

Abstract
The emergence of direct-acting antiviral agents (DAAs) for HCV infection represents a major advance in treatment. The NS3 protease inhibitors, boceprevir and telaprevir, were the first DAAs to receive regulatory approval. When combined with PEG-IFN and ribavirin, these agents increase rates of sustained virologic response in HCV genotype 1 to ~70%. However, this treatment regimen is associated with several toxicities. In addition, both boceprevir and telaprevir are substrates for and inhibitors of the drug transporter P-glycoprotein and the cytochrome P450 enzyme 3A4 and are, therefore, prone to clinically relevant drug interactions. Several new DAAs for HCV are in late stages of clinical development and are likely to be approved in the near future. These include the protease inhibitors, simeprevir and faldaprevir, the NS5A inhibitor, daclatasvir, and the nucleotide polymerase inhibitor, sofosbuvir. Herein, we review the clinical pharmacology and drug interactions of boceprevir, telaprevir and these investigational DAAs. Although boceprevir and telaprevir are involved in many interactions, these interactions are manageable if health-care providers proactively identify and adjust treatments. Emerging DAAs seem to have a reduced potential for drug interactions, which will facilitate their use in the treatment of HCV.
Introduction
Worldwide, ~150 million people are living with chronic HCV infection.1 Without treatment, two-thirds of HCV-infected individuals will develop chronic liver disease and many will progress to cirrhosis and hepatocellular carcinoma.1 These complications can be prevented with antiviral treatment, but not all patients are eligible for, able to access, tolerate, or respond to current therapies. Thus, new agents are desperately needed. Fortunately, multiple agents are in various stages of clinical development for the treatment of HCV.
Compounds that target each of the proteins encoded by the single-stranded HCV RNA genome include inhibitors of three structural proteins (core, E1 and E2), the ion channel protein p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B).2 Boceprevir and telaprevir, NS3 protease inhibitors, were the first direct-acting antiviral agents (DAAs) to receive regulatory approval for the treatment of HCV. These drugs are specific for HCV genotype 1, which is the most common genotype in the world, but also the most difficult to treat.3 So-called triple therapy—that is, PEG-IFN plus ribavirin plus either boceprevir or telaprevir—has increased cure rates (sustained virologic response; SVR) by roughly 30% in both treatment-naive and treatment-experienced patients with HCV genotype 1.4–9 However, there is still room for improvement. Multiple contraindications and toxicities are associated with PEG-IFN plus ribavirin, and with triple therapy. Boceprevir and telaprevir can cause anaemia and gastrointestinal effects. Boceprevir causes a bitter, earthy, or metallic taste10 and telaprevir can cause rash (even severe or life-threatening rash) and anorectal discomfort.11 Both are substrates and inhibitors of the cytochrome P450 3A (CYP3A) enzyme and several drug transporters, which predisposes them to drug–drug interactions.
Emerging DAAs will probably overcome many of the shortcomings of current therapies. The next wave of DAAs will include two new NS3 protease inhibitors, simeprevir and faldaprevir; the NS5A inhibitor daclatasvir; and the nucleotide NS5B polymerase inhibitor, sofosbuvir. In some cases, these agents will be added to PEG-IFN plus ribavirin therapy in much the same way as boceprevir and telaprevir, but others, such as sofosbuvir, might be approved in an interferon-free regimen. Subsequent advances will see the emergence of additional DAAs and novel combinations ultimately leading to a new treatment standard of interferon-free multi-DAA treatment, with or without ribavirin. Studies suggest SVR rates of ~80–90% in treatment-naive patients when faldaprevir, simeprevir, daclatasvir or sofosbuvir are added to PEG-IFN plus ribavirin combination therapy.12-15 Several all-oral DAA combinations are showing similarly high SVR rates in phase II studies.16-18 Addition of two or more DAAs to PEG-IFN plus ribavirin provides almost 100% SVR even in historically difficult-to-treat patient populations.19-21 These studies suggest new treatments will certainly increase SVR rates. Many new DAAs also have longer half-lives than current therapies, which facilitates less frequent dosing that should enhance compliance. For instance, faldaprevir, simeprevir, daclatasvir and sofosbuvir are all being given once daily in phase III studies. Most investigational agents seem to offer improved genotype coverage, and also seem to have fewer adverse effects. Available data suggest investigational DAAs might also have a reduced potential for drug–drug interactions. However, several of these agents are substrates of CYP3A and drug transporters and some inhibit and induce enzymes and transporters, implying that potential drug–drug interactions will remain an important aspect of management into the foreseeable future.
In this Review, we describe the principles and clinical consequences of drug interactions in patients with chronic HCV, summarize the pharmacology and drug interaction potential of boceprevir, telaprevir and the investigational DAAs faldaprevir, simeprevir, daclatasvir and sofosbuvir, and examine the magnitude and management of specific drug–drug interactions with these agents.
Principles of drug interactions
Drug–drug interactions can be pharmacokinetic or pharmacodynamic in nature. Pharmacokinetic drug interactions result in a change in drug concentrations. Pharmacodynamic drug interactions do not result in a change in drug concentrations, but can result in additive, synergistic or antagonistic effects. For instance, in the treatment of HCV, two drugs that both cause anaemia would be considered to have a pharmacodynamic interaction. The focus of this Review is on potential pharmacokinetic interactions.
Pharmacokinetic drug interactions occur at the level of drug absorption, metabolism, distribution or elimination. In terms of interactions at the level of metabolism, the CYP enzymes are responsible for the breakdown of many drugs. Specifically, the CYP3A4 isoform metabolizes the majority of marketed medications.22 CYP enzymes are capable of being induced and inhibited. Induction of CYP enzymes results in a reduction in concentrations of substrates; conversely, inhibition of CYP enzymes results in an increase in concentrations of substrates. CYP enzymes are not the only site of drug–drug interactions however. An increasing number of drug–drug interactions are attributed to membrane transporters. Unlike CYP enzymes, our understanding of membrane transporters is in its infancy because analytical techniques to identify transporters were not available until the early 1990s. Although thousands of membrane transporters have been identified, only a few have been implicated in clinically relevant drug–drug interactions. Examples include P-glycoprotein (P-gp, also known as multidrug resistance protein 1), solute carrier organic anion transporter family member 1B1 (SLCO1B1, also known as organic anion transporting polypeptide 1B1 or OATP1B1), multidrug resistance-associated proteins (MRP), and ATP-binding cassette sub-family G member 2 (ABCG2, also known as breast cancer resistance protein or BCRP). As with the CYP enzymes, membrane transporters can be induced or inhibited.
For every drug, there exists of concentrations that balances the likelihood of efficacy with the probability of toxicity (Figure 1). For some drugs, this range of concentrations is wide, for others the range of concentrations is narrow. Many factors can affect drug concentrations, including organ dysfunction, body weight, diet, host genetics and, the topic of this Review, drug interactions. Drugs with a wide therapeutic range have a high tolerance to drug–drug interactions because concentration shifts are unlikely to increase the probability of toxicities or decrease the likelihood of efficacy. However, for drugs with a narrow therapeutic range, drug–drug interactions can have important clinical implications. Drug interactions that increase concentrations (for example, CYP or transporter inhibition) can lead to an increase in concentration-dependent toxicities. Subtherapeutic concentrations can lead to the development of drug resistance, which can compromise the success of current and future treatments. Some antiviral drugs are considered to have a narrow therapeutic range, but whether boceprevir, telaprevir or investigational DAAs fall into this category is currently unclear.23
Figure 1.
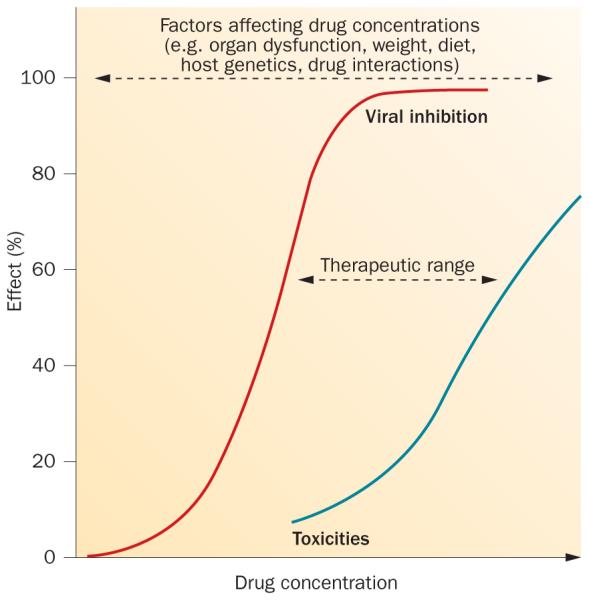
Concept of a therapeutic range. For every drug, there exists a range of concentrations that balances the likelihood of efficacy with the probability of toxicity.
The potential for pharmacokinetic interactions between drugs is often tested first in healthy volunteers. This approach is logical considering the consequences of an interaction (that is, toxicity or viral resistance). However, additional pharmacokinetic considerations exist in people with HCV. First, HCV infection itself has been shown to impair drug metabolism by reducing microsomal enzymatic activity.24 Second, CYP enzyme activity is impaired as liver disease progresses.25 Furthermore, with increased fibrosis, a higher likelihood of portal-systemic shunting exists.26 Thus, the magnitude of a drug interaction could differ in individuals with HCV based on the degree of hepatic impairment or stage of fibrosis, and might be particularly problematic in those with advanced liver disease.
DAA pharmacology
Table 1 shows the pharmacology and interaction potential of current and late-phase investigational agents for HCV. The pharmacology of boceprevir, telaprevir, simeprevir, faldaprevir, daclatasvir and sofosbuvir are described in greater detail below.
Table 1.
Pharmacology and interaction potential of currently approved and investigational DAAs in late-phase clinical development
| Drugs | Route of metabolism or excretion |
CYP effects | Transporter substrate |
Transporter effects | Comments |
|---|---|---|---|---|---|
| Protease inhibitors | |||||
|
| |||||
| ABT450/ ritonavir |
CYP3A108 | CYP3A inhibition by ritonavir | ND | Inhibits OATP1B1109 | NA |
|
| |||||
| Asunaprevir | ND | Moderate inhibitor of CYP2D6; weak inducer of CYP3A4110 |
OATP1B1/2B1111 | Weak P-gp and OATP1B1/1B3111 inhibitor |
NA |
|
| |||||
| Boceprevir10 | CYP3A, AKR | Moderate CYP3A inhibitor | P-gp | Weak P-gp inhibitor112 | NA |
|
| |||||
| Danoprevir/ ritonavir |
CYP3A | CYP3A inhibition by ritonavir | ND | ND | A study with midazolam (CYP3A probe) and warfarin (CYP2C9 probe) showed that danoprevir did not change the effect of ritonavir on these probes (midazolam increased, warfarin decreased)113 |
|
| |||||
| Faldaprevir | CYP3A | Moderate inhibitor of hepatic and intestinal CYP3A; weak inhibitor of CYP2C9;40 inhibitor of UGT1A141 |
P-gp, MRP242 | Inhibits OATP1B1, OATP1B3, OATP2B142 |
NA |
|
| |||||
| Simeprevir | CYP3A | Mild inhibitor of CYP1A2 and intestinal CYP3A37 |
ND | OATP1B1 and MRP2 inhibitor38 |
NA |
|
| |||||
| Telaprevir11 | CYP3A | Strong CYP3A inhibitor | P-gp | Moderate P-gp inhibitor | NA |
|
| |||||
| NS5A inhibitors | |||||
|
| |||||
| ABT267 | ND | ND | ND | ND | AUC and Cmax increased 62% and 67%, respectively, by ritonavir114 |
|
| |||||
| Daclatasvir | CYP3A44 | ND | P-gp44 | Moderate P-gp and OATP1B1 inhibitor46 |
NA |
|
| |||||
| Ledipasvir49 | ND | Not a CYP inhibitor or inducer |
P-gp | Weak inhibitor of P-gp, BCRP, OATP1B1, OATP1B3 |
NA |
|
| |||||
| Nucleos(t)ide polymerase inhibitors | |||||
|
| |||||
| Mericitabine | Renal115 | ND | ND | ND | Cytidine and uridine analogue |
|
| |||||
| Sofosbuvir | Renal50 | ND | P-gp | ND | Uridine analogue116 |
|
| |||||
| Non-nucleoside polymerase inhibitors | |||||
|
| |||||
| ABT333 | CYP2C8, CYP3A4 and CYP2D6 contribute approximately 60%, 30%, and 10% to ABT-333 metabolism, respectively117 |
ND | ND | ND | NA |
|
| |||||
| BI 207127 | ND | ND | P-gp, BCRP, OATP1B1, OATP1B342 |
ND | NA |
Abbreviations: AKR, aldoketoreductase; BCRP, breast cancer resistance protein; CYP, cytochrome P450; DAA, direct-acting antiviral agent; MRP, multidrug resistance protein; NA, not applicable; ND, no data; OATP1, organic anion transporting polypeptide; P-gp, P-glycoprotein; UGT, uridine glucuronyl transferase.
Protease inhibitors
Boceprevir, telaprevir, simeprevir and faldaprevir prevent the NS3 viral protease from cleaving the enzymes responsible for viral replication. Boceprevir and telaprevir are linear peptide mimetics with a ketoamide group that covalently binds with a serine in the catalytic triad.27 Simeprevir and faldaprevir are noncovalent peptide mimetic inhibitors that have a macrocyclic structure.27 All four protease inhibitors are metabolized by CYP3A, and are therefore affected by potent CYP3A inhibitors (for example, ketoconazole) and inducers (for example, rifampin). These HCV protease inhibitors also inhibit CYP3A, to varying degrees, and thus might increase concentrations of other CYP3A substrates. Ritonavir is an HIV protease inhibitor. However, in the field of HIV, it is no longer used for its antiviral effects, but is instead used for its ability to potently inhibit CYP3A. Ritonavir increases the concentrations of CYP3A substrates, reducing their dosing requirements and prolonging the dosing interval.28 The concept of pharmacokinetic enhancement or ‘boosting’ of CYP3A substrates with ritonavir is now being used in the field of HCV. Two HCV protease inhibitors, danoprevir and ABT450, are being studied in combination with a boosting dose of ritonavir.29,30
Boceprevir
Boceprevir is administered at a dose of 800 mg every 7–9 h. Boceprevir exposures are increased by 65% with food, thus the drug should be taken with food, but bioavailability is similar regardless of dietary fat content. Omeprazole does not affect boceprevir absorption.31 Boceprevir is metabolized by aldoketoreductase enzymes and CYP3A, and it is also a substrate for P-gp. Boceprevir is a moderate CYP3A inhibitor and a weak P-gp inhibitor. In vitro, no evidence exists that boceprevir inhibits other CYP enzymes or induces any CYP enzymes. Boceprevir inhibits the drug transporters OATP1B1 and BCRP32 and is 75% protein bound. After a single 400 mg dose of boceprevir (which is lower than the approved 800 mg dose), the area under the curve (AUC) and maximum concentration (Cmax) of the active form of boceprevir (SCH534128) were increased 32% and 28%, respectively, in those with moderate hepatic impairment (Child–Pugh B) and 45% and 62%, respectively, in those with severe hepatic impairment (Child–Pugh C), relative to individuals with no impairment.33 No dosage adjustment is necessary for patients with renal impairment.33 Boceprevir AUC is only 10% lower in patients with end-stage renal disease requiring haemodialysis than in individuals with normal renal function.33
Telaprevir
The recommended dose of telaprevir is 750 mg every 7–9 h with a high (≥20g) fat meal, but telaprevir 1,125 mg twice daily dosing was noninferior to thrice daily dosing in a phase III trial.34 Telaprevir is metabolized by CYP3A and is a strong CYP3A inhibitor and moderate P-gp inhibitor.11 Telaprevir has been shown in vitro to inhibit several hepatic and renal transporters.35 It is 59–76% protein bound. In vitro, telaprevir did not inhibit CYP enzymes other than CYP3A and has a low potential to induce CYP2C, CYP3A or CYP1A. Surprisingly, telaprevir AUC and Cmax are reduced by 46% and 49%, respectively, in those with moderate (Child–Pugh B) hepatic impairment.36 The mechanism for this finding is unclear, but could relate to decreased protein binding or reduced absorption. Thus, the appropriate dose of telaprevir in those with moderate or severe hepatic impairment has not been determined. The reduction in telaprevir AUC and Cmax was less for those with Child–Pugh A hepatic impairment: 15% and 10%, respectively, so no dose adjustment is necessary in these patients. A single-dose study of telaprevir in individuals with creatinine clearances <30 ml/min/1.73 m2 (equivalent to 0.50 ml/s/m2) found a 10% higher Cmax and 21% higher AUC than those without renal impairment, thus no dosage adjustment is necessary for those with mild, moderate, or severe renal impairment, but telaprevir has not been studied in persons with end-stage renal disease or those requiring haemodialysis.11
Simeprevir
Simeprevir is being evaluated at a dose of 150 mg once daily in phase III trials. Simeprevir is metabolized by CYP3A, and is a mild inhibitor of CYP3A and CYP1A237 as well as an inhibitor of OATP1B1 and MRP2.38 In eight volunteers with Child–Pugh B cirrhosis, simeprevir AUC and Cmax were increased 2.62-fold and 1.76-fold, respectively, compared with eight volunteers without hepatic impairment, but similar to those observed in persons with Child–Pugh A cirrhosis.39 No data are available at this time on whether simeprevir needs to be taken with food or with a certain type of meal. The degree of protein binding is also unknown.
Faldaprevir
The highest faldaprevir dose being evaluated in phase III trials is 240 mg once daily. Faldaprevir does not alter caffeine (CYP1A2), efavirenz (CYP2B6), or dextromethorphan (CYP2D6) exposures, but oral midazolam was increased 2.92-fold and S-warfarin increased 1.29-fold, which suggests that faldaprevir moderately and weakly inhibits CYP3A and CYP2C9, respectively.40 Faldaprevir inhibits uridine glucuronosyltransferase 1A1, which causes hyperbilirubinaemia.41 Faldaprevir is a substrate for P-gp and MRP2.42 Faldaprevir pharmacokinetics are not altered in patients with Child–Pugh A cirrhosis.42 No data are currently available on food requirements or protein binding for faldaprevir.
NS5A inhibitors
Daclatasvir inhibits NS5A, a protein essential to the replication machinery of HCV and critical in the assembly of new infectious viral particles.43 Daclatasvir is dosed at 60 mg once daily in phase III trials. Daclatasvir is a substrate for CYP3A and a substrate and inhibitor of P-gp;44 the drug is 99% protein bound.45 Daclatasvir did not have a clinically relevant effect on the CYP3A probe midazolam.46 Dose adjustments of daclatasvir do not seem to be necessary in the setting of hepatic impairment.47 Total daclatasvir plasma AUC and Cmax are lower in patients with hepatic impairment than healthy controls, but unbound drug exposures are similar. Although studies are lacking, dose adjustment for renal impairment might not be needed owing to the mainly hepatic route of elimination of this agent. Food requirements for daclatasvir have not been reported.
Polymerase inhibitors
NS5B RNA-dependent RNA polymerase is essential for HCV replication as it catalyses the synthesis of the complementary minus-strand RNA and subsequent genomic plus-strand RNA.48 Two types of polymerase inhibitors exist, nucleos(t)ide and non-nucleoside analogues. The nucleos(t)ide analogues are prodrugs, requiring activation by host phosphorylation enzymes for activity; the phosphorylated nucleos(t)ide drug competes with endogenous nucleotide bases for incorporation into replicating HCV. Sofosbuvir undergoes phosphorylation by host enzymes to a uridine triphosphate analogue, which is responsible for its antiviral effects; it is not metabolized by CYP enzymes.49 Measuring the phosphorylated anabolite in hepatocytes would be challenging, thus the pharmacokinetic and interaction data available with sofosbuvir describe concentrations of sofosbuvir itself (that is, the prodrug) and the uridine metabolite in blood plasma (GS-331007) and might not necessarily reflect the active form of the drug. The recommended dose of sofosbuvir is 400 mg once daily. Dose adjustments of sofosbuvir are necessary in patients with renal impairment.50 Sofosbuvir exposures are doubled in people with Child–Pugh B and C hepatic impairment, but GS-331007 concentrations are unchanged.51 Interestingly, despite the increase in parent drug concentration and no change in metabolite concentrations, these HCV-infected individuals with cirrhosis (n = 16) had a 3.4-log reduction in HCV RNA after 7 days of sofosbuvir monotherapy versus the 4.7-log reduction observed in HCV-infected individuals without cirrhosis. This finding suggests that impaired phosphorylation or portal-systemic shunting could influence viral responses to this DAA. At this time, no data on food requirements or protein binding for sofosbuvir are available.
Specific drug interactions
In the era of DAAs, health-care providers involved in the treatment of patients with HCV must consider potential drug interactions between DAAs and other drugs and supplements. Figure 2 provides an algorithm for screening, adjusting, and monitoring for potential drug interactions with DAAs. The following section is a summary of established and theoretical interactions of DAAs with other agents. We have highlighted those interactions that have been identified as clinically important or within therapeutic classes commonly used in people with HCV. New data are limited for interactions with antidepressants52 and no new data are available for interactions with antipsychotics, anxiolytics, sleep aids and antihypertensive agents, thus the reader is referred to a prior review for interactions with these classes of drugs.53 We also outlined in detail in a prior review the interaction potential of boceprevir and telaprevir with HMG-CoA reductase inhibitors (that is, statins), which are classic substrates for the transporter OATP1B1.53
Figure 2.

An algorithm for screening, adjusting and monitoring for potential drug interactions with DAAs. Abbreviation: DAA, direct-acting antiviral.
Immunosuppressants
Ciclosporin and tacrolimus are CYP3A and P-gp substrates. Telaprevir and boceprevir are both inhibitors of CYP3A and P-gp, with telaprevir being a more potent inhibitor of both. The effects of boceprevir and telaprevir on ciclosporin and tacrolimus have been evaluated in healthy volunteers. Boceprevir increases ciclosporin and tacrolimus concentrations by 2.7-fold and 17.1-fold, respectively.54 Telaprevir increases ciclosporin and tacrolimus concentrations by 4.64-fold and 70.3-fold, respectively.55 Simeprevir seems to have a much smaller influence on ciclosporin and tacrolimus than either boceprevir or telaprevir. Ciclosporin and tacrolimus AUC are increased 19% and decreased 17%, respectively, by simeprevir.56 Thus, doses of ciclosporin and tacrolimus do not require initial adjustment when administered with simeprevir, but immunosuppressant concentrations should be monitored during treatment. The decrease in tacrolimus, although small, might be magnified by clearance of HCV RNA, which further enhances metabolism and reduces trough levels of tacrolimus and can increase risk of allograft rejection.57 Sofosbuvir does not affect ciclosporin or tacrolimus concentrations. Interestingly, ciclosporin increases sofosbuvir AUC 353%, but the uridine metabolite in blood plasma (GS-331007) is unchanged.58 The mechanism and clinical importance of the increased sofosbuvir concentration is unknown. A healthy volunteer drug–drug interaction study has not been performed with DAAs and sirolimus, but one group reported a 24-fold higher sirolimus AUC than values previously reported in the literature when telaprevir was used to treat 16 patients with HCV after liver transplantation.59
Although telaprevir and boceprevir do not currently have regulatory approval in the post-transplant setting, these individuals are arguably the patients in greatest need of treatment. Several groups have reported their initial experience with PEG-IFN plus ribavirin in combination with telaprevir or boceprevir post-transplantation.60–62 SVR data are limited at this time, but experts are forecasting a 50% cure rate63 and a few trends have emerged. First, treatment of HCV post-transplantation is a major endeavour with our current therapies and should be undertaken only by experienced providers with appropriate infrastructure. Second, the interactions between immunosuppressants and boceprevir or telaprevir require vigilance from health-care providers and require resources, but do seem manageable. Third, toxicities are common, such as anaemia requiring blood transfusions and growth factors. Death, although rare, has occurred during this treatment.
Figure 3 summarizes our protocol at the University of Colorado Denver, USA, for using triple therapy in patients with recurrent HCV after liver transplantation. We use ciclosporin plus mycophenolate mofetil during HCV protease-inhibitor-based treatment. Therapeutic drug monitoring of ciclosporin is performed before, during and after protease inhibitor treatment to refine doses. During triple therapy, we utilize a 2 h post-dose ciclosporin level (C2) with a goal of C2 of approximately 500 ng/ml.64 Telaprevir is preferentially used to minimize the time on the HCV protease inhibitor. Ciclosporin is preferentially used because of the smaller magnitude of the drug interactions with HCV protease inhibitors relative to tacrolimus or sirolimus and augmentation of the antiviral activity of HCV treatment.63
Figure 3.

Protocol for using triple therapy in patients with recurrent HCV after liver transplantation. *All treatment discontinued if HCV RNA >1,000 IU/ml at 4–12 weeks of triple therapy with PEG-IFN-α, ribavirin and a protease inhibitor or detectable at/after 24 weeks. Abbreviations: LADR, low accelerated dose regimen; MMF, mycophenolate mofetil.
Antiretroviral drugs
Owing to shared routes of transmission, ~30% of people infected with HIV are co-infected with HCV.65 A critical consideration in people with HIV–HCV co-infection is the potential for drug interactions. Many antiretroviral drugs are substrates for or otherwise affect CYP enzymes and drug transporters. Several drug–drug interaction studies have been performed with DAAs and antiretroviral agents in healthy volunteers. A limited number of antiretroviral agents seem to be safe to administer with telaprevir and boceprevir. Tenofovir disoproxil fumarate (TDF) is a frequently prescribed antiretroviral agent. Boceprevir does not alter tenofovir AUC.66 Telaprevir increases tenofovir AUC by 30%.67 In isolation, this finding is unlikely to have clinical relevance, but in combination with other agents that might increase tenofovir concentrations, renal function should be monitored. Raltegravir, an HIV integrase inhibitor, can be safely combined with both boceprevir and telaprevir.67,68 In healthy volunteers, interactions occur between telaprevir and boceprevir and several ritonavir-boosted HIV protease inhibitors, whereby concentrations of both the HIV and HCV protease inhibitors are reduced.67,69 The mechanism(s) for these interactions are unclear and the focus of current investigation. In the meantime, only the ritonavir-boosted HIV protease inhibitor, atazanavir, can be safely combined with telaprevir.70 Ritonavir-boosted darunavir, fosamprenavir and lopinavir should not be used with telaprevir or boceprevir. Non-nucleoside reverse transcriptase inhibitors are primarily inducers of CYP3A enzymes, with efavirenz being the most potent inducer of the class. Efavirenz reduces the AUC of boceprevir and telaprevir by ~50% in healthy volunteers.66,67 Telaprevir has been studied at a higher dose than usual (1,125 mg every 8 h) with efavirenz and this dose increase seems to overcome the inductive effects of efavirenz. Telaprevir AUC is reduced 16% by etravirine, but etravirine concentrations are unchanged.71 By contrast, boceprevir is not substantially affected by etravirine, but boceprevir reduces etravirine AUC by 23%.72 The AUC for rilpivirine is increased 79% by telaprevir71 and 39% by boceprevir.73 Telaprevir and boceprevir increase maraviroc AUC by 9.5-fold and 3-fold, respectively.74 Thus, a reduced dose of maraviroc, 150 mg twice daily, should be used in combination with these protease inhibitors. A study with elvitegravir and cobicistat is underway.
The interaction potential with several antiretroviral agents and faldaprevir, daclatasvir, simeprevir and sofosbuvir has been explored. Faldaprevir has been studied with TDF, efavirenz and ritonavir-boosted darunavir.75 Darunavir and tenofovir AUC were increased by 15% and 22%, respectively, when administered with faldaprevir. Faldaprevir AUC was increased by 130% with ritonavir-boosted darunavir, decreased by 22% with TDF and decreased by 35% with efavirenz. In this phase III trial, patients co-infected with HIV and HCV taking ritonavir-boosted darunavir received faldaprevir 120 mg daily and those taking efavirenz received faldaprevir 240 mg daily.75 Daclatasvir has been studied with TDF, efavirenz and ritonavir-boosted atazanavir. The daclatasvir dose should be increased from 60 mg to 90 mg daily when combined with efavirenz and decreased to 30 mg daily with ritonavir-boosted atazanavir.76 Simeprevir has been studied with TDF, rilpivirine, efavirenz, raltegravir and ritonavir-boosted darunavir. Efavirenz reduced simeprevir exposure by 71% and co-administration is not advised. Ritonavir-boosted darunavir increased simeprevir exposure 2.6-fold, even after dose reduction of simeprevir from 150 mg to 50 mg; thus, co-administration of simeprevir with ritonavir-boosted protease inhibitors is not recommended.77 Sofosbuvir has been studied with ritonavir-boosted darunavir, raltegravir, rilpivirine and the combination antiretroviral product containing TDF, emtricitabine and efavirenz.78 After a single dose of sofosbuvir given before and 14 days after the antiretroviral agent(s), the pharmacokinetics of the antiretroviral compounds and sofosbuvir and its uridine metabolite in blood plasma were largely unchanged. Sofosbuvir seemed to decrease raltegravir AUC by 27% and increase tenofovir Cmax by 25% (AUC was unchanged), whereas sofosbuvir increased ritonavir-boosted darunavir AUC by 34%.78 The mechanisms and clinical importance (if any) of these interactions are unknown.
Table 2 provides a summary of available interaction data of DAAs and antiretroviral agents. In brief, telaprevir and boceprevir have many interactions with antiretroviral agents that might preclude safe combination. Simeprevir seems to have similar contraindications to telaprevir and boceprevir (that is, no addition of efavirenz or ritonavir-boosted protease inhibitors). The interactions of daclatasvir and faldaprevir with antiretroviral agents seem to be manageable with DAA dose modification. Sofosbuvir has the most benign interaction profile of the DAAs studied with antiretroviral agents to date, with the caveat that data regarding the active form of the drug (that is, the uridine analogue triphosphate) has not been reported.
Table 2.
Direct-acting antiviral and antiretroviral scorecard
| HIV therapy | HCV therapy |
|||||
|---|---|---|---|---|---|---|
| Boceprevir | Telaprevir | Simeprevir | Faldaprevir | Daclatasvir | Sofosbuvir | |
| Atazanavir/ritonavir | × | ✓ | No data | No data | ✓* | No data |
|
| ||||||
| Darunavir/ritonavir | × | × | × | ✓ | No data | ✓ |
|
| ||||||
| Fosamprenavir/ritonavir | No data | × | No data | No data | No data | No data |
|
| ||||||
| Lopinavir/ritonavir | × | × | No data | No data | No data | No data |
|
| ||||||
| Nelfinavir | No data | No data | No data | No data | No data | No data |
|
| ||||||
| Efavirenz | × | ✓* | × | ✓* | ✓* | ✓ |
|
| ||||||
| Rilpivirine | ✓ | ? | ✓ | No data | No data | ✓ |
|
| ||||||
| Etravirine | ? | ✓ | No data | No data | No data | No data |
|
| ||||||
| Raltegravir | ✓ | ✓ | ✓ | No data | No data | ✓ |
|
| ||||||
| Elvitegravir/cobicistat | No data | No data | No data | No data | No data | No data |
|
| ||||||
| Maraviroc | ✓* | ✓* | No data | No data | No data | No data |
× indicates the presence of an interaction, ✓ indicates the absence of a clinically important interaction, ✓* indicates that the combination is acceptable, but requires dose adjustment (see main text), ? indicates the presence of an interaction with uncertain clinical importance, ‘No data’ indicates no interaction data are currently available with the combination.
Oral contraceptives
Ribavirin is teratogenic. Thus, prevention of pregnancy is of paramount importance during ribavirin-based HCV treatment. Boceprevir and telaprevir reduce ethinyl oestradiol AUC by 26% and 28%, respectively.79,80 Boceprevir and telaprevir reduce norethindrone AUC by 4% and 11%, respectively.79,80 With telaprevir, the reductions in oral contraceptive exposures affected serum gonadotropin concentrations, suggesting loss of contraceptive efficacy.80 However, this phenomenon was not observed for boceprevir.79 Despite the lack of effect of boceprevir on norethindrone pharmacokinetics, drosperinone AUC is doubled by boceprevir.66 Thus, this progestin should be avoided due to the potential for hyperkalaemia and increased likelihood of progestin-related adverse effects. Ethinyl oestradiol and norethindrone were increased by 12% and 15%, respectively, by simeprevir.81 Daclatasvir did not alter the concentrations of ethinyl oestradiol or norgestimate.44 Faldaprevir increases ethinyl oestradiol and levonorgestrel AUC by 40%.42 Thus, oral contraceptive efficacy might be compromised with boceprevir and telaprevir, but not with simeprevir, faldaprevir, or daclatasvir. Data for sofosbuvir are not yet available.
Phosphodiesterase inhibitors
No formal drug interaction studies have been undertaken with DAAs and phosphodiesterase inhibitors, but the HIV protease inhibitor ritonavir has been shown to markedly increase exposures to phosphodiesterase inhibitors. Sildenafil AUC is increased 11-fold with ritonavir 500 mg twice daily,82 vardenafil AUC is increased 49-fold with ritonavir 600 mg twice daily83 and tadalafil AUC is increased 2.2-fold with ritonavir 200 mg twice daily.84 On the basis of these interactions, phosphodiesterase inhibitors when used for pulmonary arterial hypertension, should not be used with boceprevir and telaprevir. When used for erectile dysfunction, phosphodiesterase inhibitor doses and dosing frequencies should not exceed the following with the HCV protease inhibitor owing to the theoretical potential for increased exposures: sildenafil 25 mg every 48 h, vardenafil 2.5 mg every 24 h, and tadalafil 10 mg every 72 h.
Corticosteroids
Systemic steroids can be used in patients post-transplantation or in those with autoimmune hepatitis. Systemic steroids have not been studied with telaprevir. With boceprevir, prednisone and prednisolone AUC were increased by 22% and 37%, respectively, after a single 40 mg dose of oral prednisone. Thus, dose adjustments of systemic prednisone are probably unnecessary with boceprevir.85 Inhaled and intranasal corticosteroid use has been associated with secondary adrenal insufficiency in the setting of HIV protease inhibitors. Due to inhibition of CYP3A, the exposures of exogenous corticosteroids are increased with subsequent inhibition of endogenous cortisol. Fluticasone is the corticosteroid that has been implicated in the majority of adrenal insufficiency reports in HIV-infected persons on HIV protease inhibitors. This agent, along with budesonide,86 should be used with caution in the setting of DAAs that inhibit CYP3A. Inhaled or intranasal beclomethasone and flunisolide are possible alternatives,87 although both agents require investigation in the setting of DAA treatment of HCV.
Opioids and opioid replacement
Oxycodone, tramadol and fentanyl are primarily metabolized by CYP3A,88 and thus might require dose reduction when used with boceprevir or telaprevir. Other opioids have a reduced potential for interaction with boceprevir, telaprevir, simeprevir, faldaprevir, daclatasvir and sofosbuvir. Hydrocodone and codeine are metabolized by CYP2D6.88 Morphine, hydromorphone and oxymorphone are glucuronidated by uridine glucuronosyltransferase 2B7.88 Individuals with a history of substance abuse might be receiving opioid replacements. Methadone and buprenorphine do not inhibit or induce CYP enzymes, but their pharmacokinetics and pharmacodynamics can be affected by drugs that do affect CYP enzymes.89,90 Telaprevir displaces methadone from its plasma protein binding sites, which causes a reduction in total drug concentrations, but concentrations of the unbound (free) form of the drug are unchanged. Thus, a methadone dose adjustment is probably unnecessary with the addition of telaprevir.91 Telaprevir has no effect on buprenorphine pharmacokinetics.92 Boceprevir reduces R-methadone AUC by 15% and increases buprenorphine AUC by 19%.93 These changes are small and unlikely to require opioid replacement dose adjustment. Methadone pharmacokinetics are unaffected by simeprevir94 and sofosbuvir.95
Foods, dietary and herbal supplements
Grapefruit juice has been implicated in several clinically important drug interactions, including with tacrolimus and ciclosporin.96 Two constituents of grapefruit juice, the furanocoumarins and flavonoids, have been associated with inhibition of intestinal CYP3A and inhibition of drug transporters, respectively.96 As boceprevir, telaprevir and some investigational DAA are substrates for CYP3A, P-gp and organic anionic transporting polypeptides, there is a theoretical potential for interactions with grapefruit juice. The likelihood and magnitude of an interaction with DAA would depend on several factors, including bioavailability of the DAA, the intrinsic level of expression of CYP3A4 or transporters in the gut, and the quantity and properties of the juice consumed. In the absence of formal interaction studies with DAA, a conservative approach would be to avoid consumption of grapefruit juice during DAA treatment.
Use of herbal supplements is common in patients with HCV.97 Preliminary results of a survey presented in 2012 revealed that 64% of drug interactions identified in patients on telaprevir or boceprevir were with herbal supplements.98 Unfortunately, no formal studies of the pharmacokinetics of DAAs when used in combination with herbal supplements have been undertaken. Top-selling herbal supplements include black cohosh, cranberry, Echinacea, garlic, Ginkgo biloba, ginseng, saw palmetto, silymarin (milk thistle), soy and St John’s wort.99 In vitro, Echinacea, garlic, ginkgo biloba, ginseng, silymarin, and St John's wort have been shown to inhibit or induce enzymes or transporters involved in the metabolism or disposition of boceprevir and telaprevir.99 In vivo, Echinacea100,101 and silymarin102,103 do not substantially alter HIV protease inhibitor exposures, but garlic and ginseng reduce CYP3A substrates by 44–51%.104,105 Reductions of a similar magnitude could result in subtherapeutic boceprevir or telaprevir exposures, thus use of garlic and ginseng supplements should be avoided. The potential influence of Ginkgo biloba on boceprevir or telaprevir is unclear. Midazolam AUC is reduced, but ritonavir-boosted lopinavir is unchanged with Ginkgo biloba co-administration.106 In the absence of data for DAAs, this supplement should be used with caution. St John's wort is a potent inducer of enzymes and transporters, which has caused therapeutic failure of many drugs.107 It is therefore contraindicated with boceprevir and telaprevir and should be avoided during antiviral treatment of HCV regardless of the specific DAA used.
Conclusions
Boceprevir and telaprevir represent major advances in the treatment of HCV, but they are unfortunately involved in a number of clinically important drug interactions; they are susceptible to the effects of potent inhibitors and inducers, but also capable of altering the pharmacokinetics of other drugs. The investigational DAAs simeprevir, faldaprevir and daclatasvir are CYP3A substrates and therefore might be altered by potent inhibitors and inducers, but they seem less likely to act as culprits in interactions. Sofosbuvir seems to have a very low potential for drug interactions. Health-care providers must be vigilant about identifying and managing interactions with DAA to ensure therapeutic success.
Key points.
■ Direct-acting antiviral agents (DAAs) represent a major advance in the treatment of chronic HCV infection
■ Boceprevir and telaprevir, the first DAAs to receive regulatory approval, are involved in many clinically important drug–drug interactions
■ Providers must proactively screen for potential drug–drug interactions with boceprevir and telaprevir before and during treatment and adjust therapies as needed
■ Many investigational DAAs have fewer, but are not devoid of, drug–drug interactions
Review criteria.
A PubMed search was performed with the following search terms: “SCH503034”, “boceprevir”, “VX-950”, “telaprevir”, “TMC435”, “simeprevir”, “BI 201,335”, “faldaprevir”, “BMS-790,052”, “daclatasvir”, “GS-7,977”, “PSI-7,977” and “sofosbuvir”. English-language articles containing information related to pharmacokinetics, pharmacology, and/or drug interaction potential were reviewed. Conference abstracts were also searched from the International Workshop on Clinical Pharmacology of Hepatitis Therapy, Annual Meetings of the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver, and Conference on Retroviruses and Opportunistic Infections 2008–2012.
Acknowledgements
J. J. Kiser acknowledges financial support from NIH grant R03 DK096121.
Footnotes
Competing interests
J. J. Kiser declares associations with the following companies: Bristol-Myers Squibb, Janssen, Merck, Vertex. J. R. Burton Jr declares associations with the following companies: Abbvie, Gilead, Vertex. G. T. Everson declares associations with the following companies: Abbott, Amgen, Biotest, Bristol-Myers Squibb, Centocor, Eisai, Gilead, GlaxoSmithKline, GlobeImmune, HepQuant LLC, Janssen-Tibotec, Kadmon, Medtronic, Merck, Novartis, Ortho Biotech, Pfizer, Pharmassett, Roche-Genentech, Schering-Plough, Source, Vertex. See the article online for full details of the relationships.
Author contributions
All authors researched data for the article, wrote the article, made substantial contributions to discussion of content, and reviewed/edited the manuscript before submission.
Contributor Information
Jennifer J. Kiser, Department of Pharmaceutical Sciences, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado, 12850 East Montview Boulevard, V20-C238
James R. Burton, Jr, Hepatology and Transplant Centre, University of Colorado, 1635 Aurora Court, B154, Aurora, CO 80045, USA.
Gregory T. Everson, Hepatology and Transplant Centre, University of Colorado, 1635 Aurora Court, B154, Aurora, CO 80045, USA
References
- 1.WHO. Hepatitis C Fact Sheet. WHO [online] 2013 http://www.who.int/mediacentre/factsheets/fs164/en/
- 2.Tang H, Grise H. Cellular and molecular biology of HCV infection and hepatitis. Clin. Sci. (Lond.) 2012;117:49–65. doi: 10.1042/CS20080631. [DOI] [PubMed] [Google Scholar]
- 3.Zein NN, et al. Hepatitis C virus genotypes in the United States: epidemiology, pathogenicity, and response to interferon therapy. Collaborative Study Group. Ann. Intern. Med. 1996;125:634–639. doi: 10.7326/0003-4819-125-8-199610150-00002. [DOI] [PubMed] [Google Scholar]
- 4.Bacon BR, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011;364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson IM, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 2011;364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 6.Poordad F, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeuzem S, et al. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 2011;364:2417–2428. doi: 10.1056/NEJMoa1013086. [DOI] [PubMed] [Google Scholar]
- 8.Bronowicki JP, et al. Sustained virologic response (SVR) in prior peginterferon/ribavirin (PR) treatment failures after retreatment with boceprevir (BOC) + PR: the PROVIDE study interim results [abstract 11] J. Hepatol. 2012;56(Suppl. 2):S6. [Google Scholar]
- 9.Sherman KE, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N. Engl. J. Med. 2011;365:1014–1024. doi: 10.1056/NEJMoa1014463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merck. Victrelis© (boceprevir) prescribing information. Merck [online] 2013 http://www.merck.com/product/usa/pi_circulars/v/victrelis/victrelis_pi.pdf.
- 11.Vertex. Incivek™ (telaprevir) prescribing information. Vertex [online] 2013 http://pi.vrtx.com/files/uspi_telaprevir.pdf.
- 12.Fried MW, et al. TMC435 in combination with peginterferon and ribavirin in treatment naive hcv genotype 1 patients: final analysis of the pillar phase IIb study [abstract LB-5] Hepatology. 2011;54(Suppl. 1):1429A. [Google Scholar]
- 13.Hezode C, et al. BMS-790052, a NS5A replication complex inhibitor, combined with peginterferon alfa-2a and ribavirin in treatmentnaive HCV genotype 1 or 4 patients: phase 2b AI444010 study interim week 12 results [abstract 227] Hepatology. 2011;54(Suppl. 1):474A. [Google Scholar]
- 14.Sulkowski MS, et al. Faldaprevir combined with peginterferon alfa-2a and ribavirin in treatmentnaive patients with chronic genotype-1 HCV: SILEN-C1 trial. Hepatology. 2013;57:2143–2154. doi: 10.1002/hep.26276. [DOI] [PubMed] [Google Scholar]
- 15.Kowdley KV, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatmentnaive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet. 2013;381:2100–2107. doi: 10.1016/S0140-6736(13)60247-0. [DOI] [PubMed] [Google Scholar]
- 16.Sulkowski MS, et al. All-ORAL Combination of DCV + SOF ± RBV in treatment-naïve patients with HCV GT 1, 2 or 3 [abstract LB2]; Presented at the 63rd Annual Meeting of the American Association for the Study of Liver Diseases.2012. [Google Scholar]
- 17.Everson GT, et al. An interferon-free, ribavirin-free 12-week regimen of daclatasvir (DCV), asunaprevir (ASV), and BMS-791325 yielded SVR4 of 94% in treatment-naïve patients with genotype (GT) 1 chronic hepatitis C virus (HCV) infection [abstract LB3]; Presented at the 63rd Annual Meeting of the American Association for the Study of Liver Diseases.2012. [Google Scholar]
- 18.Kowdley KV, et al. A 12-week interferon-free treatment regimen with ABT-450/r, ABT-267, ABT-333 and ribavirin achieves SVR12 rates (OBSERVED Data) of 99% in treatment-naïve patients and 93% in prior null responders with HCV genotype 1 infection [abstract LB1]; Presented at the 63rd Annual Meeting of the American Association for the Study of Liver Diseases.2012. [Google Scholar]
- 19.Lok AS, et al. Sustained virologic response in chronic HCV genotype (GT) 1-infected null responders with combination of daclatasvir (DCV; NS5A Inhibitor) and asunaprevir (ASV; NS3 Inhibitor) with or without peginterferon alfa-2a/ribavirin (PEG/RBV) [abstract 79] Hepatology. 2012;56(Suppl. 6):230A. [Google Scholar]
- 20.Feld JJ, et al. Up to 100% SVR4 rates with ritonavir-boosted danoprevir (DNVr), mericitabine (MCB), and ribavirin (R) ± peginterferon alfa-2a (40KD) (P) in HCV genotype 1-infected partial and null responders: results from the MATTERHORN study [abstract 81] Hepatology. 2012;56(Suppl. 6):231A. [Google Scholar]
- 21.Thompson AJ, et al. Six weeks of a NS5A Inhibitor (GS-5885), protease inhibitor (GS-9451) plus peginterferon/ribavirin (PR) achieves high SVR4 rates in genotype 1 IL28B CC treatment naive HCV patients: interim results of a prospective, randomized trial [abstract 759] Hepatology. 2012;56(Suppl. 6):556A. [Google Scholar]
- 22.Flockhart DA, Tanus-Santos JE. Implications of cytochrome P450 interactions when prescribing medication for hypertension. Arch. Intern. Med. 2002;162:405–412. doi: 10.1001/archinte.162.4.405. [DOI] [PubMed] [Google Scholar]
- 23.Burger D, et al. Clinical management of drug–drug interactions in HCV therapy: challenges and solutions. J. Hepatol. 2013;58:792–800. doi: 10.1016/j.jhep.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 24.Herold C, et al. Quantitative testing of liver function in relation to fibrosis in patients with chronic hepatitis B and C. Liver. 2001;21:260–265. doi: 10.1034/j.1600-0676.2001.021004260.x. [DOI] [PubMed] [Google Scholar]
- 25.Frye RF, et al. Liver disease selectively modulates cytochrome P450-mediated metabolism. Clin. Pharmacol. Ther. 2006;80:235–245. doi: 10.1016/j.clpt.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Helmke SM, DeSanto J, Herman A, Lauriski S, Everson GT. Alteration of the portal circulation across the entire spectrum of fibrosis in patients with chronic Hepatitis C as measured by dual cholate clearances [abstract 1005] Hepatology. 2012;56(Suppl.1):678A. [Google Scholar]
- 27.Ciesek S, von Hahn T, Manns MP. Secondwave protease inhibitors: choosing an heir. Clin. Liver Dis. 2011;15:597–609. doi: 10.1016/j.cld.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 28.Hull MW, Montaner JS. Ritonavir-boosted protease inhibitors in HIV therapy. Ann. Med. 2011;43:375–388. doi: 10.3109/07853890.2011.572905. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch M, Papatheodoridis GV. Danoprevir, a small-molecule NS3/4A protease inhibitor for the potential oral treatment of HCV infection. Curr. Opin. Investig. Drugs. 2010;11:951–963. [PubMed] [Google Scholar]
- 30.Lawitz E, et al. A phase 2a trial of 12-week interferon-free therapy with two direct-acting antivirals (ABT-450/r, ABT-072) and ribavirin in IL28B C/C patients with chronic hepatitis C genotype 1. J. Hepatol. 2013;59:18–23. doi: 10.1016/j.jhep.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 31.de Kanter CT, et al. Lack of a clinically significant drug-drug interaction in healthy volunteers between the HCV protease inhibitor boceprevir and the proton pump inhibitor omeprazole. J. Antimicrob. Chemother. 2013;68:1415–1422. doi: 10.1093/jac/dkt032. [DOI] [PubMed] [Google Scholar]
- 32.Merck. Boceprevir Clinical Pharmacology and Biopharmaceutics Review FDA [online] 2013 http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202258Orig1s000ClinPharmR.pdf.
- 33.Treitel M, et al. Single-dose pharmacokinetics of boceprevir in subjects with impaired hepatic or renal function. Clin. Pharmacokinet. 2012;51:619–628. doi: 10.1007/BF03261935. [DOI] [PubMed] [Google Scholar]
- 34.Buti M, et al. OPTIMIZE Trial: Noninferiority of twice-daily telaprevir versus administration every 8 hours in treatment-naive, genotype 1 HCV-infected patients [abstract LB-8]; Presented at the 63rd Annual Meeting of the American Association for the Study of Liver Diseases.2012. [Google Scholar]
- 35.Kunze A, Huwyler J, Camenisch G, Gutmann H. Interaction of the antiviral drug telaprevir with renal and hepatic drug transporters. Biochem. Pharmacol. 2012;84:1096–1102. doi: 10.1016/j.bcp.2012.07.032. [DOI] [PubMed] [Google Scholar]
- 36.Adiwijaya B, et al. Effect of mild and moderate hepatic impairment on telaprevir pharmacokinetics [abstract PK1] Reviews in Antiviral Therapy & Infectious Diseases. 2011;6:3. [Google Scholar]
- 37.Sekar V, et al. TMC435 and Drug Interactions: Evaluation of metabolic interactions for TMC435 via cytochrome P450 (CYP) enzymes in healthy volunteers [abstract 1076] J. Hepatol. 2010;52(Suppl.1):S416. [Google Scholar]
- 38.Huisman MT, et al. In vitro studies investigating the mechanism of interaction between TMC435 and hepatic transporters [abstract 278] Hepatology. 2010;52(Suppl. 1):461A. [Google Scholar]
- 39.Sekar V, et al. Pharmacokinetics of TMC435 in subjects with moderate hepatic impairment [abstract 472] J. Hepatol. 2011;54(Suppl.1):S193. [Google Scholar]
- 40.Sabo JP, et al. Cytochrome P450 (CYP) interactions with the HCV protease inhibitor faldaprevir (BI 201335) in healthy volunteers [abstract A-1248]; Presented at the 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy.2012. [Google Scholar]
- 41.Sane R, et al. Mechanism of isolated unconjugated hyperbilirubinemia induced by the HCV NS3/4A protease inhibitor BI 201335 [abstract 1236] J. Hepatol. 2011;54(Suppl.1):S488. [Google Scholar]
- 42.Kort J. Clinical pharmacology update on the direct acting antivirals faldaprevir and BI 207127; Presented at the 14th International Workshop on Clinical Pharmacology of HIV Therapy.2013. [Google Scholar]
- 43.Gish RG, Meanwell NA. The NS5A replication complex inhibitors: difference makers? Clin. Liver Dis. 2011;15:627–639. doi: 10.1016/j.cld.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 44.Bifano M, et al. BMS-790052 has no clinically significant effect on the pharmacokinetics ofa combined oral contraceptive containing ethinyl estradiol and norgestimate in healthy female subjects [abstract 1340] Hepatology. 2011;54(Suppl. 1):991A–992A. [Google Scholar]
- 45.Nettles RE, et al. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology. 2011;54:1956–1965. doi: 10.1002/hep.24609. [DOI] [PubMed] [Google Scholar]
- 46.Bertz R. Bristol Myers Squibb HCV full development portfolio overview; Presented at the 14th International Workshop on Clinical Pharmacology of HIV Therapy.2013. [Google Scholar]
- 47.Bifano M, et al. Single-dose pharmacokinetics of daclatasvir in subjects with hepatic impairment compared with healthy subjects [abstract 1362] Hepatology. 2011;54(Suppl. 1):1004A. [Google Scholar]
- 48.Powdrill MH, Bernatchez JA, Gotte M. Inhibitors of the hepatitis C virus RNA-dependent RNA polymerase NS5B. Viruses. 2010;2:2169–2195. doi: 10.3390/v2102169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathias A. Clinical Pharmacology of DAAs for HCV: what’s new and what’s in the pipeline; Presented at the 14th International Workshop on Clinical Pharmacology of HIV Therapy.2013. [Google Scholar]
- 50.Cornpropst M, et al. The effect of renal impairment and end stage renal disease on the single-dose pharmacokinetics of GS-7977 [abstract 1101] J. Hepatol. 2012;56(Suppl.2):S433. [Google Scholar]
- 51.Lawitz E, et al. The effect of hepatic impairment on the safety, pharmacokinetics, and antiviral activity of GS-7977 in hepatitis C infected subjects treated for seven days [abstract 1130] J. Hepatol. 2012;56(Suppl.1):S445–S446. [Google Scholar]
- 52.Beumont-Mauviel M, et al. The pharmacokinetic interaction between the investigational HCV NS3/4A protease inhibitor TMC435 and escitalopram [abstract 1353] Hepatology. 2011;54(Suppl. 1):1000A. [Google Scholar]
- 53.Kiser JJ, Burton JR, Anderson PL, Everson GT. Review and management of drug interactions with boceprevir and telaprevir. Hepatology. 2012;55:1620–1628. doi: 10.1002/hep.25653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hulskotte E, et al. Pharmacokinetic interaction between the hepatitis C virus protease inhibitor boceprevir and cyclosporine and tacrolimus in healthy volunteers. Hepatology. 2012;56:1622–1630. doi: 10.1002/hep.25831. [DOI] [PubMed] [Google Scholar]
- 55.Garg V, et al. Effect of telaprevir on the pharmacokinetics of cyclosporine and tacrolimus. Hepatology. 2011;54:20–27. doi: 10.1002/hep.24443. [DOI] [PubMed] [Google Scholar]
- 56.Ouwerkerk-Mahadevan S, Simion A, Mortier S, Peeters M, Beumont-Mauviel M. No clinically significant interaction between the investigational HCV protease inhibitor TMC435 and the immunosuppressives cyclosporine and tacrolimus [abstract 80] Hepatology. 2012;56(Suppl.1):213A. [Google Scholar]
- 57.Kugelmas M, et al. Hepatitis C virus therapy, hepatocyte drug metabolism, and risk for acute cellular rejection. Liver Transpl. 2003;9:1159–1165. doi: 10.1053/jlts.2003.50233. [DOI] [PubMed] [Google Scholar]
- 58.Mathias A, Cornpropst M, Clemons D, Denning J, Symonds W. No clinically significant pharmacokinetic drug-drug interactions between sofosbuvir (GS-7977) and the immunosuppressants, cyclosporine a or tacrolimus in healthy volunteers [abstract 1869] Hepatology. 2012;56Suppl.1:1063A–1064A. [Google Scholar]
- 59.O’Leary JG, McKenna GJ, Klintmalm GB, Davis GL. Effect of telaprevir on the pharmacokinetics of sirolimus in Liver transplant recipients. Liver Transpl. 2013;19:463–465. doi: 10.1002/lt.23623. [DOI] [PubMed] [Google Scholar]
- 60.Burton JR, et al. A multicenter study of protease inhibitor-triple therapy in HCV-infected liver transplant recipients: report from the CRUSH-C Group [abstract 211] Hepatology. 2012;56(Suppl.1) [Google Scholar]
- 61.Coilly A, et al. Efficacy and safety of protease inhibitors for hepatitis C recurrence after liver transplantation: a first multicentric experience [abstract 9] Hepatology. 2012;56(Suppl.1):194A–195A. [Google Scholar]
- 62.Werner CR, et al. Telaprevir-based triple therapy in liver transplant patients with hepatitis C virus: a 12-week pilot study providing safety and efficacy data. Liver Transpl. 2012;18:1464–1470. doi: 10.1002/lt.23542. [DOI] [PubMed] [Google Scholar]
- 63.>Burton JR, Jr, Everson GT. Management of the transplant recipient with chronic hepatitis C. Clin. Liver Dis. 2013;17:73–91. doi: 10.1016/j.cld.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 64.Cantarovich M, et al. Comparison of neoral dose monitoring with cyclosporine through levels versus 2-hr postdose levels in stable Liver transplant patients. Transplantation. 1998;66:1621–1627. doi: 10.1097/00007890-199812270-00009. [DOI] [PubMed] [Google Scholar]
- 65.Sulkowski MS. Viral hepatitis and HIV coinfection. J. Hepatol. 2008;48:353–367. doi: 10.1016/j.jhep.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 66.Kasserra C, Hughes E, Treitel M, Gupta S, O’Mara E. Clinical pharmacology of boceprevir: metabolism, excretion, and drug–drug interactions [abstract 118]; Presented at the 18th Conference on Retroviruses and Opportunistic Infections.2011. [Google Scholar]
- 67.van Heeswijk RP, Beumont M, Kauffman R, Garg V. Review of drug interactions with telaprevir and antiretrovirals. Antivir. Ther. doi: 10.3851/IMP2527. http://dx.doi.org/10.3851/IMP2527. [DOI] [PubMed]
- 68.de Kanter CT, Blonk MI, Colbers AP, Schouwenberg BJ, Burger DM. Lack of a clinically significant drug–drug interaction in healthy volunteers between the hepatitis C virus protease inhibitor boceprevir and the HIV integrase inhibitor raltegravir. Clin. Infect. Dis. 2013;56:300–306. doi: 10.1093/cid/cis824. [DOI] [PubMed] [Google Scholar]
- 69.Hulskotte EG, et al. Pharmacokinetic interactions between the hepatitis C virus protease inhibitor boceprevir and ritonavirboosted HIV-1 protease inhibitors atazanavir, darunavir, and lopinavir. Clin. Infect. Dis. 2012;56:718–726. doi: 10.1093/cid/cis968. [DOI] [PubMed] [Google Scholar]
- 70.Thomas DL, et al. Provisional guidance on the use of hepatitis C virus protease inhibitors for treatment of hepatitis C in HIV-infected persons. Clin. Infect. Dis. 2012;54:979–983. doi: 10.1093/cid/cir882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kakuda TN, et al. Pharmacokinetic interaction between etravirine or rilpivirine and telaprevir in healthy volunteers: a randomised, two-way crossover trial [abstract O-18]; Presented at the 13th International Workshop on Clinical Pharmacology of HIV Therapy; 2012. [DOI] [PubMed] [Google Scholar]
- 72.Hammond KP, et al. Pharmacokinetic interaction between boceprevir and etravirine in HIV/HCV seronegative volunteers. J. Acquir. Immune Defic. Syndr. 2013;62:67–73. doi: 10.1097/QAI.0b013e318275da93. [DOI] [PubMed] [Google Scholar]
- 73.Rhee EG, et al. Absence of a significant pharmacokinetic interaction between the HCV protease inhibitor boceprevir and HIV-1 NNRTI rilpivirine [abstract 537]; Presented at the 20th Conference on Retroviruses and Opportunistic Infections.2013. [Google Scholar]
- 74.Vourvahis M, Plotka A, Kantaridis C, Fang A, Heera J. The effect of boceprevir or telaprevir on the pharmacokinetics of maraviroc: an open-label, fixed sequence study in healthy volunteers [abstract O-17]; Presented at the 14th International Workshop on Clinical Pharmacology of HIV Therapy.2013. [Google Scholar]
- 75.Sabo JP, et al. Pharmacokinetic interactions of darunavir/ritonavir, efavirenz, and tenofovir with the HCV protease inhibitor faldaprevir in healthy volunteers [abstract 35]; Presented at the 20th Conference on Retroviruses and Opportunistic Infections.2013. [Google Scholar]
- 76.Bifano M, et al. Assessment of HIV antiretroviral drug interactions with the HCV NS5A replication complex inhibitor daclatasvir demonstrates a PK profile which supports coadministration with tenofovir, efavirez, and atazanavir/r [abstract 61B]; Presented at the 19th Conference on Retroviruses and Opportunistic Infections.2012. [Google Scholar]
- 77.Ouwerkerk-Mahadevan S, Sekar V, Simion A, Peeters M, Beumont-Mauviel M. The pharmacokinetic interactions of the HCV protease inhibitor simeprevir (TMC435) with HIV antiretroviral agents in healthy volunteers [abstract 36620]; Presented at Infectious Diseases Week.2012. [Google Scholar]
- 78.Kirby B, et al. No clinically significant pharmacokinetic drug interactions between sofosbuvir (GS-7977) and HIV antiretroviral atripla, rilpivirine, darunavir/ritonavir, or raltegravir in healthy volunteers [abstract 1877] Hepatology. 2012;56(Suppl. 1):1067A. [Google Scholar]
- 79.Lin WH, et al. Pharmacokinetic interaction between the HCV protease inhibitor boceprevir and ethinyl estradiol/norethindrone [abstract 1901] Hepatology. 2012;56(Suppl. 1):1078A–1079A. [Google Scholar]
- 80.Garg V, et al. The pharmacokinetic interaction between an oral contraceptive containing ethinyl estradiol and norethindrone and the HCV protease inhibitor telaprevir. J. Clin. Pharmacol. 2012;52:1574–1583. doi: 10.1177/0091270011419855. [DOI] [PubMed] [Google Scholar]
- 81.Ouwerkerk-Mahadevan S, Simion A, Spittaels K, Peeters M, Beumont-Mauviel M. No pharmacokinetic interaction between the investigational HCV protease inhibitor simeprevir (TMC435) and an oral contraceptive containing ethinylestradiol and norethindrone [abstract 773] Hepatology. 2012;56(Suppl. 1):565A–566A. [Google Scholar]
- 82.Pfizer. Viagra® (sildenafil citrate) prescribing information Pfizer [online] 2013 http://www.pfizer.com/files/products/uspi_viagra.pdf.
- 83.GlaxosSmithKline. Levitra (vardenafil hydrochloride) prescribing information GlaxoSmithKline [online] 2013 http://www.levitra.com/assets/pdf/PI.pdf.
- 84.Lilly Cialis (tadalafil) prescribing information. Lilly [online] 2013 http://pi.lilly.com/us/cialis-pi.pdf.
- 85.Jumes P, et al. Pharmacokinetic interaction between the HCV protease inhibitor boceprevir and prednisone in healthy volunteers [abstract 1896] Hepatology. 2012;56(Suppl. 1):1076A. [Google Scholar]
- 86.Frankel JK, Packer CD. Cushing’s syndrome due to antiretroviral-budesonide interaction. Ann. Pharmacother. 2011;45:823–824. doi: 10.1345/aph.1P731. [DOI] [PubMed] [Google Scholar]
- 87.Tseng A, Foisy M. Important drug–drug interactions in HIV-infected persons on antiretroviral therapy: an update on new interactions between HIV and non-HIV drugs. Curr. Infect. Dis. Rep. 2012;14:67–82. doi: 10.1007/s11908-011-0229-1. [DOI] [PubMed] [Google Scholar]
- 88.Smith HS. Opioid metabolism. Mayo Clin. Proc. 2009;84:613–624. doi: 10.1016/S0025-6196(11)60750-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gerber JG, Rhodes RJ, Gal J. Stereoselective metabolism of methadone N-demethylation by cytochrome P4502B6 and 2C19. Chirality. 2004;16:36–44. doi: 10.1002/chir.10303. [DOI] [PubMed] [Google Scholar]
- 90.Cone EJ, Gorodetzky CW, Yousefnejad D, Buchwald WF, Johnson RE. The metabolism and excretion of buprenorphine in humans. Drug Metab. Dispos. 1884;12:577–581. [PubMed] [Google Scholar]
- 91.van Heeswijk R, et al. Pharmacokinetic interaction between telaprevir and methadone. Antimicrob. Agents Chemother. 2013;57:2304–2309. doi: 10.1128/AAC.02262-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luo X, Trevejo J, Van Heeswijk R, Garg V. No significant effect of the HCV protease inhibitor telaprevir on the pharmacokinetics and pharmacodynamics of buprenorphine in HCV-negative volunteers [abstract 132]; Presented at HepDART Koloa, Hawaii.2011. [Google Scholar]
- 93.Hulskotte EGJ, et al. Pharmacokinetic interaction between HCV protease inhibitor boceprevir and methadone or buprenorphine in subjects on stable maintenance therapy [abstract 771LB]; Presented at the 7th International Workshop on Clinical Pharmacology of Hepatitis Therapy; 2012. [DOI] [PubMed] [Google Scholar]
- 94.Beumont-Mauviel M, De Smedt G, Peeters M, Akuma SH, Sekar V. The pharmacokinetic interaction between the investigational NS3–4A HCV protease inhibitor TMC435 and methadone [abstract 1353] Hepatology. 2011;54(Suppl.1):1000A. [Google Scholar]
- 95.Denning J, et al. Lack of effect of the nucleotide analog polymerase inhibitor PSI-7977 on methadone pharmacokinetics and pharmacodynamics [abstract 372] Hepatology. 2011;54(Suppl.1):544A. [Google Scholar]
- 96.Pirmohamed M. Drug–grapefruit juice interactions: two mechanisms are clear but individual responses vary. BMJ. 2013;346:f1. doi: 10.1136/bmj.f1. [DOI] [PubMed] [Google Scholar]
- 97.Seeff LB, et al. Herbal product use by persons enrolled in the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) Trial. Hepatology. 2008;47:605–612. doi: 10.1002/hep.22044. [DOI] [PubMed] [Google Scholar]
- 98.Kipp G, Mohammed R, Lin A, Johnson H. Evaluation of pharmacist identified and mitigated drug–drug interactions in hepatitis C virus infected patients starting telaprevir or boceprevir [abstract 1741] Hepatology. 2012;56(Suppl.1) [Google Scholar]
- 99.van den Bout-van den Beukel CJ, Koopmans PP, van der Ven AJ, De Smet PA, Burger DM. Possible drug-metabolism interactions of medicinal herbs with antiretroviral agents. Drug Metab. Rev. 2006;38:477–514. doi: 10.1080/03602530600754065. [DOI] [PubMed] [Google Scholar]
- 100.Penzak SR, et al. Echinacea purpurea significantly induces cytochrome P450 3A activity but does not alter lopinavir–ritonavir exposure in healthy subjects. Pharmacotherapy. 2010;30:797–805. doi: 10.1592/phco.30.8.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Molto J, et al. Herb–drug interaction between Echinacea purpurea and darunavir–ritonavir in HIV-infected patients. Antimicrob. Agents Chemother. 2011;55:326–330. doi: 10.1128/AAC.01082-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Piscitelli SC, et al. Effect of milk thistle on the pharmacokinetics of indinavir in healthy volunteers. Pharmacotherapy. 2002;22:551–556. doi: 10.1592/phco.22.8.551.33205. [DOI] [PubMed] [Google Scholar]
- 103.Molto J, et al. Effect of milk thistle on the pharmacokinetics of darunavir–ritonavir in HIVinfected patients. Antimicrob. Agents Chemother. 2012;56:2837–2841. doi: 10.1128/AAC.00025-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Piscitelli SC, Burstein AH, Welden N, Gallicano KD, Falloon J. The effect of garlic supplements on the pharmacokinetics of saquinavir. Clin. Infect. Dis. 2002;34:234–238. doi: 10.1086/324351. [DOI] [PubMed] [Google Scholar]
- 105.Malati CY, et al. Influence of Panax ginseng on cytochrome P450 (CYP)3A and P-glycoprotein (P-gp) activity in healthy participants. J. Clin. Pharmacol. 2012;52:932–939. doi: 10.1177/0091270011407194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robertson SM. Effect of ginkgo biloba extract on lopinavir, midazolam and fexofenadine pharmacokinetics in healthy subjects. Curr. Med. Res. Opin. 2008;24:591–599. doi: 10.1185/030079908x260871. [DOI] [PubMed] [Google Scholar]
- 107.Henderson L, Yue QY, Bergquist C, Gerden B, Arlett P. St John’s wort (Hypericum perforatum): drug interactions and clinical outcomes. Br. J. Clin. Pharmacol. 2002;54:349–356. doi: 10.1046/j.1365-2125.2002.01683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bernstein B, et al. Pharmacokinetics, safety and tolerability of the HCV protease inhibitor ABT-450 with ritonavir following multiple ascending doses in healthy adult volunteers [abstract 58]; Presented at HepDART Kohala Coast, Hawaii.2009. [Google Scholar]
- 109.Menon R, et al. Pharmacokinetics, safety and tolerability following multiple dosing of polymerase inhibitor, ABT-333 and protease inhibitor, ABT-450 with ritonavir [abstract O-13]; Presented at the 7th International Workshop on Clinical Pharmacology of Hepatitis Therapy.2012. [Google Scholar]
- 110.Eley T, et al. Evaluation of drug interaction potential of the HCV protease inhibitor asunaprevir (ASV; BMS-650032) at 200 mg twice daily (BID) in metabolic cocktail and p-glycoprotein (P-gp) probe studies in health volunteers [abstract 381] Hepatology. 2011;54(Suppl.1):548A. [Google Scholar]
- 111.Eley T, et al. In vivo and in vitro assessment of asunaprevir (ASV; BMS-650032) as an inhibitor and substrate of organic anion transport polypeptide (OATP) transporters in healthy volunteers [abstract O-04]; Presented at the 7th International Workshop on Clinical Pharmacology of Hepatitis Therapy.2012. [Google Scholar]
- 112.Jumes P, et al. Pharmacokinetic interaction between the HCV protease inhibitor boceprevir and digoxin in healthy adult volunteers [abstract O-05]; Presented at the 7th International Workshop on Clinical Pharmacology of Hepatitis Therapy.2012. [Google Scholar]
- 113.Marcos PN, et al. Danoprevir.
- 114.Dumas E, et al. Pharmacokinetics, safety and tolerability of the HCV NS5A inhibitor ABT-267 following single and multiple doses in healthy adult volunteers [abstract 1204] J. Hepatol. 2011;54:S475. [Google Scholar]
- 115.Moreira S, et al. The effect of mild to moderate renal impairment on the pharmacokinetics (PK) of the hepatitis C virus (HCV) polymerase inhibitor mericitabine (MCB, RG7128) [abstract 358] Hepatology. 2011;54(Suppl.1):537A–538A. [Google Scholar]
- 116.Ma H, et al. Characterization of the metabolic activation of hepatitis C virus nucleoside inhibitor beta-D-2'-Deoxy-2'-fluoro-2’Cmethylcytidine (PSI-6130) and identification of a novel active 5'-triphosphate species. J. Biol. Chem. 2007;282:29812–29820. doi: 10.1074/jbc.M705274200. [DOI] [PubMed] [Google Scholar]
- 117.Maring C, et al. Preclinical potency, pharmacokinetic and ADME characterization of ABT-333, a novel non-nucleoside HCV polymerase inhibitor [abstract 953] J. Hepatol. 2009;50(Suppl.1):S346–S347. [Google Scholar]