preparing for a possible pandemic: influenza A/H5N1 vaccine development. Curr Opin Pharmacol 2007; 7:484–490. [DOI] [PubMed] [Google Scholar]
- 52. Stevens TL, Bossie A, Sanders VM et al. Regulation of antibody isotype secretion by subsets of antigen‐specific helper T cells. Nature 1988; 334:255–258. [DOI] [PubMed] [Google Scholar]
- 53. Szyszko E, Brokstad K, Cox RJ, Hovden AO, Madhun A, Haaheim LR. Impact of influenza vaccine formulation with a detailed analysis of the cytokine response. Scand J Immunol 2006; 64:467–475. [DOI] [PubMed] [Google Scholar]
- 54. Hauge S, Madhun AS, Cox RJ, Brokstad KA, Haaheim LR. A comparison of the humoral and cellular immune responses at different immunological sites after split influenza virus vaccination of mice. Scand J Immunol 2007; 65:14–21. [DOI] [PubMed] [Google Scholar]
- 55. Lindblad EB. Aluminium adjuvants – in retrospect and prospect. Vaccine 2004; 22:3658–3668. [DOI] [PubMed] [Google Scholar]
- 56. Hovden AO, Brokstad KA, Major D, Wood J, Haaheim LR, Cox RJ. A pilot study of the immune response to whole inactivated avian infuenza H7N1 virus vaccine in mice. Infuenza and Other Respiratory Viruses 2009; 3:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huber VC, McKeon RM, Brackin MN et al. Distinct contributions of vaccine‐induced immunoglobulin G1 (IgG1) and IgG2a antibodies to protective immunity against influenza. Clin Vaccine Immunol 2006; 13:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Suguitan AL Jr, McAuliffe J, Mills KL et al. Live, attenuated influenza A H5N1 candidate vaccines provide broad cross‐protection in mice and ferrets. PLoS Med 2006; 3:e360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mahmood K, Bright RA, Mytle N et al. H5N1 VLP vaccine induced protection in ferrets against lethal challenge with highly pathogenic H5N1 influenza viruses. Vaccine 2008; 26:5393–5399. [DOI] [PubMed] [Google Scholar]