

Abstract
Aberrant DNA methylation is a hallmark of cancer but mechanisms contributing to the abnormality remain elusive. We have previously shown that ∆DNMT3B is the predominantly expressed form of DNMT3B. In this study, we found that most of the lung cancer cell lines tested predominantly expressed DNMT3B isoforms without exons 21, 22 or both 21 and 22 (a region corresponding to the enzymatic domain of DNMT3B) termed DNMT3B/∆DNMT3B-del. In normal bronchial epithelial cells, DNMT3B/ΔDNMT3B and DNMT3B/∆DNMT3B-del displayed equal levels of expression. In contrast, in patients with non-small cell lung cancer NSCLC), 111 (93%) of the 119 tumors predominantly expressed DNMT3B/ΔDNMT3B-del, including 47 (39%) tumors with no detectable DNMT3B/∆DNMT3B. Using a transgenic mouse model, we further demonstrated the biological impact of ∆DNMT3B4-del, the ∆DNMT3B-del isoform most abundantly expressed in NSCLC, in global DNA methylation patterns and lung tumorigenesis. Expression of ∆DNMT3B4-del in the mouse lungs resulted in an increased global DNA hypomethylation, focal DNA hypermethylation, epithelial hyperplastia and tumor formation when challenged with a tobacco carcinogen. Our results demonstrate ∆DNMT3B4-del as a critical factor in developing aberrant DNA methylation patterns during lung tumorigenesis and suggest that ∆DNMT3B4-del may be a target for lung cancer prevention.
Keywords: DNMT3B, ΔDNMT3B, ΔDNMT3B-del, DNA methylation, Lung cancer, Mouse model, Tumorigenesis
Highlights
-
•
DNMT3B/∆DNMT3B-del is the predominantly expressed isoform in a large number of lung cancers.
-
•
∆ DNMT3B4-del can cause aberrant DNA methylation patterns similar to tumorigenesis.
-
•
∆ DNMT3B4-del facilitates carcinogen-induced lung tumorigenesis in a mouse model.
Lung cancer is the leading cause of cancer-related deaths in the United States. Epigenetic alterations, particularly alterations in DNA methylation patterns, play critical roles in lung tumorigenesis. We show that DNMT3B/ΔDNMT3B-del is predominantly expressed in a significant percentage of lung cancers including both cell lines and primary tumors. We demonstrate that ΔDNMT3B4-del is critical in the formation of aberrant DNA methylation patterns in mouse lungs similar to human lung cancers and contributes to neoplasia formation when exposed to carcinogens, supporting ΔDNMT3B4-del as a novel target for lung cancer prevention.
1. Introduction
DNA methylation is a major form of inheritable epigenetic mechanism that regulates the genome. It is important in the dosage compensation of sex chromosome, the repression of retrotransposons, the maintenance of genome stability, and the coordinated expression of imprinted genes during development and cellular differentiation (Edwards et al., 2010, Hansen et al., 2011, Jones, 2002, Robertson, 2001). These epigenetic modifications direct the establishment of cell lineage through maintenance of inheritable gene expression profile and genomic stability (Messerschmidt et al., 2014). Most sites of DNA methylation occur at cytosine residuals of CpG dinucleotides. In normal adult tissues, the CpG sites at the repetitive regions of the genome are heavily methylated, while most of the CpG islands located at or near the promoter regions are unmethylated. DNA methylation is catalyzed by various DNA methyltransferases. DNMT1 is the enzyme critical for maintaining DNA methylation patterns during DNA replication whereas DNMT3A and DNMT3B can methylate DNA in de novo manner to develop new methylation patterns in the genome (Bestor, 2000, Li et al., 1992, Okano et al., 1999). While DNA demethylation may occur in either a passive or active manner (Chen et al., 2003, Tahiliani et al., 2009), mechanisms of these actions remain largely unclear.
In tumorigenesis, the landscape of DNA methylation patterns changes dramatically, with the general observation of reduced global DNA methylation and an increase of DNA methylation at certain promoter CpG islands (Jones, 2012). While extensive efforts have been devoted to study promoter DNA hypermethylation as a common epigenetic alteration in cancers (Jones and Baylin, 2007, Suzuki et al., 2002), global DNA hypomethylation is recognized as an epigenetic abnormality in human tumorigenesis as well (Feinberg and Vogelstein, 1987). It is believed that DNMTs, particularly DNMT3B, are involved in the promoter hypermethylation in tumorigenesis as DNMT3B is often overexpressed in cancer tissues (Van Emburgh and Robertson, 2011). However, mechanisms causing the changes in the DNA methylation landscape during tumorigenesis due to variant expression of DNMT3B remain unresolved.
DNMT3B has two subfamilies, DNMT3B and ΔDNMT3B. DNMT3B has a number of transcription variants resulting from alternative pre-mRNA splicing, including at least 7 DNMT3Bs and 7 ΔDNMT3Bs (Robertson and Jones, 1999, Wang et al., 2006b), but the actual number of DNMT3B variants involved in methylation regulation are likely greater (Ostler et al., 2007). Interestingly, proteins predicted to be encoded from some of the variants (DNMT3B3–3B7 and ΔDNMT3B-del) lack the conserved enzymatic motifs or possess different peptide sequences due to frame shifts following the alternative splicing (Robertson and Jones, 1999, Wang et al., 2006a, Wang et al., 2006b). Certain functional consequences of DNMT3Bs without the enzymatic domain have been reported in an isoform or cell context specific manner. For example, overexpression of DNMT3b4 is a frequent event in early hepatocarcinogenesis and has been linked to an increased satellite DNA hypomethylation (Saito et al., 2002). Forced expression of DNMT3B7 resulted in an attenuated global DNA methylation pattern and an increased incidence of mediastinal lymphomas in a Myc overexpressing transgenic mouse model (Shah et al., 2010). Paradoxically, when DNMT3B7 was overexpressed in a neuroblastoma model, it increased global DNA methylation and induced cell differentiation resulting in a tumor growth inhibition (Ostler et al., 2012). No significant phenotype has, however, been reported when DNMT3B3 and DNMT3B6 were overexpressed in transgenic mouse models (Linhart et al., 2007).
In previous studies, we showed that the predominant forms of DNMT3B transcripts are from ΔDNMT3B in both non-small cell lung cancer (NSCLC) cell lines and primary NSCLC tissues (Wang et al., 2006a, Wang et al., 2006b). However, we only analyzed the 5 prime end of the transcripts in these studies and therefore could not distinguish whether these transcripts include sequences (exons 21 and 22) required for the predicted enzymatic domain. Here we report that ΔDNMT3B transcripts detected in NSCLC are predominantly those lacking of exons 21, 22 or both 21 and 22 termed ΔDNMT3B-del in both NSCLC cell lines and primary NSCLC tumors. In a transgenic mouse model with lung specific expression of ΔDNMT3B4-del, we demonstrated a role of the DNMT3B isoform in shaping DNA methylation landscape and early lung tumorigenesis.
2. Materials & Methods
2.1. Cell Lines and Primary Tissues
Human NSCLC lines H157, H226, H292, H358, H460, H522, H596, H1299, H1792, H1944, Calu-1, SK-MES, and A549, were purchased from the American Type Cell Culture (Rockville, MD). Cells were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM l-glutamine, 100 IU/ml penicillin, and 100 mg/ml streptomycin at 37 °C in the presence of 5% CO2. The normal human bronchial epithelial cell lines HBE1, HBE2, HBE3, HBE4 and HBE5 (kindly provided by Dr. John Minna of The University of Texas Southwestern Medical Center, Dallas, TX) were maintained in Keratinocyte-SFM basal medium (GIBCO) supplemented fresh at the time of use with 0.1–0.2 ng/ml EGF human recombinant and 20–30 μg/ml bovine pituitary extract. The cells were cultured and treated at 37 °C in a humidified incubator containing 5% CO2 and maintained free of mycoplasma as determined by Hoechst staining.
2.2. RNA Extraction and RT-PCR
Total RNA for each cell line and clinical sample was isolated using Tri-Reagent (Molecular Research Center, Inc., Cincinnati, OH) according to the manufacturer's instructions. Approximately 1 μg of total RNA from each sample was used to conduct reverse transcription reaction in 20 μl volume using Superscript II RNase H-Reverse Transcriptase (Invitrogen Life Technologies, Carlsbad, CA). The synthesized DNA was either used immediately for PCR amplification or was stored at − 20 °C for further analysis.
PCR reaction was performed in a 12.5 μl volume containing 0.5 μl RT products, 7% DMSO, 1.5 mM dNTPS, 6.7 mM MgCl2, 16.6 mM (NH4)2SO4, 67 mM tris, 10 mM β-mercaptoethanol, 6.7 μM EDTA, 0.5 μM of both the forward and the reverse primer, and 0.625 units of Hotstar Taq DNA polymerase (Qiagen, Inc., Chatsworth, CA). Amplification was carried out with the initial denaturing step at 95 °C for 15 min, followed by 35 cycles of 95 °C for 30 s, primers specific temperature for 30 s, and 72 °C for 1 min in thermal cycler (Hybaid; PCR Express, Middlesex, UK) with a last extension step of 72 °C for 7 min. The PCR products were mixed with 6 × loading buffer containing 0.5-mg/ml of ethidium bromide and separated by electrophoresis on a 2% agarose gel. Oligonucleotide primer set used to amplify mRNA fragment franking exons 19–23 of DNMT3B with reverse primer 5′- AAG AGG TGT CGG ATG ACA GG- 3′ locates in exon 23 and forward primer 5′- ATC TCA CGG TTC CTG GAG TG- 3′ locates in exon 19. An alignment of DNMT3B isoforms surrounding the region is provided in Supplementary Material I with the primers highlighted with green color.
2.3. Transgenic Mice Generation
EcoRI and NheI (end blunted) fragment from pcDNA6 V5His-ΔDNMT3Bdel expression vector was inserted into the SP-C 3.7-SV40 vector. A Not I and ClaI (end blunted) fragment of IRES-EGFP from pMIG vector was engineered into EcoRI site of intermediate SP-C/ΔDNMT3Bdel to generate ΔDNMT3Bdel transgenic constructs. Transgene construct was verified by sequencing analysis. For microinjection ΔDNMT3B-del transgene was released from cloning vector by Ndel/DrdI digestion and agarose gel purified. Transgenic lines were engineered in C57Bl/6 hybrid strain by the Transgenic/Knockout Core Facility at the University of Maryland School of Medicine. Transgenic integration was confirmed by using a PCR assay of genomic DNA from tails of individual mice. In NNK challenge experiment, mice were injected intraperitoneally with NNK (Toronto Research Chemicals, On, Canada) at 150 mg/kg beginning at 8 weeks of age with one injection per week for two weeks. 14 month after the first injection, mice were euthanized by CO2, lungs inflated with 10% neutral-buffered formalin, and tissue fixed in the same solution for histology examination.
2.4. Lentivirus Production and Cell Transduction
DNA fragment of DNMT3B-del-IRES-EGFP from transgenic construct was cloned into MCS of pLVX-Tight-Puro vector of Lenti-X™ Tet-On® Advanced inducible expression system (Clotech, Mountain View, CA). Tet-on Virus packaging was performed by transient transfection of 293FT cells with a transfer plasmid and packaging vectors as described in the manufacturer's user manual. Seventy-two hours after transfection, the lentiviral particles were collected and filtered, and then concentrated using Amicon ultra-15 (Merck Millipore Ltd., Darmstadt, Germany) by centrifugation at 3000 g for 1 h at 4 °C. Final concentrated viral particles were aliquot at desired volume into freezer tubes and stored at − 80 °C.
2.5. Flow Cytometry Analysis
HBE4 cells were seeded in a 6-well plate and cultured for 24 h before virus transduction. The transduction with ΔDNMT3B-del viruses was carried out with 6 μg hexadimethrine bromide (Polybrene) for 8 h. The infected cells were then cultured under normal culture medium with or without 1 μg/ml doxycycline (Dox, a tetracycline derivative) to induce protein expression for 48 h. Cells were harvested, washed in PBS, and fixed in 70% ethanol for 24 h at 4 °C. Cells were then washed twice with PBS, incubated with 500 μl PI/RNase staining buffer (BD Pharmingen, San Jose, CA, USA) for 45 min, and passed through the meshed blue capped falcon tubes (BD Biosciences, San Jose, CA). Cell cycle was analyzed on a FACSCalibur flow cytometer (Becton-Dickinson, UK). Ten thousand events were acquired per sample and data were analyzed using CellQuest software (Becton-Dickinson, UK).
2.6. Expression Analysis of Transgenic Mouse Lines by Real-Time PCR
Allprep RNA/DNA/Protein Mini kit (Qiagen, Inc.) was used to prepare total RNA from cell culture. Total RNA was treated with DNase according to the manufacturer's instructions (Qiagen). One microgram of total RNA were used to converted cDNA by using the SuperScript II reverse transcriptase kit (Invitrogen, Grand island, NY, USA). Gene expression of exogenous hΔDNMT3B-del and endogenous mDNMT3B were determined by TaqMan gene expression assay with a 7900HT Real-Time PCR System detector (Applied Biosystems). Primer set for hΔDNMT3B-del is: Forward primer: 5′-TGACCGGCCGTTCTTCTG-3′; Reverse primer: 5′-CCGTGAGATGTCCCTCTTGTC-3′; FAM probe: 5′ 5′-TGTTTGAGAATGTTGTAGCCAT-3′. Other TaqMan gene expression assays used included mouse Dnmt3B (Mm01240113); nkx2.1 (Mm00447558); Gata6 (Mm00802636); Gapdh (Mm99999915) (Applied Biosystems, Foster City, CA). The specific primers and probe designed by Applied Biosystems were used for amplification of target genes and detection. Mouse GAPDH was used as an internal control for normalization of gene expression.
2.7. Western Blot Analysis
Proteins from transgenic mice lung tissues or whole cell extractions were quantified using the BCA assay (Thermo Scientific, Rockford, IL). A total of 50 μg protein extracts were subjected to electrophoresis on a NuPAGE 10% Bis-Tris gel (Invitrogen) and transferred to a polyvinylidene difluoride (PVD) membrane by iBlot gel transfer system (Invitrogen, Grand island, NY). The membranes were blocked for 1 h with 5% non-fat dry milk in TBST (10 mM Tris–HCl, pH 7.4, 100 mM NaCl, 0.1% (V/V) Tween −-20), and successively incubated with primary antibodies overnight at 4 °C. Anti-EGFP (Sigma, St. Louis, MO) and anti-DNMT3B (Imgenex, San Diego, CA, USA) antibodies were used to detect DNMT3B expression in transgenic mice lung tissue, while β-actin was used as an internal control. DNA chromosome aneuploidy related proteins were probed using the following antibodies: anti-pH2Ax, anti-53BP and anti-pCHK2 purchased from Cell Signaling technology (Danvers MA), and Anti-Mad2L2 obtained from Abcam (Cambridge MA). The reaction was followed by probing with peroxidase-coupled secondary antibodies including anti-mouse IgG or anti-Rabbit IgG at dilution 1:5000 (Santa Cruz Biotechnology, Dallas, TX, USA), binding results were visualized using enhanced chemiluminescence (GE Healthcare).
2.8. Immunohistochemistry
5 μm thick formalin-fixed, paraffin embedded tissue sections from transgenic mice lung were used for immunohistochemistry staining. Briefly, sections were deparaffinized in a series of xylene baths and then rehydrated using a graded alcohol series. The sections were then microwaved for 20 min in 10 mmol/L citrate buffer (pH 6.0) for antigen retrieval. The slides were blocked against endogenous peroxidase activity using 3% hydrogen peroxide solution for 10 min and then with normal blocking buffer (Vectastain Kit) for 30 min, sections were incubated overnight at 4 °C with primary antibodies (anti-53BP1, anti-pH2AX and anti-pCHK2, 1:200 dilution). After washing with PBS (pH 7.4), the sections were then processed using standard avidin–biotin immunochemical techniques (ABC) according to the manufacturer's instruction (Vector Laboratories). Staining was visualized with 3,3-diaminobenzidine tetrahydrochloride (Vector Laboratories) as a chromogen. Hematoxylin was used for counterstaining.
2.9. DNA Extraction and MBD-Seq Library Preparation
Three animals per group – wild type and transgenic – were randomly selected. The Wizard Genomic DNA purification kit (Promega, A1120) was employed to extract the genomic DNA from the previous selected individuals. The concentration of the DNA was determined by the Qubit dsDNA Broad-Range Assay (Invitrogen, Q32850). The methyl binding domain protein-enriched genome sequencing (MBD-Seq) approach was used to identify methylated DNA regions. MethylCap kit (Diagenode, C02020010) was employed for the acquisition of methylated DNA. Firstly, the extracted DNA was dissolved to a concentration of 0.1 μg/μl for a final volume of 40 μl in a 1.5 ml tube. Then, the DNA was fragmented into 300–500 base pairs segments in a Bioruptor Sonicator. The resultant DNA fragments were filtered in an agarose gel to visualize and confirm their length. Secondly, 141.8 μl of capture reaction mix containing 12 μl of sheared DNA and 129.8 μl of buffer B was prepared. From this preparation, 119 μl was incubated with 1 μl of diluted MethylCap protein on a rotating wheel at 40 rpm for 2 h and at 4 °C to let the reaction occurs. The remaining (22.8 μl) was conserved to use later as input sample. GSH-coated magnetic beads captured the methylated DNA. The free DNA was washed off and elutes collected. Three serially increased concentrations of elution buffer (150 μl of low, medium and high) were used for the elution of samples. Purification of all fractions and inputs was achieved by employing the MiniElute PCR Purification Kit (QIAGEN, 28006). Lastly, to ascertain the enrichment efficiency, qPCR (iCycler iQ PCR system, Bio-Rad) was performed in duplicates for each sample. For relative fold enrichment calculation, the applied 2-∆∆Ct method compared enrichment values of a positive to a negative primer pair, between experimental (methyl DNA) and reference (input DNA) samples.
The library for sequencing was assembled following the next steps. The end repair was achieved using the NEBNext® End Repair Module (NEB, E6050S). Then, the DNA Polymerase I, Large (Klenow) Fragment (NEB, M0210L) incorporated a 3′ A into the DNA segments. Additionally, a pair of Solexa adaptors (Illumina) was added to the repaired ends (T4 ligase, Promega, M1801). The target fragments – 200 to 500 bp including adaptors – were selected through filtration in a 2% agarose gel. PCR, using Phusion® Hot Start High-Fidelity DNA Polymerase (NEB, M0530S), amplified purified DNA templates. Next, DNA quality was checked. The DNA library was diluted and the concentration verified using the Qubit assay (Life Technology, Q32850). Finally, the sequencing was performed in a Solexa 1G Genome Analyzer (Illumina) according to the manufacturer protocol.
2.10. MBD-Seq Data Analysis
Sequence FASTQ files were checked for quality assurance. After confirming a satisfactory quality, alignment of the files to the mm10 reference genome, obtained from the UCSC browser (http://genome.ucsc.edu), was performed employing Bowtie, an ultrafast, memory-efficient short read aligner software. Initial sequence reads were 50 base pair long however, the first twelve 5′ and three 3′ reads were trimmed to conserved high sequence quality. Different procedures included in SAMtools and BEDtools were implemented for data manipulation, filtering and format transformation. The abundance of reads in a determined genomic region represents its correspondent methylation level. Accordingly, the peak-calling step was implemented independently for each sample using Model Based Analysis of ChIP-Seq (MACS). The program models empirically the shift of the tags and based on a dynamic Poisson distribution estimates the local bias, yielding more accurate predictions. The implementation of the DiffBind R package permitted the identification of differentially methylated regions (DMRs) between transgenic and wild type mice (Ross-Innes et al., 2012). The software identifies distinctly bound sites based on affinity data. The input for DiffBind consisted of two files per sample; the output from MACS containing the called-peaks and the bam files constituted by the aligned reads. DiffBind builds a matrix with the consensus peaks, which have been obtained with a “minimum overlap” of 3, considering the replicates number for each condition. Next, and after the corresponding contrast is defined, DiffBinf performs an empirical Bayes method, the edgeR analysis. For this case, the default method trimmed mean of M-values (TMM) was used for normalization. This approach subtracts the control reads and accounts for the effective library size as well. The threshold was set to 0.1 for the false discovery rate (FDR).
2.11. DMRs Annotation
The ChIPpeakAnno software was employed for the annotation of the regions with distinct methylation levels (Zhu et al., 2010). The package reports overlaps, distance and relative position for the inquired entities. The dataset “mmusculus_gene_ensembl” was used for annotation. It was acquired from biomart and corresponds to the genome GRCm38 (mm10), the same one used for the alignment. Information regarding CpG islands was retrieved from UCSC and later used as feature input in the ChIPpeakAnno package.
3. Results
3.1. Predominant Expression of DNMT3B/ΔDNMT3B-del in NSCLC Cell Lines
DNMT3B may have various expression forms and Fig. 1A illustrates the possible transcription variants of DNMT3B4 as an example. We used primers flanking exons 19 to 23 of DNMT3B and ΔDNMT3B to determine the expression patterns of the DNMT3B and ΔDNMT3B region corresponding to the enzymatic domain in 4 immortalized normal bronchial epithelial (HBE) cell lines and 12 NSCLC cell lines. To our surprise, 9 (75%) of the 12 NSCLC cell lines expressed only or predominantly DNMT3B and ΔDNMT3B forms lacking exons 21, 22 or both 21 and 22 whereas the HBE cells expressed equal quantity of DNMT3B and ΔDNMT3B transcripts with and without the exons 21–22 (Fig. 1B). All of the RT-PCR products were verified by direct sequencing of the excised bands. The sequences obtained are consistent with the predicted DNMT3B and ΔDNMT3B splicing variants. We termed the transcripts lacking exons 21, 22 or both 21 and 22 as DNMT3B/ΔDNMT3B-del.
Fig. 1.

Expression of DNMT3B variants. (A) Illustration of major DNMT3B isoforms (3B1–3B5) with the excluded exon(s) with brown numbers. (B) “Δ” is for DNMT3B subfamily with an alternative transcription initiation site and lack of N-terminal 200 amino acids and “del” represents isoforms excluding exons 21, 22 or both 21 and 22 (shaded boxes). (C) Expression of DNMT3B/ΔDNMT3B-del in lung cancer cell lines by RT-PCR. (D) DNMT3B/ΔDNMT3B-del in bronchial brushing samples from smokers without lung cancer. C1 represents control cells expressing both DNMT3B/ΔDNMT3B and DNMT3B/ΔDNMT3B-del. C2 represents control cells expressing only DNMT3B/ΔDNMT3B-del. Expression patterns of additional samples are shown in Supplementary Fig. 1. (E) Expression of DNMT3B/ΔDNMT3B-del in surgically resected NSCLC tissues. *, represent tumors with either equal expression levels of DNMT3B/ΔDNMT3B and DNMT3B/ΔDNMT3B-del or with greater of DNMT3B/ΔDNMT3B. Expression patterns of additional bronchial brushing and primary NSCLC tissues are shown in Supplementary Fig. 1.
The transcripts lacking one or two of exons 21 and 22 would encode truncated proteins lacking motifs VII and VIII, the TRD (target recognition domain), or a part of motif IX (Robertson et al., 1999), thus eliminating the DNA methyltransferase activity. The results, therefore, indicate that the majority of NSCLC cell lines have either no or low levels of DNMT3B enzymatic activity and suggest that the enzymatic activity of ΔDNMT3B or DNMT3B is not required for creating the altered DNA methylation landscapes in these NSCLC cells. Because ΔDNMT3B is the major transcripts of DNMT3B in NSCLC (Wang et al., 2006b), it was previously assumed that ΔDNMT3B-del is involved in the formation of aberrant DNA methylation patterns observed during lung tumorigenesis.
To rule out the possibility that the observed DNMT3B/ΔDNMT3B-del expression patterns were a cell culture artifact, we analyzed bronchial epithelial cells obtained from 30 healthy heavy smokers during bronchoscopy procedure. Twenty-eight (93%) of the 30 samples had adequate quality of RNA allowing accurate RT-PCR analysis and data interpretation. All the 28 samples exhibited expression patterns consistent with either equal quantity of DNMT3B/ΔDNMT3B and DNMT3B/ΔDNMT3B-del or a greater quantity of DNMT3B/ΔDNMT3B (Fig. 1C and Supplementary Fig. 1A), which is the similar pattern as observed in the HBE cells. We then analyzed 119 primary NSCLC tumors from patients with surgically resected NSCLC for DNMT3B/ΔDNMT3B expression patterns using the same primer set used for cell lines and the primary bronchial epithelial cells. We observed that 111 (93%) of the 119 tumors expressed predominantly DNMT3B/ΔDNMT3B-del including 47 (39%) tumors with no detectable DNMT3B/ΔDNMT3B (Fig. 1D and Supplementary Fig. 1B), which is consistent with the results observed in the NSCLC cell lines. Together, these results indicate that the loss of DNMT3B/ΔDNMT3B expression or overexpression of the enzymatic domain lacking DNMT3B/ΔDNMT3B-del is a common abnormality not only in cultured NSCLC cells but also in primary NSCLC tumors.
Supplementary Fig. 1.

(A) Complete list of DNMT3B/ΔDNMT3B-del expression patterns in all bronchial brushing samples from chronic smokers without lung cancer. (B) Complete list of DNMT3B/ΔDNMT3B-del expression patterns in all primary NSCLC tissues. C1 is control sample with equal amount of DNMT3B/ΔDNMT3B and DNMT3B/ΔDNMT3B-del expression. C2 is control samples with only DNMT3B/ΔDNMT3B-del expression.
To determine if DNMT3B/ΔDNMT3B-del transcripts can be translated into proteins, we attempted to measure the corresponding protein products using Western blot analysis. Unfortunately, all the commercially available antibodies against DNMT3B we tested as well as antibodies generated in our own laboratory, while effective in detecting DNMT3B expressed and purified in bacteria, were found to be non-specific to DNMT3B when used for total cellular protein extracts. We therefore tested plasmids containing either ΔDNMT3B or ΔDNMT3B-del isoforms by transfecting H1299 cells with these plasmids and analyzed the expression levels of the corresponding recombinant proteins using an anti-tag antibody. We detected both ΔDNMT3B and the shortened ΔDNMT3B-del isoforms by Western blot analysis using an anti-V5-tag antibody (Supplementary Fig. 2A), indicating that the transcripts of these DNMT3B isoforms can be translated into proteins in cells. We further transfected HEK293 cells with ΔDNMT3B4-del-GFP fusion protein construct and found ΔDNMT3B4-del was predominantly located in nucleus (Supplementary Fig. 2B).
Supplementary Figure 2.

Expression of ΔDNMT3B isoforms and the cellular localization of ΔDNMT3B4. (A), Western blot analysis using antibody against V5 tag in cells transfected with various ΔDNMT3B plasmid constructs with V5 tag sequence. Cells transfected with pcDNA6/V5–His-ΔDNMT3B4 (Lanes 1 and 2); lipofectamine only (Lane 3); pcDNA6/V5–His control vector (Lane 4); pcDNA6/V5–His-ΔDNMT3B4-del (Lane 5); pcDNA6/V5–His-ΔDNMT3B3-del (Lane 6); pcDNA6/V5–His-ΔDNMT3B2-del (Lane 7) and pcDNA6/V5–His-ΔDNMT3B1-del (Lane 8). (B), HEK293 cells were transfected with ΔDNMT3B4-del-GFP fusion protein construct. The expression and cellular location of ΔDNMT3B4-del was detected under fluorescence microscope.
3.2. Transgenic Mice with Lung-Specific Expression of ΔDNMT3B4-del
To determine the causal relationship between ΔDNMT3B-del and the landscape changes in global DNA methylation patterns in lung tumorigenesis, we constructed ΔDNMT3B4-del transgenes (Δ3B4DG) under the control of surfactant protein-C promoter (SPC) aiming to overexpress ΔDNMT3B4-del specifically in mouse lung epithelial and alveolar cells which express endogenous SPC (Fig. 2A). The reason of selecting ΔDNMT3B4-del was based on our earlier observation that ΔDNMT3B4 is the DNMT3B isoform expressed at the highest level among the NSCLC cell lines and primary NSCLC tissues examined (Wang et al., 2007). Microinjection of the transgene into pro-nuclei of fertilized eggs gave rise to total 30 live pups, 6 of them carried SPC/Δ3B4DG transgene with 4 of them achieved germ-line transmission. These founder lines were named as: Tg(SPC-DNMTΔ3B/DG)1lm, Tg(SPC-DNMTΔ3B/DG)3lm, Tg(SPC-DNMTΔ3B/DG)25lm and Tg(SPC-DNMTΔ3B/DG)27lm according to MGI-Guidelines for Nomenclature of Gene, Genetic Markers, Alleles, and Mutations in Mouse and Rat: http://www.informatics.jax.org/mgihome/nomen/gene.shtml.
Fig. 2.

Generation of transgenic mouse lines overexpressing ΔDNMT3B4-del in lungs. (A) Structure of the ΔDNMT3B4-del transgenic construct. Major components of the transgene and the primer sets used for genotyping. (B) Results of real time RT-PCR determining expression of the exogenous human ΔDNMT3B4-del and the endogenous mouse Dnmt3B in lungs of the transgenic mouse lines. Relative expression levels were normalized with endogenous control GAPDH.
To verify expression of the transgene, we analyzed F1 litters of these transgenic founder lines using real-time RT-PCR. The ΔDNMT3B4-del transgene was found expressed at variable levels in the mouse lung tissues of Tg(SPC-DNMTΔ3B/DG)1lm, Tg(SPC-DNMTΔ3B/DG)3lm and Tg(SPC-DNMTΔ3B/DG)27lm litters but not in Tg(SPC-DNMTΔ3B/DG)25lm mice (Fig. 2B). Tg(SPC-DNMTΔ3B/DG)25lm mice were excluded for further analysis. The lack of the transgene expression in Tg(SPC-DNMTΔ3B/DG)25lm mice was likely due to a positional effect of the transgene insertion. For more accurate interpretation of the data, we measured the endogenous levels of mouse Dnmt3b expression in the lungs using real-time RT-PCR. As expected, endogenous Dnmt3b expression levels were comparable among different transgenic mouse lines but lower compared to the expression levels of the transgene, ranging from less than 10% for Tg(SPC-DNMTΔ3B/DG)3lm mice to about 50% for Tg(SPC-DNMTΔ3B/DG)27lm mice (Fig. 2B). Thus, we generated transgenic mice overexpressing ΔDNMT3B4-del in the mouse lung tissues.
3.3. Phenotypes in ΔDNMT3B-del Overexpressing Mice
Grossly, we found no apparent developmental defects in the transgenic animals. The animals lived normally and were able to breed. To determine the impact of ΔDNMT3B4-del in lungs of the animals, we examined the lungs from 43 Tg(SPC-DNMTΔ3B/DG) mice with age ranging from 12 to 16 months and 20 age matched wild-type mice. No gross abnormalities in the lungs were observed comparing the transgenic mice with wild-type mice during necropsy. We then examined the lung tissues of each mouse in a series of sections of paraffin-embedded formalin-fixed lung tissues. Under microscope magnification, we observed several morphological changes including enlarged lung alveoli in the distal bronchi, increased bronchial epithelium hyperplasia (Fig. 3A up right panel), enhanced parenchymal inflammatory aggregates (Fig. 3A low left panel) and tumor-like structures (Fig. 3A low right panel) compared to the lungs from wild-type mice (Fig. 3A up left panel).
Fig. 3.

Microscopic changes in lungs in the transgenic mice overexpressing ΔDNMT3B4-del. (A) Three major morphological changes were observed: bronchial epithelial hyperplasia, parenchymal inflammatory cell aggregates and tumor-like structure. WT: wild-type mouse; TG: transgenic mouse. (B) Accumulative incidence of microscopic alterations observed in lungs of the transgenic animals. (C) pH 3 staining in lungs of wild-type mouse (WT) and transgenic mouse (TG).
Among the 43 Tg(SPC-DNMTΔ3B/DG) mice, 17 were from Tg(SPC-DNMTΔ3B/DG)1lm, 13 from Tg(SPC-DNMTΔ3B/DG)3lm and 13 from Tg(SPC-DNMTΔ3B/DG)27lm. We observed bronchial epithelial hyperplasia in the lungs from 25 (58%) of the 43 mice compared to 1 (5%) of the 20 wild-type mice. The difference of presenting bronchial epithelial hyperplasia is statistically significant between the ΔDNMT3B-del overexpressing transgenic animals and the wild-type animals (P < 0.0001 by Fisher's Exact Test, Fig. 3B). The hyperplastic lesions in the transgenic lungs showed increased number of epithelial cells positive for phosphorylated Histone H3 protein (pH 3), indicating augmented proliferation potentials of the epithelial cells in lungs of the transgenic animals (Fig. 3C). Ten (23%) of the transgenic animals presented with parenchymal inflammatory aggregates and 5 (12%) developed tumor-like structures in the lungs compared to 3 (15%) and none (0%) for the 20 wild-type mice, respectively (Fig. 3B), but the differences are not statistically significant.
3.4. Changes of DNA Methylation Landscape in Lungs of the Transgenic Mice
We first examined expression levels of two major transcriptional factors, Nkx2.1 and Gata6 which regulate the expression of SPC gene and are critical in lung development, in lungs of the transgenic mice. As expected, lungs were the only tissue that expressed both Nkx2.1 and Gata6 at a high level in both transgenic and wild-type mice (Supplementary Fig. 3), suggesting that ΔDNMT3B4-del transgene expression in the lungs did not impact SPC promoter activity in lungs of the transgenic mice. Hence, ΔDNMT3B4-del transgene expression itself was also unlikely affected by the resulting morphological changes in the lungs.
Supplementary Figure 3.
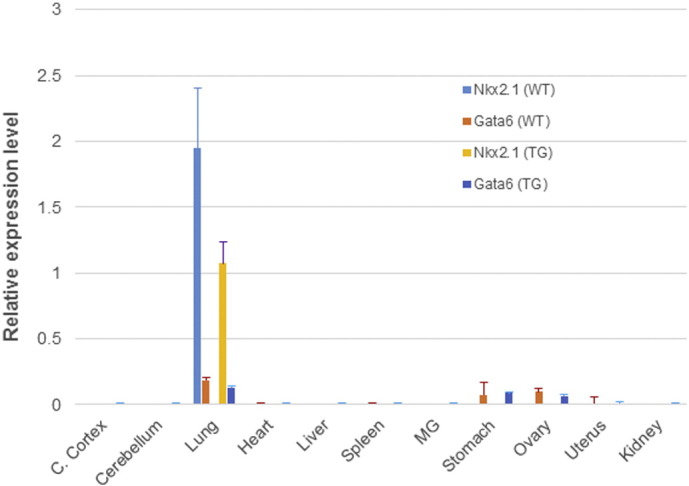
Relative expression levels of Nkx2.1 and Gata6 transcripts in lungs from transgenic and wild-type animals by real time RT-PCR. Expression levels were normalized with mouse GAPDH mRNA for each sample.
To determine potential impact in global DNA methylation patterns in the lungs of the transgenic mice elicited by ΔDNMT3B4-del overexpression, we first analyzed the overall levels of DNA methylation in the genome of the mouse lungs using Imprint® Methylated DNA Quantification kit (Sigma). Compared with DNA samples obtained from lungs of wild type animals, DNA samples obtained from lungs of ΔDNMT3B4-del expressing mice exhibited a significantly reduced amount of DNA methylation (P < 0.05) but not DNA samples obtained from heart tissues where no transgene expression was detected (Supplementary Fig. 4), indicating that DNA samples from lungs of the transgenic mice were globally hypomethylated. Such DNA hypomethylation may be used to explain the observed phenotypes in lung tissues of the transgenic mice as this type of abnormalities has been reported in the early tumorigenesis (Feinberg and Vogelstein, 1983; Jones and Baylin, 2007).
Supplementary Figure 4.
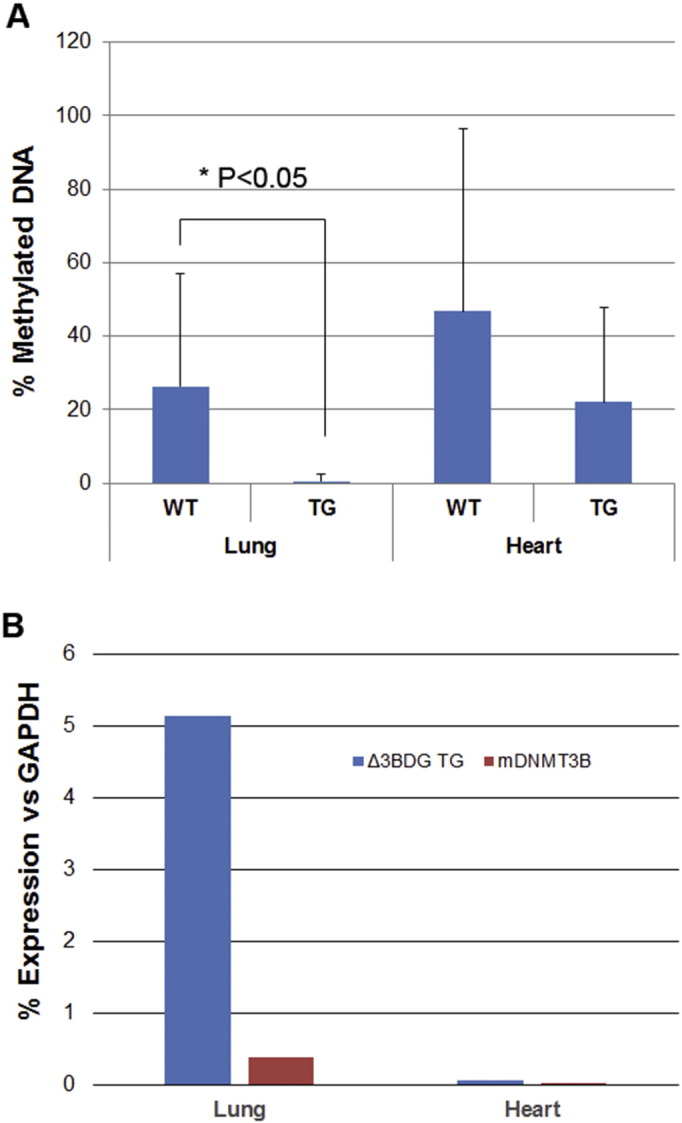
Global DNA methylation levels in ΔDNMT3B4-del transgenic and wild-type mice. (A) Genomic DNA extracted from lung and heart tissues of ΔDNMT3B4-del transgenic and wild-type mice was measured for relative levels of methylated gene promoters using ImprintTM Methylated DNA Quantification kit (MDQ1, Sigma). Heart tissues were included as negative control because very low to none transgene expression was detected in this organ of the transgenic mice. The difference was statistically significant (p < 0.05 by Student’s t Test with sample size 6 for each group). (B). Relative expression levels of ΔDNMT3B4-del transgene in lung and heart tissues of the transgenic and wild-type mice.
We then wanted to determine the impact of the transgene expression on global DNA methylation patterns in a more specific manner. To do this, we used methyl binding domain protein-enriched genome sequencing (MBD-Seq) approach (Decock et al., 2012, Lan et al., 2011, Li et al., 2010) to analyze methylated DNA regions in the genomic DNA samples with base pair resolution. For our analysis, we constructed 6 libraries of genomic DNA samples from mouse lung tissues obtained from both wild-type and transgenic mice (for each group, n = 3 biological replicates).
More than 76 million reads were obtained ranging from 8.6 million to > 17 million reads for each library. The raw data has been deposited in NCBI database and can be access through: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68922. Among the reads, 91.5 to 96.5% of the reads for each MBD-Seq sample were mapped to mouse genome (Supplementary Table 1). We used ChIPpeakAnno software for annotating the regions with distinct methylation levels (Zhu et al., 2010). Ignoring the total number of reads, the samples derived from transgenic and wild-type mice demonstrated similar alignment levels (Supplementary Table 1), an indication of the robustness of the method. We identified 92,205 methylated regions that overlap in at least three samples and these regions were used for further analyses. The overall read counts aligned to a normalized gene body are shown in Fig. 4A. Each gene mapped with reads was converted its gene length from transcription start site (TSS) to transcription end site (TES) as 100% of the gene body in order to make the measurements comparable.
Fig. 4.

DNA methylation patterns in lungs of wild-type and transgenic animals measured by MBD-Seq. (A) Reads of DMR aligned to normalized gene bodies for samples from wild-type (green) and transgenic (brown) animals. The genes were transformed to a body percentage from its length to make the measurements comparable. (B) Overall reads aligned to normalized gene bodies with extended intergenic regions and CpG island shores for samples from wild-type and transgenic animals. (C) Highlight of overall reads aligned to intergenic regions for samples from wild-type and transgenic animals. (D) Clustering of genomic DNA samples from transgenic and wild-type animals based on DMRs. (E) Depiction of DMR methylation for each sample group (wild-type and transgenic). Signs + and − illustrate the regions with incremented affinity in wild-type and transgenic group, respectively. * (p < 0.001 by Wilcoxon test). (F) Summary of the locations of DMRs regarding features, genes and CpG islands. (G) Distribution of DMRs of both hyper-methylated and hypo-methylated in transgenic mice relative to transcription starting site (TSS).
We observed that the DNA methylation level was consistently lower in the gene bodies of DNA samples from lungs of the wild-type mice compared with DNA samples from the transgenic mice (Fig. 4A). However, this pattern reversed at the CpG island shores (Fig. 4B), where DNA samples from the wild-type mice exhibited a significantly higher cytosine methylation than DNA samples from the transgenic mice. The scale for the y-axis is much smaller than the observed in chart correspondent for gene body but the shades from the standard error do not overlap, except in the peaks observed inside the − 1000 bp of the left of the CpG islands. Interestingly, the intergenic regions also showed a clear opposite pattern compared to the gene body (Fig. 4C). Although the differences between the two sample groups are small in values for the normalized counts, the differences are statistically highly significant (P < 0.0001).
The DiffBind R package was applied for the identification of differentially methylated regions (DMRs) between the transgenic and wild type mice (Ross-Innes et al., 2012). A total of 407 genomic regions were identified to have distinct methylation levels (Supplementary Table 2). When these DMRs were used to classify the animals, they perfectly clustered the transgenic and wild-type samples together (Fig. 4D), which validated the reliability of DMR detection method. Among the 407 DMRs, 103 (25%) were hypomethylated and 304 (75%) were hypermethylated in samples from the transgenic mice. As depicted in Fig. 4E, DNA methylation patterns from the transgenic mice showed a higher methylation but also a broader variability compared to those from wild-type mice. The locations of the DMRs in genomic DNA regions as well as in CpG islands are summarized in Fig. 4F. Approximately 40% of the DMRs are located inside genes whereas most of the remaining DMRs are equally distributed in the downstream or upstream of genes. Interestingly, only 6% of the DMRs contain a CpG island. The rest remains in similar proportion upstream or downstream of the CpG islands. The hypermethylated DMRs in tissues from the transgenic mice tend closer to transcriptional starting sites of genes whereas the hypomethylated DMRs more widespread and located downstream of transcription start sites (Fig. 4G).
3.5. Hyperplasia and Genome Instability in Lungs of the Transgenic Mice
It has been suggested that cells with altered DNA methylation are susceptible to undergoing chromosomal loss, gain, or rearrangement (Dodge et al., 2005, Tuck-Muller et al., 2000). We examined the expression of DNA damage response genes including 53BP1, pCHK2 and pH2AX in lung tissue sections. Comparing to lungs of wild-type mice, the hyperplastic lung tissues from the transgenic mice appear to express higher levels of 53BP1 and pH2AX (Fig. 5A).
Fig. 5.

Expression of genes associated with DNA damage induced by overexpression of ΔDNMT3B4-del. (A) Immunohistochemistry for proteins as indicated in a series of sections from lungs of wild-type and transgenic animals. (B) Scheme of major components in inducible expression of ΔDNMT3B4-del in lentiviral expression system. (C) EGFP expression driven by tet-op promoter through IRES fragment in the presence of 1 μg/ml Dox, an indirect proof of ΔDNMT3B4-del expression. (D) Cell cycle distributions measured in the immortalized normal bronchial epithelial (HBE4) cells infected with lentivirus hosting the inducible expression vectors of ΔDNMT3B4-del before and after 48 h treatment with 1 μg/ml Dox. (E) Levels of aneuploidy and DNA damage associated proteins in HBE4 cells infected with lentiviral particles before and after 48 h treatment with Dox.
To substantiate the findings observed in the transgenic mice, we overexpressed ΔDNMT3B4-del in immortalized normal bronchial epithelial (HBE4) cells by infecting the cells with lentivirus containing ΔDNMT3B4-del expressing cassette as depicted in Fig. 5B. The ΔDNMT3B4-del expression in the cells could be induced by treating the cells with tetracycline for 48 h as shown in Fig. 5C. Expression of ΔDNMT3B4-del significantly increased the percentage of cells in G2/M phase (Fig. 5D). The percentage of the cells with abnormal DNA content (> G2 cells) were also increased, indicating that the overexpression of ΔDNMT3B4-del might have caused DNA damage and formation of aneuploidy, which are frequently found in the early stage of tumorigenesis (Kops et al., 2005, Sugai et al., 2003). Expression of DNA damage response genes 53BP1, pCHK2 and pH2AX were evaluated in the inducible ΔDNMT3B4-del expression cells. Consistent with the findings observed in lungs of the transgenic mice, pH2AX and 53BP1 proteins were significantly elevated with no notable change in the expression of pCHK2 (Fig. 5E). Protein Mad2L2 that is associated with cell aneuploidy was also increased after induction of ΔDNMT3B4-del expression (Fig. 5E).
3.6. Promoting Tumor Formation in Lungs of Carcinogen Treated Transgenic Mice
Genomic instability caused by aberrant expression of ΔDNMT3B4-del could prime cells for acquisition of genetic mutations, resulting in an increased rate of tumor formation. To test this possibility, we treated the transgenic mice with tobacco carcinogen 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) to determine whether overexpression of ΔDNMT3B4-del can confer an increased rate of lung cancer formation. C57Bl/6 mouse strain, from which ΔDNMT3B-del transgenic lines were made, is generally resistant to NNK challenge (Ryan et al., 1987). Both transgenic mice and their wild-type littermates were treated with NNK (150 mg/kg) beginning at 8 weeks of age with one injection per week for two weeks. Fourteen months following the first injection, no surface lung tumor was observed by gross examination in both groups of mice. Nevertheless, 5 out of 6 transgenic mice treated with NNK showed microscopic lung adenocarcinoma or tumor-like structures (Fig. 6) whereas no notable morphological change was observed in the lungs of 5 wild-type mice (P = 0.015 by Fisher's Exact Test).
Fig. 6.

Microscopic tumors in lungs of the transgenic animals treated with tobacco carcinogen NNK. Typical morphology of lung sections from wild-type (WT) animals and transgenic (TG) animals with tumor-like lesions (TG-tumor-like) and adenocarcinomas (TG-tumor).
4. Discussion
Genomic DNA methylation is established and maintained by two distinct processes: de novo DNA methylation which is mediated by DNMT3A, DNMT3B and DNMT3L (isoform lacking the characteristic C-terminal catalytic domain) and DNA methylation maintenance which is catalyzed by DNMT1. Overexpression of DNMT3B has been reported in many cancer types including NSCLC. Furthermore, different DNMT3B transcripts derived from alternative splicing or alternative use of transcription initiation site have been reported (Ostler et al., 2007, Wang et al., 2006b), which may impact the pattern of DNA methylation and gene expression (Wang et al., 2007). In this study, we focused on DNMT3B variants with alternative splicing at the 3-prime region without exons 21, 22 or both 21 and 22. Such exclusions result in a disruption of the catalytic units of the methyltransferase and therefore DNA methyltransferase activity. Our analysis showed that almost half of NSCLC cell lines and primary NSCLC tumors examined expressed only the DNMT3B variants lacking exons 21, 22 or both 21 and 22 (termed ΔDNMT3B-del), whereas the normal bronchial epithelial cells expressed DNMT3B variants with and without the exons in a ratio of approximately 1:1. This finding is somewhat surprising because of the common belief that increased DNA methyltransferase activity contributes to abnormal DNA methylation patterns in cancers. While exact mechanisms remain to be determined, our finding suggests that a proper ratio of DNMT3B/ΔDNMT3B and DNMT3B/ΔDNMT3B-del is important for maintaining DNA methylation landscapes, wherein reduced levels of DNMT3B/ΔDNMT3B and overexpression of catalytically deactivated DNMT3B/ΔDNMT3B-del contribute to altered DNA methylation patterns in lung tumorigenesis.
To substantiate our hypothesis, we generated transgenic mice overexpressing ΔDNMT3B4-del specifically in lung epithelial and alveoli cells. As expected, induced overexpression of ΔDNMT3B4-del caused an increased lung ductal epithelial hyperplasia and inflammatory aggregates in most of the transgenic mice. However, none of the lesions progressed to tumors even animals aged up to year and a half, suggesting this factor alone is a modifier and not sufficient to transform lung epithelial cells to malignancy. It is possible that overexpressed ΔDNMT3B4-del, which lacks the catalytic domain of DNA methyltransferase, competes with de novo DNA methyltransferase 3A and 3B to counteract the activity of the de novo DNA methylases. This dominant negative effect could lead to altered DNA methylation patterns in the cancer cell genome, an effect similarly caused by DNMT3L observed in embryonic stem cells (Neri et al., 2013). Consistent with this argument, overall CpG sites methylated in genomic DNA from lungs of transgenic mice were significantly lower as determined by the ELISA-based assay. Interestingly, genomic DNA from lungs of the transgenic mice exhibited an increased methylation in the gene bodies but hypomethylation in intergenic regions, similar to genomic DNA methylation patterns found in lung cancer cells. Bearing in mind that changes of DNA methylation pattern were observed in genomic DNA from lungs of the transgenic animals rather than tumor cells, our results suggest that overexpression of ΔDNMT3B4-del is able to disturb the normal DNA methylation pattern to facilitate early stage molecular alterations and morphologic changes consistent with initiation of tumorigenesis.
The global DNA methylation patterns observed in lung tissues from the transgenic mice overexpressing ΔDNMT3B4-del showed some similarity to those observed in human cancers, particularly the global DNA hypomethylation and regional hypermethylation in certain CpG islands. It has been reported that DNA hypermethylation of CpG islands causes inactivation of tumor suppressor genes through gene promoter suppression (Baylin and Herman, 2000). In addition, global genomic hypomethylation has been reported to be associated with genomic instability (Putiri and Robertson, 2011). Of the proteins associated with DNA damage response we examined, 53BP1 and pH2AX were significantly increased in lungs of the transgenic animals as well as in normal lung epithelial cells overexpressing ΔDNMT3B4-del. Consistent with this observation, immortalized normal bronchial epithelial cells with induced ΔDNMT3B-del expression displayed increased cell populations accumulated in G2 phase (48 h post-induction) as well as increased activation of MDM2L, a gene associated with aneuploidy. These results support the role of ΔDNMT3B4-del in reducing genomic instability.
In a previous study, we have shown that ΔDNMT3B4 may preferentially target certain CpG enriched promoters (Wang et al., 2007) but no consensus targeting sequence was identified. In this study, while overexpressing ΔDNMT3B4-del resulted in an altered DNA methylation pattern in lungs of the transgenic mice, we did not seek to identify consensus sequences preferentially targeted by the DNMT3B isoform due to the complexity in formation of DNA methylation landscape and the heterogeneity nature of the lung tissues where various cell types were pooled together for the analysis. Such attempts will be necessary in future studies by analyzing data obtained from micro-dissected bronchial epithelial cells and tumor cells of the mice.
It is important to note that expression of ΔDNMT3B4-del alone was insufficient to transform lung epithelial cells in our animal model. This is not surprising because aberrant DNA methylation changes are early events in tumorigenesis and multiple factors are necessary for development of clinically relevant cancer. In fact, the transgenic mice became susceptible for lung cancer development and developed lung cancer when challenged with NNK treatment. While ΔDNMT3B4 is the most abundant isoform expressed in NSCLC (Wang et al., 2006a, Wang et al., 2006b), abnormal expression of other isoforms are also observed in NSCLC. It will be important to determine the potential impact of other isoforms in DNA methylation patterns and lung tumorigenesis both alone and in combination with ΔDNMT3B4.
Taken together, our study demonstrates an important role of DNMT3B/ΔDNMT3B-del in lung tumorigenesis and developed a transgenic mouse model valuable for future in vivo studies to better understand mechanisms regarding how the DNA methylation changes during lung tumorigenesis as well as develop strategies to prevent lung cancer by targeting DNMT3B/ΔDNMT3B-del induced tumorgenesis. Because current demethylation agents used or tested in clinic are non-specific to isoforms of DNA methyltransferases, our findings raise the possibility of selectively targeting individual isoforms, at least DNMT3Bs, to improve efficiency and reduce potential side effects. Further studies focusing on comparative analysis of individual isoforms will improve our understanding of the biological impacts of these isoforms and facilitate the translation of the knowledge into development of clinical applications.
The following are the supplementary data related to this article
Supplementary Figure 5.

Promoter methylation status of selected genes in lungs of the transgenic mice (A) and gene expression levels of the selected corresponding genes (B).
Sequencing and mapping details of the samples.
Differntially methylated regions (DMRs).
Sequence Alignment of RT-PCR analysis of DNMT3B/deltaDNMT3B-del form.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2015.09.002.
Authorship Contributions
Mark Ma: participated in study design; generated transgenic animals; performed experiments, data analysis and manuscript writing.
Ruxian Lin: performed experiments and data analysis.
Jose Carrillo, Yaokun Li, and Jiuzou Song: generated and analyzed DNA demethylation data; participated in manuscript writing.
Manisha Bhutani and Ashatosh Pathak: performed experiments and data analysis.
Hening Ren: participated in experiments and data interpretation.
Li Mao: developed concept; supervised experimental designs; participated in data interpretation and manuscript writing with final approval.
Acknowledgments
The work is supported by NIH grants R01 R01CA136635 and P30CA134274. We would like to thank Sayaka Hanada, Hong Pan and Jun Zhao for their excellent technical assistance for this project.
References
- Baylin S.B., Herman J.G. DNA Hypermethylation in Tumorigenesis — Epigenetics Joins Genetics. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- Bestor T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Chen T., Ueda Y., Dodge J.E., Wang Z., Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock A., Ongenaert M., Hoebeeck J., De Preter K., Van Peer G., Van Criekinge W., Ladenstein R., Schulte J.H., Noguera R., Stallings R.L. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol. 2012;13:R95. doi: 10.1186/gb-2012-13-10-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge J.E., Okano M., Dick F., Tsujimoto N., Chen T., Wang S., Ueda Y., Dyson N., Li E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- Edwards J.R., O'Donnell A.H., Rollins R.A., Peckham H.E., Lee C., Milekic M.H., Chanrion B., Fu Y., Su T., Hibshoosh H. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20:972–980. doi: 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A.P., Vogelstein B. Alterations in DNA methylation in human-colon neoplasia. Semin. Surg. Oncol. 1987;3:149–151. doi: 10.1002/ssu.2980030304. [DOI] [PubMed] [Google Scholar]
- Hansen K.D., Timp W., Bravo H.C., Sabunciyan S., Langmead B., McDonald O.G., Wen B., Wu H., Liu Y., Diep D. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.A. DNA Methylation and Cancer. Oncogene. 2002;21:5358–5360. doi: 10.1038/sj.onc.1205597. [DOI] [PubMed] [Google Scholar]
- Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Jones P.A., Baylin S.B. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops G.J., Weaver B.A., Cleveland D.W. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Lan X., Adams C., Landers M., Dudas M., Krissinger D., Marnellos G., Bonneville R., Xu M., Wang J., Huang T.H. High resolution detection and analysis of cpg dinucleotides methylation using MBD-Seq technology. PLoS One. 2011;6 doi: 10.1371/journal.pone.0022226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E., Bestor T.H., Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Li N., Ye M., Li Y., Yan Z., Butcher L.M., Sun J., Han X., Chen Q., Zhang X., Wang J. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods. 2010;52:203–212. doi: 10.1016/j.ymeth.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Linhart H.G., Lin H., Yamada Y., Moran E., Steine E.J., Gokhale S., Lo G., Cantu E., Ehrich M., He T. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21:3110–3122. doi: 10.1101/gad.1594007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerschmidt D.M., Knowles B.B., Solter D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014;28:812–828. doi: 10.1101/gad.234294.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F., Krepelova A., Incarnato D., Maldotti M., Parlato C., Galvagni F., Matarese F., Stunnenberg H.G., Oliviero S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell. 2013;155:121–134. doi: 10.1016/j.cell.2013.08.056. [DOI] [PubMed] [Google Scholar]
- Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Ostler K.R., Davis E.M., Payne S.L., Gosalia B.B., Exposito-Cespedes J., Le Beau M.M., Godley L.A. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene. 2007;26:5553–5563. doi: 10.1038/sj.onc.1210351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostler K.R., Yang Q., Looney T.J., Zhang L., Vasanthakumar A., Tian Y., Kocherginsky M., Raimondi S.L., DeMaio J.G., Salwen H.R. Truncated DNMT3B isoform DNMT3B7 suppresses growth, induces differentiation, and alters DNA methylation in human neuroblastoma. Cancer Res. 2012;72:4714–4723. doi: 10.1158/0008-5472.CAN-12-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putiri E.L., Robertson K.D. Epigenetic mechanisms and genome stability. Clin. Epigenetics. 2011;2:299–314. doi: 10.1007/s13148-010-0017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson K.D. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- Robertson K.D., Jones P.A. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene. 1999;18:3810–3820. doi: 10.1038/sj.onc.1202737. [DOI] [PubMed] [Google Scholar]
- Robertson K.D., Uzvolgyi E., Liang G., Talmadge C., Sumegi J., Gonzales F.A., Jones P.A. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J., Barker P.E., Nesbitt M.N., Ruddle F.H. KRAS2 as a genetic marker for lung tumor susceptibility in inbred mice. J. Natl. Cancer Inst. 1987;79:1351–1357. [PubMed] [Google Scholar]
- Saito Y., Kanai Y., Sakamoto M., Saito H., Ishii H., Hirohashi S. Overexpression of a splice variant of DNA methyltransferase 3b, DNMT3b4, Associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10060–10065. doi: 10.1073/pnas.152121799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M.Y., Vasanthakumar A., Barnes N.Y., Figueroa M.E., Kamp A., Hendrick C., Ostler K.R., Davis E.M., Lin S., Anastasi J. DNMT3B7, a truncated DNMT3B isoform expressed in human tumors, disrupts embryonic development and accelerates lymphomagenesis. Cancer Res. 2010;70:5840–5850. doi: 10.1158/0008-5472.CAN-10-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugai T., Takahashi H., Habano W., Nakamura S., Sato K., Orii S., Suzuki K. Analysis of genetic alterations, classified according to their DNA ploidy pattern, in the progression of colorectal adenomas and early colorectal carcinomas. J. Pathol. 2003;200:168–176. doi: 10.1002/path.1340. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Gabrielson E., Chen W., Anbazhagan R., van Engeland M., Weijenberg M.P., Herman J.G., Baylin S.B. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002;31:141–149. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck-Muller C.M., Narayan A., Tsien F., Smeets D.F., Sawyer J., Fiala E.S., Sohn O.S., Ehrlich M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet. Cell Genet. 2000;89:121–128. doi: 10.1159/000015590. [DOI] [PubMed] [Google Scholar]
- Van Emburgh B.O., Robertson K.D. Modulation of Dnmt3b function in vitro by interactions with Dnmt3L, Dnmt3a and Dnmt3b splice variants. Nucleic Acids Res. 2011;39:4984–5002. doi: 10.1093/nar/gkr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Walsh G., Liu D.D., Lee J.J., Mao L. Expression of delta DNMT3B variants and its association with promoter methylation of p16 and RASSF1A in primary non-small cell lung cancer. Cancer Res. 2006;66:8361–8366. doi: 10.1158/0008-5472.CAN-06-2031. [DOI] [PubMed] [Google Scholar]
- Wang L., Wang J., Sun S., Rodriguez M., Yue P., Jang S.J., Mao L. A novel DNMT3B subfamily, DeltaDNMT3B, is the predominant form of DNMT3B in non-small cell lung cancer. Int. J. Oncol. 2006;29:201–207. [PubMed] [Google Scholar]
- Wang J., Bhutani M., Pathak A.K., Lang W., Ren H., Jelinek J., He R., Shen L., Issa J.P., Mao L. Delta DNMT3B variants regulate DNA methylation in a promoter-specific manner. Cancer Res. 2007;67:10647–10652. doi: 10.1158/0008-5472.CAN-07-1337. [DOI] [PubMed] [Google Scholar]
- Zhu L.J., Gazin C., Lawson N.D., Pages H., Lin S.M., Lapointe D.S., Green M.R. ChIPpeakAnno: a bioconductor package to annotate ChIP-seq and ChIP–Chip data. BMC Bioinforma. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequencing and mapping details of the samples.
Differntially methylated regions (DMRs).
Sequence Alignment of RT-PCR analysis of DNMT3B/deltaDNMT3B-del form.