

Abstract
Over 160 chiral vicinal bromochlorinated natural products have been identified, however a lack of synthetic methods for the selective incorporation of halogens into organic molecules has hindered their synthesis. Here we disclose the first total synthesis and structural confirmation of isoplocamenone and plocamenone, as well as the first selective and scaleable synthesis of the preclinical anticancer natural product halomon. The synthesis of these interhalogenated compounds has been enabled by our recently developed chemo-, regio-, and enantioselective dihalogenation reaction.
Over the course of the past few decades, a plethora of halogenated natural products have been isolated, primarily from marine sources.1a–c Roughly 240 contain chiral vicinal dihalide motifs, with the majority (ca. 70%) being bromochlorinated terpenes.1d Many of these interhalogenated compounds are reported to possess promising biological activity including unique cytotoxicity profiles. Additionally, such molecules pose exquisite challenges to selective synthesis and structural characterization. This is highlighted by the lack of strategies that provide stereocontrolled access to relevant halogenated motifs and by the repeated misassignment of such natural products.
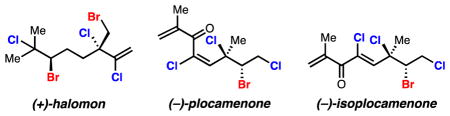
The most-studied molecule of this class, halomon (1, Fig. 1), was originally isolated in 1975 from the red algae Portieria hornemannii.2a Full structural data as well as relative and absolute stereochemical assignments were presented seventeen years later by the National Cancer Institute upon its reisolation and crystallization. In this study, 1 produced “one of the most extreme cases of differential cytotoxicity”2b in the NCI-60 human tumor cell line screen. Preliminary in vivo studies were so encouraging that the compound was selected by the NCI for preclinical drug development, but was dropped explicitly due to lack of sufficient material.3a Despite intense interest in 1 from the scientific community,3 efforts to isolate sufficient material from natural sources have failed,3a,h likely due to local and seasonal variations in terpene content.3g,h Although no comprehensive study on the mechanism of action has been reported, insight is gained by the discovery that halomon inhibits DNA methyltransferase-1,4 an important target for cancer research and therapy.5 Surprisingly, based on computational analysis of its cytotoxicity profile, researchers have concluded that the activity of 1 is unlikely to stem from it acting as a non-specific alkylating agent.3b,6 Experimental investigations into the validity of this statement have not been conducted.
FIGURE 1.

Structures of halomon, plocamenone, and isoplocamenone and our approach to such motifs.
Two non-selective total syntheses of halomon have been reported by Mioskowsky and Hirama.7 Both efforts afford halomon as mixtures of all possible diastereo- and enantiomers and rely on silica gel chromatography and two consecutive semi-preparative HPLC separations with achiral followed by chiral columns to afford pure (+)-1 in undisclosed amounts. Hirama reported a discrepancy in optical rotation between synthetic and natural material, leading them to posit the originally isolated halomon may have been impure. While this could call into question the original NCI-60 screen, subsequent NCI-60 screens with halomon from different sources have revealed similar cytotoxicity profiles.7g,h,i
In addition to providing material for biological evaluation, total synthesis is an invaluable tool for the structural assignment of natural produts.8 Noncrystalline linear polyhalogenated terpenes are notoriously difficult to characterize, as illustrated by numerous NMR studies relying on comparison to synthetic halogenated model systems for structure elucidation.2a,9 For example, the cytotoxic agent plocamenone (E)-2 has been repeatedly misassigned in the literature,9b,10 with the closest proposed structure being the (Z)-isomer.10b More recently, Urban isolated and characterized both double bond isomers from Plocamium angustum and assigned the previously isolated compound, plocamenone, as the (E)-isomer ((E)-2). The minor isomer, termed isoplocamenone ((Z)-2) was reportedly prone to decomposition.11
A recent advance made in this laboratory allows for the chemo-, regio-, and enantioselecive bromochlorination, dibromination, and dichlorination of allylic alcohols (Fig. 1, bottom).12 Here, this method is used strategically to achieve the first total syntheses, structural confirmation, and determination of the natural enantiomer of isoplocamenone and plocamenone. Further, we report the first total synthesis of (+)-halomon with essentially complete stereocontrol.
Selective syntheses of (−)-plocamenone and (−)-isoplocamenone
We began our syntheses of plocamenone and isoplocamenone with (S,R)-Schiff base 3-catalyzed bromochlorination12b of known allylic alcohol 4 (Scheme 1), which can be synthesized in one step from commercially available isoprene oxide.13 Absolute configuration was assigned through X-ray crystallographic analysis (see Supporting Information). The resulting alcohol 5 was then oxidized with Dess–Martin periodinane and subjected to Horner–Wadsworth–Emmons olefination with 614 to yield ketone 7 in a 13:1 ratio of double bond isomers (see Supporting Information for nOe structural assignment). Treatment with Eschenmoser’s chloride salt (8) and subsequent activation with iodomethane and elimination15 yielded a separable 1:2 mixture of plocamenone ((E)-2) and isoplocamenone ((Z)-2).16a The use of Eschenmoser’s iodide salt produced a significantly messier reaction mixture but showed no signs of double bond isomerization. This leads us to surmise that conjugate addition of chloride followed by C–C bond rotation and elimination may be responsible for this isomerization of the trisubstituted olefin.
SCHEME 1. Synthesis of (−)-plocamenone and (−)-isoplocamenone.

Reagents and conditions: a. NBS (1.05 equiv), ClTi(Oi-Pr)3 (1.10 equiv), 20 mol % (S,R)-3, hexanes, −20 °C, 64%; b. Dess–Martin periodinane (DMP) (1.5 equiv), NaHCO3 (10.0 equiv), CH2Cl2, r.t.; c. diethyl (1-chloro-2-oxobutyl)phosphonate (6) (2.0 equiv), sodium hexamethyl-disilazide (NaHMDS) (1.4 equiv), THF, −78 °C to 0 °C, 70% from 5; d. trimethylsilyl trifluoromethanesulfonate (TMSOTf) (2.0 equiv), N,N-diisopropylethylamine (3.0 equiv), CH2Cl2, −78 °C to r.t.; 8 (5.0 equiv), −78 °C to r.t.; e. iodomethane (5.0 equiv), N,N-diisopropylethylamine (3.6 equiv), THF, r.t., 54% from 7; f. floodlamp, Et2O, 5 days, r.t.
In its most recent isolation and structural report,11 isoplocamenone was reported to be unstable, but no mention of decomposition products was made. In light of the apparent isomerization during the α-methylenation, we suspected that isoplocamenone could possibly convert into its double bond isomer plocamenone. Indeed, when irradiated with a flood lamp for five days, a pure sample of isoplocamenone was found to isomerize to a 3:1 mixture of plocamenone/isoplocamenone.16b These syntheses have also established the absolute configuration of the natural products as the shown enantiomers.17
Selective synthesis of (+)-halomon
Our highly selective route to halomon starts with known allylic alcohol 9,18 which was bromochlorinated according to reported conditions12b with (R,S)-Schiff base 3 to provide 10 in 90% ee on multigram scale (Scheme 2). It was found that the resulting bromochloroalcohol could be deoxygenated in nearly quantitative yield via activation as the trifluoromethanesulfonate ester followed by treatment with L-selectride.19 The resulting bromochlorinated myrcene derivative 11 was taken on without purification and subjected to a formal 1,4-bromohydration sequence,20 which occurs via dibromide 13 in moderate overall yield but excellent selectivity for the trans double bond isomer 12 which is readily isolable in pure form. Several aspects of this sequence are of note. The initial 1,4-dibromination to 13 is highly selective for the E-isomer, presumably due to dibromination occuring on the s-trans conformational isomer of 11. Bromide displacement with acetate occurs with high selectivity for the less-hindered terminal allylic site, but at high conversion subsequent displacement of the second allylic bromide occurs, prompting us to stop this reaction prior to full conversion. In our hands, direct bromoacetylation of 11 with acetyl hypobromite gave mixtures of 1,4- and 1,2-bromoacetylated products.
SCHEME 2. Synthesis of (+)-halomon.

Reagents and conditions: a. NBS (1.05 equiv), ClTi(Oi-Pr)3 (1.10 equiv), 20 mol % (R,S)-3, hexanes, −20 °C, 73%; b. trifluoromethanesulfonic anhydride (1.2 equiv), 2,6-lutidine (1.5 equiv), CH2Cl2, −78 °C; c. L-selectride (2.2 equiv), THF, −78 °C, crude 95% from 10; d. Br2 (1.0 equiv), K2CO3 (0.1 equiv), hexanes, −78 °C; e. Lithium acetate (1.0 equiv), DMF, 0 °C; f. K2CO3 (1.0 equiv), methanol, 0 °C to r.t., 39% 12 + 22% 13; g. t-BuOCl (1.5 equiv), ClTi(Oi-Pr)3 (1.10 equiv), 30 mol % (S,R)-3, hexanes, −20 °C, 82%; h. trifluoromethanesulfonic anhydride (1.2 equiv), 2,6-lutidine (1.5 equiv), CH2Cl2, −78 °C; i. sodium hydroxide, H2O, THF, 0 °C to r.t., 69% from 14.
Bromohydrin 12 was then subjected to dichlorination conditions12b with (S,R)-Schiff base 3 to give alcohol 14 in >20:1 diastereomeric ratio. In the absence of chiral ligand an inseperable 1:1 mixture of dichloride diastereomers was obtained. Other dichlorination conditions such as Et4NCl3 also resulted in no diastereoselectivity. At this point, the absolute and relative configuration was confirmed by X-ray crystal structure analysis of ferrocenecarboxylate derivative 14a (Scheme 3, top). We have identified this derivatization as being particularly valuable in cases where common benzoate and sulfonate derivatives fail to induce adequate crystallinity. Alcohol 14 was then converted to the primary trifluoromethanesulfonate ester and selectively eliminated with aqueous hydroxide in the presence of five alkyl halides to afford (+)-halomon (1). The route thus developed has allowed for the synthesis of 400 mg of pure (+)-1. In initial biological activity investigations (K562 human leukemia), we have repeatedly measured 50% growth inhibitory concentrations of 1.3–2.3 μM, closely matching the originally reported 1.2 μM.2b We also have used this chemistry to synthesize unnatural enantiomer (−)-1 demonstrating the power of the developed dihalogenation reaction in this context.21 The stage is now set for the synthesis of other unnatural stereoisomers as well as analogues for structure–activity relationship and target-identification studies.22
Scheme 3. Stereochemical confirmation of 14 and a first generation approach to halomon.

Our first strategy toward a selective synthesis of halomon involved attempted double bromochlorination of bis-allylic alcohol 15 (Scheme 3, bottom). Likely due to poor solubility of the substrate in the reaction solvent, the diastereoselectivity of the reaction was found to be poor. Whereas a 20 mol % ligand loading afforded a 1:1 mixture of diastereomers, the best result was obtained with 100 mol %, giving a meager ratio of 3:1 favoring the desired diastereomer 16 (confirmed via X-ray crystallographic analysis, Scheme 3). The identity of the minor diastereomer was not unambiguously determined.
The chemo-, enantio-, and regioselective di- and interhalogenation developed in this laboratory has proven to be an invaluable tool for the synthesis of stereo-complex polyhalogenated natural products. It has enabled the first total synthesis of isoplocamenone and plocamenone, as well as the first selective and scaleable total synthesis of halomon. The syntheses have made possible the structural confirmation of isoplocamenone and plocamenone, the determination of their natural enantiomers, and the finding of an interesting photoisomerization between the two compounds. In addition, the synthesis reported here proved to be a reliable source of pure (+)-halomon, allowing a total of more than 400 mg to have been synthesized to date. This material will be used in future studies toward answering important questions pertinent to the mechanism of action of halomon.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by generous funds from Stanford University and the National Institutes of Health (R01 GM114061 & R01 GM087934).
We are grateful to Professor C. Khosla for helpful discussions as well as to Dr. A. Oliver (University of Notre Dame) for X-ray crystallographic analysis and Dr. S. Lynch (Stanford University) for assistance with NMR spectroscopy.
Footnotes
Supporting Information. Experimental procedures, characterizations, and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gribble GW. Naturally Occurring Organohalogen Compounds – A Comprehensive Survey. Springer-Verlag; Wien: 1996. Gribble GW. Naturally Occurring Organohalogen Compounds – A Comprehensive Update. Springer-Verlag; Wien: 2010. Gribble GW. Environ Chem. 2015;12:396–405.(d) Estimation based on analysis of Ref. 1a–c.
- 2.(a) Burreson JB, Woolard FX, Moore RE. Chem Lett. 1975;4:1111–1114. [Google Scholar]; (b) Fuller RW, Cardellina JH, Kato Y, Brinen LS, Clardy J, Snader KM, Boyd MR. J Med Chem. 1992;35:3007–3011. doi: 10.1021/jm00094a012. [DOI] [PubMed] [Google Scholar]
- 3.Fuller RW, Cardellina JH, Jurek J, Scheuer PJ, Alvarado-Lindner B, McGuire M, Gray GN, Steiner JR, Clardy J, Menez E, Shoemaker RH, Newman DJ, Snader KM, Boyd MR. J Med Chem. 1994;37:4407–4411. doi: 10.1021/jm00051a019.(b) In the previous reference the authors noted a 40% apparent “cure” rate in xenografts of the aggressive U251 brain tumor cell line. Egorin MJ, Sentz DL, Rosen DM, Ballesteros MF, Kearns CM, Callery PS, Eiseman JL. Cancer Chemother Pharmacol. 1996;39:51–60. doi: 10.1007/s002800050537.Egorin MJ, Rosen DM, Benjamin SE, Callery PS, Sentz DL, Eiseman JL. Cancer Chemother Pharmacol. 1997;41:9–14. doi: 10.1007/s002800050701.Jaspars M. In: Advances in Drug Discovery Techniques. Harvey AL, editor. John Wiley & Sons; New York: 1998. pp. 65–84.Kladi M, Vagias C, Roussis V. Phytochem Rev. 2004;3:337–366.Vogel CV, Pietraszkiewicz H, Sabry OM, Gerwick WH, Valeriote FA, Vanderwal CD. Angew Chem Int Ed. 2014;53:12205–12209. doi: 10.1002/anie.201407726.Payo DA, Colo J, Calumpong H, de Clerck O. Mar Drugs. 2011;9:2438–2468. doi: 10.3390/md9112438.
- 4.Andrianasolo EH, France D, Cornell-Kennon S, Gerwick WH. J Nat Prod. 2006;69:576–579. doi: 10.1021/np0503956. [DOI] [PubMed] [Google Scholar]
- 5.For reviews, see: Dhanak D, Jackson P. Biochem Biophys Res Commun. 2014;455:58–69. doi: 10.1016/j.bbrc.2014.07.006.Gnyszka A, Jastrzebski Z, Flis S. Anticancer Res. 2013;33:2989–2996.
- 6.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiem DA, Rubinstein L, Plowman J, Boyd MR. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 7.Schlama T, Baati R, Gouverneur V, Valleix A, Falck JR, Mioskowski C. Angew Chem Int Ed. 1998;37:2085–2087. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2085::AID-ANIE2085>3.0.CO;2-J.Sotokawa T, Noda T, Pi S, Hirama M. Angew Chem Int Ed. 2000;39:3430–3432. doi: 10.1002/1521-3773(20001002)39:19<3430::aid-anie3430>3.0.co;2-3.Hirama M, Togawa N. 3589592. JP. 1999For studies towards the synthesis of 1, see: Braddock DC, Gao AX, White AJP, Whyte M. Chem Commun. 2014;50:13725–13728. doi: 10.1039/c4cc06234e.Also see: Boyes AL, Wild M. Tetrahedron Lett. 1998;39:6725–6728.(c) For the first syntheses of acyclic polyhalogenated Plocamium monoterpenes, see Ref. 3g. [accessed August 18, 2015];Data obtained directly from the NCI-60 database via: CellMiner Database Version 1.6.1. http://discover.nci.nih.gov/cellminer.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, Pommier Y. Cancer Res. 2012;72:3499–3511. doi: 10.1158/0008-5472.CAN-12-1370. See Supporting Information for halomon NCI-60 data.
- 8.Nicolaou KC, Snyder SA. Angew Chem Int Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]
- 9.(a) Crews P, Naylor S, Hanke FJ, Hogue ER, Kno E, Braslau R. J Org Chem. 1984;49:1371–1377. [Google Scholar]; (b) Naylor S, Manes LV, Crews P. J Nat Prod. 1985;48:72–75. [Google Scholar]; (c) Faulkner DJ, Stallard MO. Tetrahedron Lett. 1973;14:1171–1174. [Google Scholar]
- 10.(a) Dunlop RW, Murphy PT, Wells RJ. Aust J Chem. 1979;32:2735–2739. [Google Scholar]; (b) Leary JV, Kfir R, Sims JJ, Fulbright DW. Mutat Res. 1979;68:301–305. doi: 10.1016/0165-1218(79)90162-9. [DOI] [PubMed] [Google Scholar]; (c) Brownlee RTC, Hall JG, Reiss JA. Org Magn Res. 1983;21:544–547. [Google Scholar]; (d) Stierle DB, Sims JJ. Tetrahedron Lett. 1984;25:153–156. [Google Scholar]
- 11.Timmers MA, Dias DA, Urban S. Mar Drugs. 2012;10:2089–2102. doi: 10.3390/md10092089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Hu DX, Shibuya GM, Burns NZ. J Am Chem Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]; (b) Hu DX, Seidl FJ, Bucher C, Burns NZ. J Am Chem Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]
- 13.Mandal AK, Schneekloth JS, Kuramoji K, Crews CM. Org Lett. 2006;8:427–430. doi: 10.1021/ol052620g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prévost S, Gauthier S, Caño do Andrade MC, Mordant C, Touati AR, Lesot P, Savignac P, Ayad T, Phansavath P, Ratovelomanana-Vidal V, Genêt J-P. Tetrahedron: Asymmetry. 2010;21:1436–1446. [Google Scholar]
- 15.(a) Schreiber J, Maag H, Hashimoto N, Eschenmosher A. Angew Chem Int Ed. 1971;10:330–331. [Google Scholar]; (b) Danishefsky S, Kitahara T, McKee R, Schuda PF. J Am Chem Soc. 1976;98:6715–6717. doi: 10.1021/ja00426a066. [DOI] [PubMed] [Google Scholar]
- 16.(a) Conducting the α-methylenation in the dark yielded the same 1:2 mixture of double bond isomers. (b) Irradiation for 12 hours produced a 2:1 plocamenone/isoplocamenone ratio.
- 17.Natural (E)-2: [α]D = −14.3° (c = 0.8) from Ref. 10a; synthetic (E)-2: [α]D = −15.6° (c = 0.5, CHCl3).
- 18.Wender PA, Croatt MP, Witulski B. Tetrahedron. 2006;62:7505–7511. [Google Scholar]
- 19.For the reduction of alkyl sulfonates with lithium triethyl-borohydride, see: Krishnamurthy S, Brown HC. J Org Chem. 1976;41:3064–3066.Krishnamurthy S. J Organomet Chem. 1978;156:171–181.For utility in synthesis, see: Hua DH, Sinai-Zingde G, Venkataraman S. J Am Chem Soc. 1985;107:4088–4090.
- 20.Conditions adapted from: Doi N, Seko S, Kimura K, Takahashi T. 20020107422. US. 2002
- 21.Both enantiomers of synthetic halomon were analyzed by reverse phase chiral HPLC according to conditions reported by Hirama (Ref. 7b) and found to be of >99% ee (see Supporting Information).
-
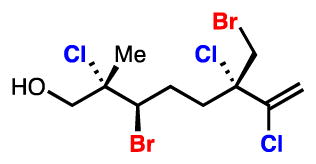
22.For example, silyl protection of 10 has allowed for the synthesis of the following derivatizable analog:
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.