

Abstract
We report the optimization of a series of metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulators (PAMs) from an acyl dihydropyrazolo[1,5-a]pyrimidinone class. Investigation of exocyclic amide transpositions with this unique 5,6-bicyclic core were conducted in attempt to modulate physicochemical properties and identify a suitable backup candidate with a reduced half-life. A potent and selective PAM, 1-(2-(phenoxymethyl)-6,7-dihydropyrazolo[1,5-a]pyrimidin-4(5H)-yl)ethanone (9a, VU0462807), was identified with superior solubility and efficacy in the acute amphetamine-induced hyperlocomotion (AHL) rat model with a minimum effective dose of 3 mg/kg. Attempts to mitigate oxidative metabolism of the western phenoxy of 9a through extensive modification and profiling are described.
Keywords: metabotropic glutamate receptor 5, mGlu5, positive allosteric modulator (PAM)
Graphical Abstract

The pursuit of positive allosteric modulators (PAMs) of mGlu5 as a promising therapeutic approach to treat cognitive deficits in schizophrenia has been stimulated by evidence from several antipsychotic and cognitive animal models.1-7 Recently, we reported identification of a mGlu5 PAM clinical candidate VU0409551/JNJ-46778212 (1)8 and subsequent efforts9 towards a backup candidate within the (2(phenoxymethyl)-6,7-dihydrooxazolo[5,4-c]pyridine-5(4H)-yl(aryl)methanone series focused on enhancing in vivo efficacy through improvements in PK and/or in vitro potency within the series. Prior findings from Merck-Addex10 and our laboratories11 unveiled a target-mediated CNS adverse-effect liability; which appeared to be driven by excessive PAM cooperativity or allosteric agonism, suggesting that PAMs with lower cooperativity and lacking allosteric agonism may be preferable for obtaining an improved therapeutic index.12,13 Interestingly, electrophysiological studies with 1 indicate that signal bias may allow a path for an enhanced therapeutic window, even for PAMs with high efficacy (cooperativity/fold-shift).14 Although no neurotoxicity was observed for 1 in 14 day oral dose-limiting studies in rat at 450 mg/kg/day (273-440 μM-h), IND-enabling studies at 360 mg/kg/day for 30 days (253-416 μM-h) induced Fluoro-Jade C staining in a small number of rats, raising concerns about potential neurotoxicity sensitivity of an mGlu5 PAM in man.8,14
As one differentiator from the lead series we undertook the development of mGlu5 PAMs with a low cooperativity profile.13 However, in addition to this approach and based on our findings with 1 we wished to pursue back-up contenders specifically within other exocyclic amide subseries (4) that might have 1) cooperativity and signaling profiles comparable to 1, and 2) support a shorter predicted elimination half-life in man (e.g. T1/2 4-6 h versus 12-24 h). Compounds with such a profile may provide a mechanism to test the hypothesis that a short-acting mGlu5 PAM with a reduced in vivo residence-time and duration of action could avoid CNS toxicity while maintaining a favorable and sufficient efficacy profile. In this case, our screening paradigm consisted of traditional in vitro testing (e.g. calcium mobilization, glutamate fold-shift, mGlu selectivity assays), amphetamine-induced hyperlocomotion (AHL) challenge in rat at a single oral dose, and preclinical PK in rat and dog followed by signally pathway profiling in vitro and in more native systems for selected compounds. Previous studies in a highly efficient ketone series exemplified by PAM 215 and the Astra-Zeneca 2,3-dihydro-1H-pyrido[2,3-b][1,4]oxazine 316 provided the rational to initiate an amide translocation as illustrated by 4. Compounds within template 2 were found to have high metabolic turnover relative to 1 despite excellent ligand efficiency and physicochemical properties. In the case of 2 and related congeners this was shown to be primarily connected to the oxidative liability at the benzylic ether. Therefore, backup efforts within 4 appeared to be an attractive starting point to identify shorter acting compounds with the target pharmacological profile. Similar to studies focused within ketone series 2, the dihydropyrazolo[1,5-a]pyrimidine core was utilized to define a preferred translocated exocyclic amide with the target profile.
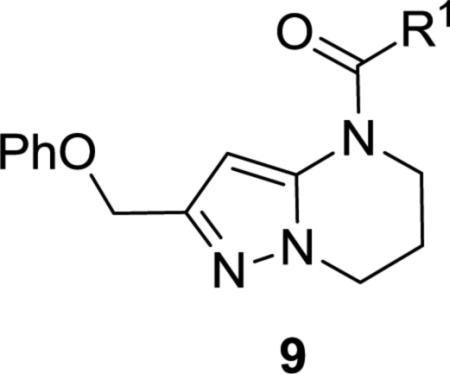
To this end we focused initially on synthesizing target 9 positioning the amide in an orientation similar to that represented by Astra-Zeneca template 3 (Scheme 1). Synthesis was envisioned to proceed in a strategy similar to that utilized in the preparation of 2, via incorporation of the western phenoxy and building the 5,6-ring system from left to right. Initial cross-Claisen condensation using ethyl 2-phenoxyacetate and acetonitrile afforded cyano ketone 5 in 24% yield. Treatment of 5 with hydrazine in MeOH generated aminopyrazole 6 in 82% yield. Using 6, a tandem Michael-lactam formation using methylacrylate in pyridine and water at elevated temperature proceeded in good yield to give key intermediate 7. Lactam 7 was reduced with borane dimethyl sulfide complex to give amine 8 and subsequently coupled with acids or acyl chlorides to afford targets 9a-r. Alternatively, treatment of lactam 7 with Boc2O, followed by Grignard addition (R2MgBr) and TFA treatment, furnished the desired 5-substituted 6,7-dihydropyrazolo[1,5-a]pyrimidine via elimination of the acyl hemiaminal. Reduction of the 6,7-dihydropyrimidine ring using Pd/C with atmospheric hydrogen followed by acylation afforded alpha 5-substituted targets 10a-b.
Scheme 1.

Reagents and conditions: (a) NaH, CH3CN, rt-55 °C, 4 h, 24%; (b) H2NNH2, EtOH, 85 °C, 16 h, 82%; (c) methylacrylate, pyridine, H2O, 135 °C, 48 h, 75%; (d) BH3-SMe2, THF, 0 – 60 °C, 16 h, 95%; (e) R1COCl, DCM, Et3N or R1CO2H, Ghosez reagent, DMF, DIPEA, 0 °C – rt, 25-70%; (f) Boc2O, THF, 76%; (g) R2MgBr, THF, 0 °C, 33-61%; (h) TFA, DCM, 0 °C, 64-71%; (i) Pd/C, H2, EtOAc, 82%; (j) R1COCl, DCM, Et3N, 0 °C – rt, 22-58%.
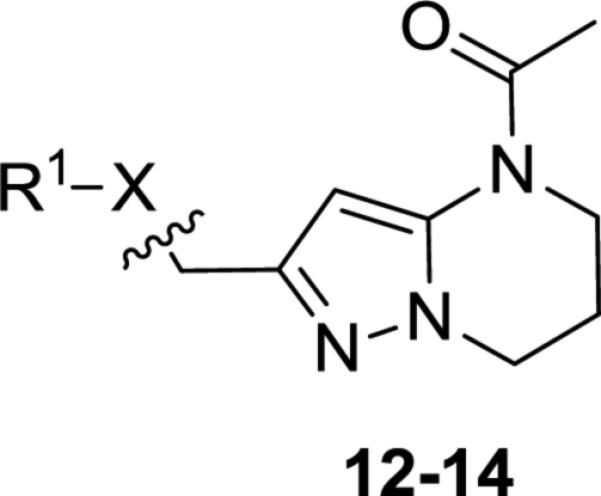
To access western phenoxy and various linker analogs in parallel, compound 9a was deprotected using BBr3 at low temperature to afford bromide 11 in moderate yield (55%, Scheme 2). Displacement of bromide 11 with alcohols, thiols, and amines proceeded smoothly to afford analogs (12-14). Sulfide 13a could be subsequently oxidized using one or two equivalents of MCPBA to afford sulfone 13b and sulfoxide 13c, respectively.
Scheme 2.

Reagents and conditions: (a) BBr3, DCM, 0°C, 3 h, 55% (b) R1OH, DMF, K2CO3, 140 °C μwave, 20 min., 33-84; (c) R1SH, CH3CN, 120 °C μwave, 20 min., 81%; (d) MCPBA 1 or 2 equiv, DCM, −20 °C – rt, 2 hr, 24-68, (e) R2R3NH, DMF, K2CO3, rt, 4-16h, 45-76%.
Gratifyingly, the translocated exocyclic amide concept 4 proved successful within the dihydropyrazolo pyrimidine core as acetylated analog 9a (Table 1), a close comparator to AZ-ether 3, was a PAM with an EC50 of 360 nM and good efficacy (65% Glu Max). Importantly, no allosteric agonism was detected in cell lines over expressing mGlu5 or in rat astrocytes, which represents a native tissue setting in which mGlu5 is endogenously expressed (vida infra Fig. 4). In addition, based on potency, MW, and physicochemical properties (cLogP = 2.1), 9a displayed excellent ligand efficiency metrics (LE = 0.44, LELP = 4.7) comparable to 2 and 3, thus making it an attractive starting point for further optimization. With exception of ethyl congener 9b (EC50 = 332 nM), higher alkyl homologs of 9a proved deleterious; with diminished potency tracking with increasing steric bulk near the amide (9c versus 9d). Saturated carbocycles revealed a similar trend (9e-9g); cyclopropyl was preferred (9e, EC50 = 559 nM) with potency comparable to methyl (9a) and ethyl (9b) congeners. Interestingly, phenyl acetate analog 9h produced a PAM with potency similar to lead 9a (EC50 = 378 nM) at the obvious cost of ligand efficiency. With respect to aryl moieties, SAR was flat (9i-9m) with no general improvements in activity. A low efficacy thiazoly amide 9r (EC50 = 257 nM, Glu Max = 24%) was identified; however, heterocyclic amides more typically displayed weak (isoxazolyl 9q, pyridyl 9n, 9o) or inactive (furan 9p) profiles. In contrast to SAR within the ketone series 2, introduction of alpha substituents within the 6,7-dihydropyrazolo[1,5-a]pyrimidine (Fig. 2) including 5-methyl (10a) and 5-phenyl (10b) were flat or lost activity versus 9a, possibly indicating disruption of a preferred amide conformation.
Table 1.
Structures and activities of analogs 9.
| ||||
|---|---|---|---|---|
| Entry | R1 | pEC50a | EC50 (nM)a | Glu Max %a |
| 9a |
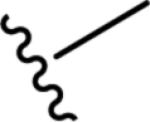
|
6.44±0.04 | 360 | 65±3 |
| 9b |
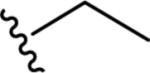
|
6.48±0.03 | 332 | 48±5 |
| 9c |

|
5.77±0.11 | 1,710 | 59±4 |
| 9d |
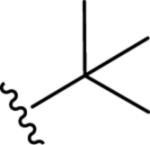
|
6.05±0.09 | 882 | 64±2 |
| 9e |
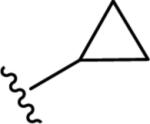
|
6.28±0.05 | 559 | 61±6 |
| 9f |
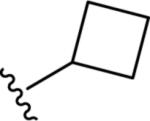
|
5.91±0.12 | 1,220 | 41±8 |
| 9g |
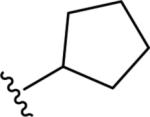
|
5.76±0.11 | 1,750 | 41±11 |
| 9h |
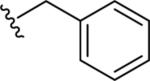
|
6.43±0.03 | 378 | 72±4 |
| 9i |
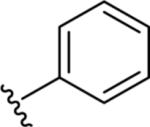
|
6.21±0.06 | 610 | 63±5 |
| 9j |

|
5.95±0.08 | 1,130 | 62±5 |
| 9k |
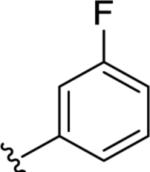
|
6.23±0.07 | 591 | 61±6 |
| 9l |
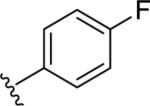
|
6.22±0.05 | 596 | 63±8 |
| 9m |
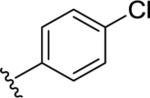
|
6.41±0.04 | 390 | 68±3 |
| 9n |
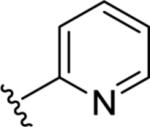
|
<5.00 | >10,000 | 44±11 |
| 9o |
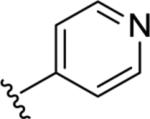
|
<5.00 | >10,000 | 51±9 |
| 9p |
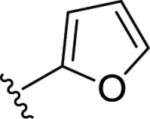
|
<4.5 | Inactive | <15 |
| 9q |
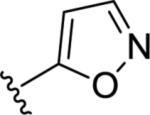
|
5.56±0.11 | 2780 | 22±7 |
| 9r |
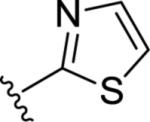
|
6.59±0.02 | 257 | 24±7 |
Calcium mobilization assay using HEK293 cells expressing human mGlu5; values are the mean±SEM of three or more independent determinations performed in triplicate.
Figure 4.

In vitro and in vivo profile summary of 9a.
Figure 2.
Structures and activities of 5-substituted analogs 10.
Utilizing 11 and the method described in Scheme 2 a thorough examination of western phenoxy SAR was undertaken and is shown in Table 2. Fluorine walk within series 12 revealed a strict preference for 3-fluoro congener (12b, EC50 = 266 nM) as 2-fluoro (12a) and 4-fluoro (12c) derivatives were weak or inactive. In addition, di-substitution as either difluoro (12d) or 3-cyano-5-fluoro (12e) afforded weak PAMs and pyridyl derivatives 12f-g were unproductive. Intermediate 11 was also utilized to revisit broader linker SAR and, in the case of thioether 13a, weak PAM activity was observed, while the corresponding sulfone (13b) and sulfoxide (13c) were inactive.
Table 2.
Structures and activities of linker analogs 12-14.
| Entry | R1X- | pEC50a | EC50 (nM)a | Glu Max %a |
|---|---|---|---|---|
| 12a |
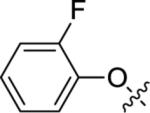
|
5.63±0.13 | 2,370 | 60±6 |
| 12b |
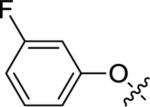
|
6.62±0.03 | 266 | 58±8 |
| 12c |
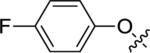
|
<5.00 | >10,000 | 56±4 |
| 12d |
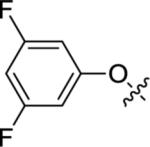
|
5.90±0.06 | 1,270 | 63±3 |
| 12e |
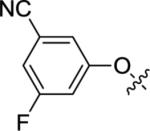
|
<5.00 | >10,000 | 39±9 |
| 12f |
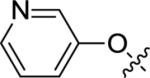
|
<4.5 | Inactive | <15 |
| 12g |
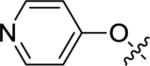
|
<4.5 | Inactive | <15 |
| 13a |
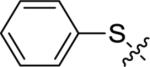
|
5.41±0.16 | 3,900 | 39±8 |
| 13b |
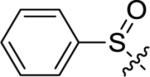
|
<4.5 | Inactive | <15 |
| 13c |
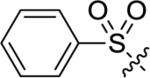
|
<4.5 | Inactive | <15 |
| 14a |
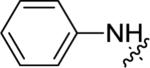
|
<5.00 | >10,000 | 20±6 |
| 14b |
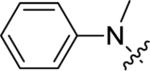
|
6.15±0.06 | 700 | 74±3 |
| 14c |
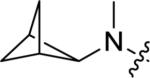
|
<4.5 | Inactive | <15 |
| 14d |
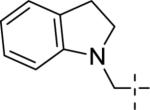
|
<4.5 | Inactive | <15 |
| 14e |
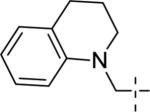
|
<5.00 | >10,000 | 48±12 |
| 14f |
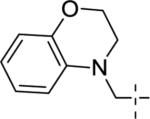
|
6.14±0.07 | 725 | 67±4 |
Calcium mobilization assay using HEK293 cells expressing human mGlu5; values are the mean±SEM of three or more independent determinations performed in triplicate.
A brief survey of amines proved more interesting as ether linkage replacements. Aniline 14a demonstrated weak PAM activity; however, the N-methyl aniline 14b displayed robust efficacy (Glu Max = 74%) and an EC50 of 700 nM. Attempts to replace the aniline moiety with a [1.1.1] propellane to afford 14c proved unsuccessful. A series of constrained anilines were then explored to expand 14b. Indoline 14d was inactive; however, tetrahydroquinoline (14e) displayed weak PAM activity. Based on the weak activity of 14e, benzoxazine 14f was prepared to further explore alternative six-member constraints. Gratifyingly, 14f demonstrated comparable potency and efficacy versus the  acyclic PAM 14b with an EC50 725 nM.
acyclic PAM 14b with an EC50 725 nM.
We next evaluated the in vitro DMPK profiles of selected analogs including 9a, 12b, 14b, and 14f (Table 3). In hepatic microsomal intrinsic clearance assays, both phenoxy PAMs 9a and 12b displayed moderate to high in vitro metabolism with predicted hepatic clearance (CLHEP) > 35 mL min−1 kg−1 in rat and > 12 mL min−1 kg−1 in human. Both amino congeners 14b and 14f displayed higher turnover in rat, with similar turnover in human near 50% maximum liver blood flow. Plasma protein binding experiments revealed high fraction unbound in both rat and human plasma for all four compounds (24-43% unbound in rat plasma, 17-46% unbound in human plasma). A rat PK study (0.2 mg/kg, intravenous cassette paradigm, male Sprague-Dawley, n = 2) using 9a and 12a confirmed high clearance in vivo with super hepatic clearance (rat CLp >70 mL min−1 kg−1). Follow up rat plasma stability studies indicated both 9a and 12a were stable (4 h incubation; 37 °C). Further stability studies in simulated gastric fluid for 1 h (SGF solubility 315 μg/mL, 37 °C) or in vehicle formulations (20% β-CD pH 6, 4 mg/mL) for up to five days also indicated the external amide remained intact with < 5% decomposition. In parallel, both 9a and 12a were evaluated in vivo in an amphetamine-induced hyperlocomotion (AHL) challenge model using an oral screening dose of 10 mg/kg.17 Interestingly as shown in Table 3, 9a demonstrated a robust 20% reversal of AHL while 12a was without effect relative to vehicle control (<10% reversal). Terminal mean brain and plasma levels (n = 6 animals, T90) were assessed from these studies to inform interpretation of the screening results. PAM 9a exhibited a terminal brain concentration of 2.25 μM and plasma concentration of 2.15 μM, corresponding to an estimated unbound brain concentration of 1.16 μM (based on a rat fu, brain = 0.51 from brain homogenate binding assay), while the corresponding 3-fluoro derivative 12a was not detected in brain samples. Thus, based on the performance of 9a in our pharmacodynamic screen and its apparent enhanced CNS penetration (calculated Kp,uu of 1.4),18 we opted to further profile 9a in additional pharmacological endpoints, discrete PK (rat and dog), and in full dose-response studies in rat AHL to determine its minimal effective dose (MED). The dose response profile for 9a in AHL with unbound terminal brain concentrations is shown in Figure 3 along with an extensive profile overview in Figure 4.
Table 3.
In vitro DMPK, rat IV pharmacokinetics, and rat amphetamine-induced locomotion % reversal from 10 mg/kg PO: 9a, 12b, 14c, and 14f.a
Figure 3.

Dose-dependent reversal effect and calculated terminal unbound brain levels of 9a in AHL in male, SD rats after oral administration (n = 8 animals/group).
As seen from Figure 3, 9a displayed a maximum reversal of 55% at a dose of 56.6 mg/kg and a 22% reversal at an MED of 3 mg/kg. At the 3 mg/kg dose, this corresponds to a terminal estimated unbound brain concentration of 199 nM, a three-fold multiple of the rat mGlu5 potency of 60 nM (Figure 4). To the best of our knowledge, only 9a and one other dihydroimidazopyrimidinone mGlu5 PAM from a lactam class19 have demonstrated an MED of 3 mg/kg (PO) in the AHL antipsychotic efficacy model; however, in the latter case, dihydroimidazopyrimidinone mGlu5 PAMs19 showed clear CNS-mediated side effects in dog and in rat modified Irwin test batteries thought to be attributed to high cooperativity and/or mixed mGlu5/mGlu3 activity. Evaluation of 9a in a rat modified Irwin study using a 30 mg/kg PO dose demonstrated no observable alterations in behavior across 30 distinct autonomic and somatomotor endpoints. In this study, 9a reached an impressive brain concentration of 71 μM 1.0 h post-dose (est. Cb,u = 36 μM). At a dose of 56.6 mg/kg, 113 μM of 9a was measured in the brain (est. Cb,u = 67 μM) and a modified Racine score of 1 noted over a 6.0 h observational period, with animals generally demonstrating decreased motor activity and coordination. Based on the concentrations achieved in the modified Irwin studies and in the AHL dose-response study at the MED, 9a appeared to provide a large safety window, in this case 100-fold or greater on the basis of these acute testing paradigms. Thus, PAM 9a represented a considerable improvement relative to candidate 1 (MED of 10 mg/kg)8 or related back-up candidates (MEDs of 30 mg/kg)9 on the basis of AHL and behavioral CNS toxicity.
Further pharmacological profiling of 9a in glutamate concentration response curve (CRC) fold-shift experiments with human or rat mGlu5 receptor expressing cell lines revealed modest maximal leftward fold-shifts (FS) of 5.0 and 4.2 respectively, using 10 μM of PAM (Fig. 4). PAM 9a was also highly selective versus other mGlu family members (mGlu1-4,6-8 FS < 1.2) and no noted activity was observed in a broad panel ancillary screen against 68 distinct GPCRs, ion channels and transporters (Eurofins Inc.). In addition, 9a had an excellent profile against the major CYP450 enzymes (>30 μM IC50s). Evaluation of 9a in discrete rat and dog pharmacokinetic studies confirmed the inherent moderate to high clearance of 9a in both species (Fig. 4); however, in rat the elimination T1/2 was 6.5 h, a profile consistent with this particular backup effort. After oral administration (10 mg/kg, 20% β-CD, 1 mg/mL) to male Sprague-Dawley rats, 9a reached an average maximal concentration (Cmax) of 2.5 μM with a corresponding time to reach Cmax (Tmax) of 1.0 h and an AUC0-8h of 3.31 μM-h, thus affording an absolute %F of 29. Unfortunately, in dog both IV and oral PK proved disappointing, with 9a exhibiting both poor exposure and low bioavailability and a short elimination T1/2 of less than 30 minutes.
Despite promising CNS penetration and acute efficacy in vivo in rat AHL, ongoing backup efforts progressing PAM 9a or related members of the series were considered challenging given the poor dog PK and steep SAR. Similar to prior studies in lead candidate series 1 and within the ketone series 2, in vitro metabolic soft-spot analysis revealed oxidative metabolism of the electron rich western phenoxy moiety, in addition to the C(4) pyrazole carbon, as major contributors to the observed metabolic instability.
In light of the limited success in addressing the metabolic soft-spot through modification of the western phenoxy ring both herein (e.g. Table 2) and in previous subseries,9,15 the medicinal chemistry campaign focused on attempting to mitigate metabolic turnover through modifications of the pyrazole core itself and through modification of the pyrazolo C(4) carbon. Efforts in this vein are briefly summarized in Figure 5. Despite considerable attempts to modify the nature of the heterocyclic core structure (Fig. 5, 15-19) or substitute the C(4) position of 9a with fluorine (20), these efforts generally resulted in compounds that suffered from diminished PAM activity or from metabolic and/or plasma instability.
Figure 5.

Isosteric replacements 15-19 and fluoro derivative 20.
In summary, we have discovered a set of potent mGlu5 PAMs with high ligand efficiency and excellent CNS penetration inspired by the exocyclic amide 3 and conceptually by the hybrid 5,6-bicyclic template 4. In an effort to identify PAMs with reduced half-lives and pharmacological target profiles most similar to candidate 1, PAM 9a (VU0462807) was identified as a selective and moderate fold-shift mGlu5 PAM having an exquisite efficacy profile in the acute AHL rat model. Despite our success in identifying a centrally penetrant compound with both improved in vivo potency and a reduced half-life, the overall metabolic instability and correspondingly poor oral F in higher species was deemed unacceptable for development and thus backup efforts in this area were suspended. Ongoing studies utilizing 9a for other indications and additional chronic studies to better understand PK/PD and duration of action are ongoing and will be communicated in due course.
Supplementary Material
Figure 1.

Structures, mGlu5 PAM activities, and ligand efficiency metrics of clinical candidate 1 (VU0409551/JNJ-46778212), Vanderbilt ketone 2, AZ exocyclic amide 3, and proposed translocated amide system 4.
Acknowledgments
Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) research was supported by grants from Janssen, The Pharmaceutical Companies of Johnson & Johnson and in part by the NIH (NS031373, MH062646).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information
Synthesis and experimental details for 9a and 20, along with routes and conditions used to prepare heterocycles 15-19 are available as supporting information.
References and Notes
- 1.Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. J Neurosc.i. 2000;20:7871. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chavez-Noriega LE, Schaffhauser H, Campbell UC. Curr Drug Targets CNS Neurol. Disord. 2002;1:261. doi: 10.2174/1568007023339337. [DOI] [PubMed] [Google Scholar]
- 3.Perroy J, Raynaud F, Homburger V, Rousset MC, Telley L, Bockaert J, Fagni LJ. Biol. Chem. 2008;283:6799. doi: 10.1074/jbc.M705661200. [DOI] [PubMed] [Google Scholar]
- 4.Conn PJ, Lindsley CW, Jones CK. Trends Pharmacol. Sci. 2009;30:25. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenbrock H, Kramer G, Hobson S, Koros E, Grundl M, Grauert M, Reymann KG, Schroder UH. Eur J Pharmacol. 2010;639:40. doi: 10.1016/j.ejphar.2010.02.057. [DOI] [PubMed] [Google Scholar]
- 6.Stauffer SR. ACS Chem. Neurosci. 2011;2:450. doi: 10.1021/cn2000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindsley CW, Stauffer SR. Pharmaceutical Patent Analyst. 2013;2:93. doi: 10.4155/ppa.12.82. [DOI] [PubMed] [Google Scholar]
- 8.Conde-Ceide S, Martinez-Viturro CM, Alcazar J, Garcia-Barrantes PM, Lavreysen H, Mackie C, Vinson PN, Rook JM, Bridges TM, Daniels JS, Megens A, Langlois X, Drinkenburg WH, Ahnaou A, Niswender CM, Jones CK, Macdonald GJ, Steckler T, Conn PJ, Stauffer SR, Bartolome-Nebreda JM, Lindsley CW. ACS Med. Chem. Letters. 2015;6:716. doi: 10.1021/acsmedchemlett.5b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y, Malosh C, Conde-Ceide S, Martinez-Viturro CM, Alcazar J, Lavreysen H, Mackie C, Bridges TM, Daniels JS, Niswender CM, Jones CK, Macdonald GJ, Steckler T, Conn PJ, Stauffer SR, Bartolome-Nebreda JM, Lindsley CW. Bioorg. Med. Chem. Lett. 2015;17:3515–3519. doi: 10.1016/j.bmcl.2015.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parmentier-Batteur S, Hutson PH, Menzel K, Uslaner JM, Mattson BA, O'Brien JA, Magliaro BC, Forest T, Stump CA, Tynebor RM, Anthony NJ, Tucker TJ, Zhang XF, Gomez R, Huszar SL, Lambeng N, Faure H, Le Poul E, Poli S, Rosahl TW, Rocher JP, Hargreaves R, Williams TM. Neuropharmacology. 2014;82:161–173. doi: 10.1016/j.neuropharm.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Rook JM, Noetzel MJ, Pouliot WA, Bridges TM, Vinson PN, Cho HP, Zhou Y, Gogliotti RD, Manka JT, Gregory KJ, Stauffer SR, Dudek FE, Xiang Z, Niswender CM, Daniels JS, Jones CK, Lindsley CW, Conn PJ. Biol. Psychiatry. 2013;73:501. doi: 10.1016/j.biopsych.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turlington M, Noetzel MJ, Chun A, Zhou Y, Gogliotti RD, Nguyen ED, Gregory KJ, Vinson PN, Rook JM, Gogi KK, Xiang Z, Bridges TM, Daniels JS, Jones C, Niswender CM, Meiler J, Conn PJ, Lindsley CW, Stauffer SR. J. Med. Chem. 2013;56:7976. doi: 10.1021/jm401028t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turlington M, Malosh C, Jacobs J, Manka JT, Noetzel MJ, Vinson PN, Jadhav S, Herman EJ, Lavreysen H, Mackie C, Bartolome-Nebreda JM, Conde-Ceide S, Martin-Martin ML, Tong HM, Lopez S, Macdonald GJ, Steckler T, Daniels JS, Weaver C, Niswender CM, Jones C, Conn JP, Lindsley CW, Stauffer SR. J. Med. Chem. 2014;57:5620. doi: 10.1021/jm500259z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rook JM, Xiang Z, Lv X, Ghoshal A, Dickerson JW, Bridges TM, Johnson KA, Foster DJ, Gregory KJ, Vinson PN, Thompson AD, Byun N, Collier RL, Bubser M, Nedelcovych MT, Gould RW, Stauffer SR, Daniels JS, Niswender CM, Lavreysen H, Mackie C, Conde-Ceide S, Alcazar J, Bartolome-Nebreda JM, Macdonald GJ, Talpos JC, Steckler T, Jones CK, Lindsley CW, Conn PJ. Neuron. 2015;86:1029. doi: 10.1016/j.neuron.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turlington M, Noetzel MJ, Bridges TM, Vinson PN, Steckler T, Lavreysen H, Mackie C, Bartolome-Nebreda JM, Conde-Ceide S, Tong HM, Macdonald GJ, Daniels JS, Jones CK, Niswender CM, Conn PJ, Lindsley CW, Stauffer SR. Bioorg. Med. Chem. Lett. 2014;24:3641. doi: 10.1016/j.bmcl.2014.04.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varnes JG, Marcus AP, Mauger RC, Throner SR, Hoesch V, King MM, Wang X, Sygowski LA, Spear N, Gadient R, Brown DG, Campbell JB. Bioorg. Med. Chem. Lett. 2011;21:1402. doi: 10.1016/j.bmcl.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 17.Kinney GG, O'Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, Smith S, Jacobson MA, Sur C, Duggan ME, Pettibone DJ, Conn PJ, Williams DL., Jr. J. Pharmacol. Exp. Ther. 2005;313:199. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 18.Di L, Rong H, Feng B. J. Med. Chem. 2013;56:2. doi: 10.1021/jm301297f. [DOI] [PubMed] [Google Scholar]
-
19.Martin-Martin ML, Bartolome-Nebreda JM, Conde-Ceide S, Alonso de Diego SA, Lopez S, Martinez-Viturro CM, Tong HM, Lavreysen H, Macdonald GJ, Steckler T, Mackie C, Bridges TM, Daniels JS, Niswender CM, Noetzel MJ, Jones CK, Conn PJ, Lindsley CW, Stauffer SR. Bioorg. Med. Chem. Lett. 2015;25:1310. doi: 10.1016/j.bmcl.2015.01.038.
- 20.Bridges TM, Rook JM, Noetzel MJ, Morrison RD, Zhou Y, Gogliotti RD, Vinson PN, Xiang Z, Jones CK, Niswender CM, Lindsley CW, Stauffer SR, Conn PJ, Daniels JS. Drug Metab. Dispos. 2013;41:1703. doi: 10.1124/dmd.113.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.