

Abstract
Background
Streptococcus agalactiae (S. agalactiae), also known as group B Streptococcus (GBS), is an important pathogen for neonatal pneumonia, meningitis, bovine mastitis, and fish meningoencephalitis. The global outbreaks of Streptococcus disease in tilapia cause huge economic losses and threaten human food hygiene safety as well. To investigate the mechanism of S. agalactiae pathogenesis in tilapia and develop attenuated S. agalactiae vaccine, this study sequenced and comparatively analyzed the whole genomes of virulent wild-type S. agalactiae strain HN016 and its highly-passaged attenuated strain YM001 derived from tilapia.
Methods
We performed Illumina sequencing of DNA prepared from strain HN016 and YM001. Sequencedreads were assembled and nucleotide comparisons, single nucleotide polymorphism (SNP) , indels were analyzed between the draft genomes of HN016 and YM001. Clustered regularly interspaced short palindromic repeats (CRISPRs) and prophage were detected and analyzed in different S. agalactiae strains.
Results
The genome of S. agalactiae YM001 was 2,047,957 bp with a GC content of 35.61 %; it contained 2044 genes and 88 RNAs. Meanwhile, the genome of S. agalactiae HN016 was 2,064,722 bp with a GC content of 35.66 %; it had 2063 genes and 101 RNAs. Comparative genome analysis indicated that compared with HN016, YM001 genome had two significant large deletions, at the sizes of 5832 and 11,116 bp respectively, resulting in the deletion of three rRNA and ten tRNA genes, as well as the deletion and functional damage of ten genes related to metabolism, transport, growth, anti-stress, etc. Besides these two large deletions, other ten deletions and 28 single nucleotide variations (SNVs) were also identified, mainly affecting the metabolism- and growth-related genes.
Conclusions
The genome of attenuated S. agalactiae YM001 showed significant variations, resulting in the deletion of 10 functional genes, compared to the parental pathogenic strain HN016. The deleted and mutated functional genes all encode metabolism- and growth-related proteins, not the known virulence proteins, indicating that the metabolism- and growth-related genes are important for the pathogenesis of S. agalactiae.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2026-y) contains supplementary material, which is available to authorized users.
Keywords: Streptococcus agalactiae, Genetic variation, Virulence attenuation, Sequence analysis, Virulence factors
Background
S. agalactiae, also known as GBS, is a Gram-positive bacterium that not only causes pneumonia and meningitis in neonates, but also induces bovine mastitis and infects reptiles, amphibians, and various fishes [1–3]. With the advances in sequencing technology and the reduction of cost, the genomes of S. agalactiae strains of different hosts and subtypes are revealed gradually. To date, 13 complete genome sequences, 19 draft genome sequences, and 282 contig sequences of S. agalactiae have been made publicly available. Studies showed that the genome of S. agalactiae can be divided into core genome, dispensable genome, and unique genome; the dispensable genome is important for the analysis of virulence differences and the development of broad-spectrum vaccines [4, 5]. Comparative genome analysis between bacterial strains that are greatly different in host specificity or virulence may help to rapidly screen for dispensable genes, gene deletions or mutations, and differentially-expressed proteins; it is also an effective way of studying the mechanisms of cross-host infection, pathogenicity, and immunogenicity of S. agalactiae [6–8].
Pridgeon et al. successfully generated an attenuated S. agalactiae strain 138spar from tilapia-derived S. agalactiae serotype Ib strain 138P in laboratory using a sparfloxacin resistance strategy; comparative genome analysis indicated that S. agalactiae 138spar had 22 deletions larger than 6 bp and 26 SNVs [7, 9]. Although S. agalactiae serotype Ib strain can cause infection and diseases in various fishes and amphibians, there is no report of its pathogenicity to humans, and comparative genome and phylogenetic studies indicate that S. agalactiae serotype Ia and Ib are distantly related [6, 10]. Currently, S. agalactiae Ia is the dominant strain causing infections and deaths in a large number of tilapia in Asia, which is also the important pathogen of early-onset neonatal meningitis [10, 11]. Comparative genome studies have demonstrated that tilapia- and trout-derived S. agalactiae type Ia strains and human-derived strains causing neonatal meningitis have a close genomic relationship [5, 6]. Our laboratory highly passaged the tilapia-derived wild-type strongly-virulent S. agalactiae Ia strain HN016 and obtained the attenuated strain YM001. To study the molecular mechanisms of S. agalactiae pathogenicity, we performed whole-genome sequencing and comparative genome analysis with HN016 and YM001 strains and found that YM001 genome had significant variations compared to HN016; in YM001 genome, there were deletions of multiple genes related to metabolism, transport, and growth. These results are of a great reference value for unraveling the pathogenesis and developing attenuated vaccine of S. agalactiae.
Results and discussion
Whole genome alignment between S. agalactiae HN016 and YM001
The assembling result indicated that the genome size of S. agalactiae YM001 was 2,047,957 bp, with a GC content of 35.61 % (GenBank accession number, CP011326), while the genome size of S. agalactiae HN016 was 2,064,722 bp, with a GC content of 35.66 % (GenBank accession number, CP011325). The similartity between both genomes was 99.69 %. Further analysis indicated that the YM001 genome contained 2044 genes and 88 RNAs, while the genome of HN016 had 2063 genes and 101 RNAs. The genome of YM001 varied significantly compared to HN016; in addition to two large deletions of 5832 and 11,116 bp respectively (see Additional file 1: Table S1; Additional file 2: Table S2), there were another ten small deletions and 28 SNVs (see Additional file 3: Table S3; Additional file 4: Table S4; Table 1).
Table 1.
YM001-specific genetic variations compared to HN016
| YM001 position | HN016 position | Gene product | Biological function | Variation | Effect on YM001 coding |
|---|---|---|---|---|---|
| Carbohydrate metabolism | |||||
| 136826 | 142595 | Fructose-bisphosphate aldolase | Fructose and mannose metabolism | SNV | K259N substitution |
| 780441 | 786056 | Phosphoenolpyruvate carboxylase | Microbial metabolism in diverse environments and Carbon metabolism | SNV | I181I substitution |
| 868738 | 874444 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | Biosynthesis of the bacterial cell wall and is critical for bacterial survival | SNV | H126R substitution |
| 1403613 | 1420411 | glycosyl transferase family 8 | glycosyl synthesis | SNV | T383I substitution |
| 1534895 | 1551880 | metallophosphoesterase | hydrolysis of phosphate | SNV | Y64C substitution |
| Lipid metabolism | |||||
| 1805677 | 1822388 | Phosphatidate cytidylyltransferase | Glycerophospholipid metabolism | SNV | P239L substitution |
| Nucleotide metabolism | |||||
| 83930 | 89762 | DNA-directed RNA polymerase subunit alpha | Primary transcript RNA production and RNA chains construction | SNV | V258A substitution |
| 192012 | 197717 | DNA-directed RNA polymerase subunit beta | Primary transcript RNA production and RNA chains construction | SNV | P360A substitution |
| 1515764 | 1532749 | Thymidylate kinase | dTDP Biosynthesis | SNV | D35D substitution |
| Amino acid metabolism | |||||
| 2033153 | 2049866 | Arginine deiminase | Acid tolerant | SNV | V362I substitution |
| Environmental information processing | |||||
| 1858831 | 1875542 | Sensor histidine kinase | Peptidoglycan metabolism | SNV | R126H substitution |
| Translation | |||||
| 393841 | 399503 | Transcription elongation factor NusA | RNA polymerase-associated protein | SNV | E116G substitution |
| 407665 | 413327 | FUR family transcriptional regulator | Peroxide stress response regulator | SNV | S42I substitution |
| 755561 | 761176 | S1 RNA-binding protein | Post-transcriptional control of RNAs | SNV | R460S substitution |
| 949470 | 955242 | chloramphenicol acetyltransferase | Hexapeptide repeat-containing transferase | SNV | G43Y substitution |
| 1264942 | 1270622 | TetR family transcriptional regulator | Transcriptional regulator | SNV | W122R substitution |
| 1967217 | 1983929 | Arginine repressor ArgR | Transcriptional regulator of arginine metabolism | SNV | 117-aa C-terminal deletion |
| DNA repair and recombination proteins | |||||
| 1022277 | 1028049 | DNA topoisomerase I | Regulation of supercoiling and maintenance of genetic stability | SNV | T644K substitution |
| Transport | |||||
| 253283 | 258943 | amino acid ABC transporter permease | Membrane transport | SNV | G147E substitution |
| 297760..297761 | 303421 | PTS system transporter subunit IIC | Starch and sucrose metabolism | Deletion | 10-aa C-terminal extension,102-aa C-terminal deletion |
| 364407 | 370068 | MarR family transcriptional regulator | Regulate multiple antibiotic resistance and the oxidative stress response | SNV | H106N substitution |
| 970865 | 976637 | Sugar ABC transporter permease | simple sugar transport system permease protein | SNV | T5T substitution |
| 1635394 | 1652379 | Multidrug transporter | Drug efflux proteins | SNV | M126V substitution |
| 1663151 | 1680136 | Glycerol uptake facilitator protein | glycerol-uptake facilitator | SNV | I44I substitution |
| 1761614 | 1778325 | PTS system ascorbate-specific transporter subunit IIC | Microbial metabolism in diverse environments | SNV | D327J substitution |
| 1828308 | 1845019 | PTS system transporter subunit IIB | Galactose metabolism | SNV | M1T substitution |
| 2020460 | 2037173 | Cobalt transporter ATP-binding subunit | energy-coupling factor transport system ATP-binding protein | SNV | E79K substitution |
| Folding, sorting and degradation | |||||
| 743279..743280 | 748941 | Recombinase RecF | Manipulate the structure of genomes | Deletion | 6-aa C-terminal extension,855-aa C-terminal deletion |
| Unknown function | |||||
| 1271532..1271533 | 1277212 | Hypothetical protein | Unknown function | Deletion | Y63N substitution, 5-aa C-terminal extension,81-aa C-terminal deletion |
Analysis of the damages and gene deletions caused by large fragment deletion
As shown in Fig. 1, YM001 genome had two large deletions compared to HN016. The deletion one was a 5832-bp sequence, which contained a repetitive sequence of 5621 bp separated by a 211-bp fragment in the genome of HN016. There were two repetitive sequences in the genome of HN016, whereas only one repetitive fragment left in the genome of YM001. This repetitive sequence contained 5S rRNA, 16S rRNA, and 23S rRNA genes, as well as other ten different tRNA genes (see Additional file 1: Table S1). The deletion two was a 11,116-bp sequence, which resulted in a truncation of two genes and deletion of 8 genes (see Additional file 2: Table S2); they were four genes of the ABC transporter family, MarR family transcriptional regulator, Ser/Thr protein phosphatase (STP), peptide deformylase, glutamate dehydrogenase, membrane protein of unknown function, and acetyltransferase.
Fig. 1.

Whole genome alignment between S. agalactiae HN016 and YM001. The genomes of HN016 and YM001 were compared with each other using progressive MAUVE with default parameters. The colinearity of the genomes and the two deletions between HN016 and YM001 are shown
The deletion or damage of ABC transporter gene
ABC transporters are integral membrane proteins that conduct transmembrane transport of various solute biomolecules using the energy of ATP hydrolysis; the substances absorbed include nutrients and osmoprotectants that range from small sugars, amino acids, and small peptides to metals, anions, iron chelators (siderophores), and vitamin B12, while the exported substances are surface components of the bacterial cell (such as capsular polysaccharides, lipopolysaccharides, and techoic acid), proteins involved in bacterial pathogenesis (such as hemolysin, heme-binding protein, and alkaline protease), peptide antibiotics, heme, drugs, and siderophores [12, 13]. Among the four ABC transporter family genes deleted in the YM001 genome, three belong to the subfamily B with efflux function, two of which encode proteins that have been demonstrated relating to the efflux of various drugs, to help bacteria obtain multidrug resistance to several antibiotics and tolerance to biocides [14, 15]; the 4th one encodes a protein of subfamily F, which involves in the intercellular communication between bacteria, and the deletion of this gene may cause growth inhibition of the mutant [16]. Comparative genome analysis of Mycoplasma hyopneumoniae (M. hyopneumoniae) pathogenic 168 strain and its highly-passaged attenuated strain 168 L showed that the ABC transporter proteins might affect the growth and survival of M. hyopneumoniae in different hosts or host tissues [17]. The growth of YM001 in both solid and liquid cultures was significantly slower than that of HN016 [18]. Fluorescent quantitative detection of tissue bacteria after oral gavage of tilapia with the two strains respectively also indicated that both the survival time and number of HN016 in vivo in tilapia were significantly greater than those of YM001 [18]. A decreased growth performance of YM001 due to ABC transporter deletion might be one of the main reasons of reduced virulence.
The deletion of MarR family transcriptional regulator
Oxidative, nitrosative, and aerobic stresses are major factors affecting the survival of pathogens in the host [19]. S. agalactiae is a facultative anaerobe, with a wide range of hosts, and may colonize in many tissues including the gastrointestinal and genitourinary tracts, brain, blood, liver, kidney, mammary gland, etc. [20]. The wide host range and colonization tissues of S. agalactiae may be associated with the ability of its regulatory systems to sense and adapt to external stimuli, such as oxidative and aerobic stress. Studies have shown that oxygen affects the infectivity and virulence of S. agalactiae [21]. MarR family transcriptional regulator has the function of regulating the oxidative stress response; therefore, deletion of MarR coding genes may result in an increased bacterial sensitivity to oxidative and aerobic stress, decreased capability of intracellular survival in macrophages, and reduced virulence [22]. The ability of S. sgalactiae to survive in macrophages is an important mechanism for its escape from the host immunity [23]. Accordingly, we speculate that the deletion of MarR family transcriptional regulator coding genes in YM001 reduced its growth adaptability and ability to survive in macrophages, decreased its ability to escape from the immune defenses of tilapia, and thereby blocked its continuous growth in and pathogenicity to tilapia.
The deletion of STP
Protein phosphorylation is essential for the regulation of cell growth, division, and differentiation in both prokaryotes and eukaryotes. Lately, bacterial homologues of eukaryotic STP have been shown to be necessary for cellular functions such as growth, differentiation, pathogenicity, and secondary metabolism. Mutations in these genes exhibited pleiotropic effects on the growth, virulence, and cell segregation of S. sgalactiae, suggesting that these enzymes may regulate the pyrophosphatase activity and other cellular functions in S. sgalactiae [24], and that these genes may have novel roles in regulating bacterial metabolic processes such as purine biosynthesis [25]. The deletion of STP in YM001 resulted in the loss of multiple purine metabolic pathways (Fig. 2). In S. sgalactiae, STP controls the function of Ser/Thr kinase, post-transcriptional regulation of hemolysin, autolysis, and virulence. Although STP is not essential for growth, it is critical for the pathogenicity of S. sgalactiae [26]. In view of its important roles in metabolism and pathogenicity of S. agalactiae, STP deletion may be one of the main causes of the reduced virulence of YM001.
Fig. 2.
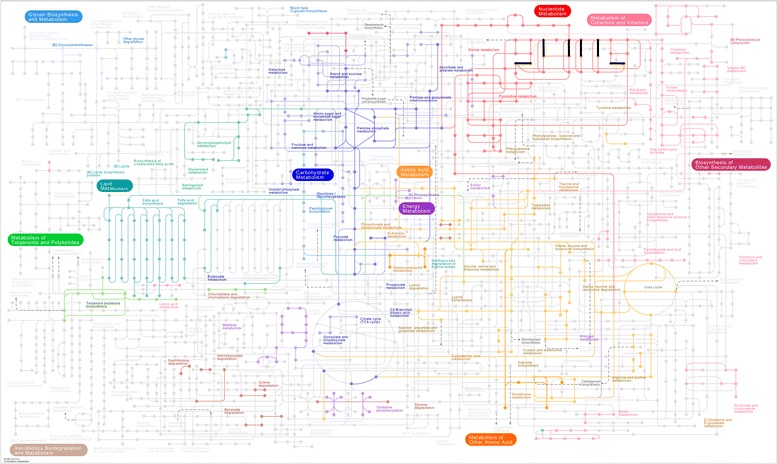
Metabolic potential. The metabolic pathways of S. agalactiae strains HN016 and YM001 were mapped and analyzed using KEGG Pathway Database. Those pathways, containing mutations affected metabolic-related genes, are shown in thick black line
The deletion of the other four genes
The other four deleted genes were peptide deformylase (PDF), glutamate dehydrogenase (GDH), acetyltransferase, and a membrane protein of unknown function. PDF is a highly conserved metalloprotease for bacterial growth and participates in bacterial protein biosynthesis and maturation [27, 28]; blocking of its function significantly inhibits the growth of Gram-positive pathogens, such as Streptococci and Staphylococci [29]. GDH is an important functional molecule in the process of energy metabolism in bacteria and is related to bacterial pathogenicity; it has been identified as a virulence factor of S. suis serotype two [30]. PDF and GDH were deleted from the genome of YM001, which might be the cause of the slow growth and low virulence of YM001. The effect of the deletions of acetyltransferase and the membrane protein of unknown function on S. agalactiae remains to be further studied.
SNV and Indels analyses between YM001 and HN016 genomes
SNV and Indels analyses between YM001 and HN016 genomes indicated that in addition to the two large deletions, there were a total of 28 SNVs (of which 1 was located in the non-coding region), and relative to HN016, YM001 had another ten deletions (including seven located in the non-coding region). Among the 27 SNVs in the coding region, 24 encoded proteins related to metabolism (ten genes), translation (six genes), and transport (eight genes) (Table 1). Two of the three deletion mutant Genes in YM001 coded for PTS system transporter subunit IIC and recombinase RecF respectively, while the third one encoded a hypothetical protein of unknown function. However, currently known S. agalactiae virulence factors such as adhesin, exoenzyme, immunoreactive antigen, metal transport, protease, toxin, etc., the majority of which are considered main antigens of S. agalactiae, did not show any variation [5]. These results indicated that genetic changes in attenuated strain generated by continuous passaging mainly affected genes related to bacterial growth and metabolism, with little effect on the virulence-related genes, which is thus conducive to the preservation of antigenicity during virulence attenuation by passaging. This may be why attenuated YM001 retained its strong immunogenicity. Current development of attenuated S. agalactiae vaccine mainly focused on the modification of its virulence factors; however, the results of this study opened a new avenue to the development of attenuated vaccine, i.e., to produce attenuated strain through modifying growth-related genes, under the premise of maximally preserving its immunogenicity and not affecting the virulence factors.
Clustered regularly interspaced short palindromic repeats (CRISPRs)
CRISPRs are a bacterial adaptive immune defense mechanism against the invasion of foreign genes. When foreign gene invades bacteria, the CRISPRs integrate and save the intruding gene fragment. Under the re-invasion of the same genes, mediated by specific RNA, CRISPRs and CRISPR-associated proteins (Cas proteins) will cut and destroy the invading foreign genes, which may include bacterial phages, plasmids, and mobile genetic elements (MGEs) [31, 32]. S. agalactiae has 2 CRISPR/Cas systems, type 1-C CRISPR2 and type 2-A CRISPR1; while the latter is ubiquitous, the former is only present in a few strains [33]. The CRISPR sequences were analyzed among the 8 S. agalactiae strains in Table 2 using the CRISPRs web server (http://crispr.u-psud.fr/Server/). The results indicated that 3 tilapia-derived S. agalactiae serotype Ib strains did not contain any CRISPR sequences, while 5 S. agalactiae serotype Ia strains all had CRISPR1 but did not contain CRISPR2. Further analysis of the CRISPR1 from the 5 S. agalactiae serotype Ia strains showed that the CRISPR sequence in S. agalactiae strain HN016 derived from tilapia in China was same as that in GD201008-001 and ZQ0910 and had eight spacers, but the attenuated strain YM001 only contained seven spacers; all other sequences were the same between both strains (Fig. 3). During the process of foreign nucleic acid invasion and bacterial evolution, to avoid overly long locus of CRISPRs, bacteria may choose to insert or remove spacer sequences between CRISPRs, and the insertion or removal of spacer is polarized, i.e., a new spacer is always inserted between the leader sequence and the following repetitive sequence, while the removed spacer is usually located at the 3’ end of CRISPRs [34]. Lopez-Sanchez et al. analyzed the CRISPRs of more than 200 wild-type S. agalactiae strains but did not find the 3’ terminal deletion [33]. Although Liu et al. showed that two spacer sequences were deleted at the 3’ end of CRISPRs in ZQ0910 stain [12], we found that the assembling of the published sequence of this fragment in ZQ0910 had certain mistake. After reanalysis and alignment of this sequence, we confirmed that the CRISPRs of ZQ0910 were exactly the same as those of HN016 and GD201008-001. Compared to other tilapia-derived wild-type virulent strains, the CRISPRs of YM001 had a deletion of 1 spacer at the 3’ end. Philippe et al. studied the CRISPR loci of S. thermophilus and found that the selective removal of spacer sequence may be caused by that these spacer sequences have little value for the survival of bacteria in the environment at the time [35]. Therefore, normal natural growth and passage are hard to cause the removal of CRISPR spacer sequence in S. agalactiae, whereas in the absence of the threat of foreign nucleic acid invasion, highly intensified continuous passage in laboratory may lead to the loss or removal of spacer sequence in CRISPRs.
Table 2.
Characteristics of sequenced S. agalactiae strains used in this study
| Strain | Serotype | MLST types | Accession | Status | Size (Mb) | Number of genes | Number of proteins | Isolatehost | Origin | Virulence description |
|---|---|---|---|---|---|---|---|---|---|---|
| No. | ||||||||||
| HN016 | Ia | ST-7 | CP011325 | Complete | 2.065 | 2063 | 1943 | Tilapia | China | virulent |
| YM001 | Ia | ST-7 | CP011326 | Complete | 2.048 | 2044 | 1929 | Tilapia | China | attenuated |
| A909 | Ia | ST-7 | NC_007432 | Complete | 2.128 | 2136 | 1996 | Human | USA | virulent |
| GD201008-001 | Ia | ST-7 | NC_018646 | Complete | 2.063 | 2088 | 1964 | Tilapia | China | virulent |
| ZQ0910 | Ia | ST-7 | NZ_AKAP00000000 | Scaffold | 2.035 | 2003 | 1970 | Tilapia | China | virulent |
| 138P | Ib | unknown | CP007482.1 | Complete | 1.839 | 1831 | 1593 | Tilapia | USA | virulent |
| 138spar | Ib | unknown | CP007565.1 | Complete | 1.838 | 1825 | 1590 | Tilapia | USA | attenuated |
| SA20-06 | Ib | ST-553 | NC_019048 | Complete | 1.821 | 1872 | 1710 | Tilapia | Brazil | virulent |
Fig. 3.

Diversity of the CRISPR1 locus in 5 S. agalactiae strains. Spacers were identified by the CRISPRtionary program, with numbers assigned to each spacer [51]. The names of strains are given on the left. R stands for Repeat, and S stands for Spacer
Prophages
Prophages are bacterial phages that integrate their genomes into the genome of host bacteria after infection. Approximately 65 % of the completely sequenced bacterial genomes carry prophage, and the content of prophage sequences in some bacteria approaches 20 % of the bacterial genomic content [36]. Studies have reported that whether a bacterial genome carries prophage often determines the difference in virulence between pathogenic and nonpathogenic strains [37, 38]. The contribution of prophage genes to the pathogenicity of S. enterica serovar Typhimurium has been demonstrated by animal experiments [39]. Prophage analysis of HN016, YM001, and GD201008-001 showed that an intact prophage sequence and an incomplete prophage sequence were detected in all the 3 strains. All coding DNA sequences (CDSs) and the locations of the prophages in these three strains were exactly the same (Fig. 4). Our results showed that there was no prophage mutations in the attenuated S. agalactiae strain YM001, which may also help to maintain the antigenic integrity of the attenuated strain.
Fig. 4.
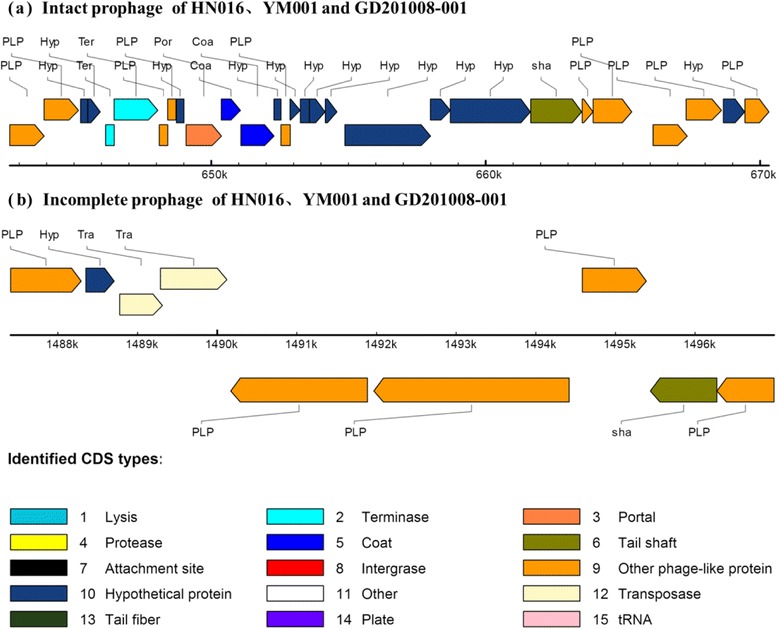
The CDS of the prophages derived from S. agalactiae HN016, YM001, and GD201008-001 respectively. A detailed view of the prophages from the 3 strains was produced using the online software PHAST (http://phast.wishartlab.com/index.html). Different colors represent various phage elements
Conclusions
In summary, compared to the parental pathogenic strain HN016, the genome of attenuated S. agalactiae YM001 showed significant variations, resulting in the deletion of 10 functional genes, which may be the main reason for the loss of YM001 virulence to tilapia. The deleted and mutated functional genes all encode metabolism- and growth-related proteins, not the known virulence proteins, indicating that the metabolism- and growth-related genes are important for the pathogenesis of S. agalactiae. The mutations in growth- and metabolism-related genes with the preservation of virulence genes reduced the virulence while retained the full antigenecity. Our results laid a foundation for the development of attenuated S. agalactiae vaccine and the study on the immune mechanism. Therefore, the present study set a basis for future investigation of the pathogenesis of S. agalactiae and facilitated the design of attenuated vaccine.
Methods
Bacterial strains
The S. agalactiae strain HN016 originally isolated in 2010 in China, from a moribund cultured tilapia with typical clinical and pathogenic characteristics of meningoencephalitis, belonged to S. agalactiae serotype Ia, multilocus sequence type seven (ST7) [17]. This field strain was gradually attenuated by 840 continuous passages in TSB medium, and the 840th passage was named strain YM001 [18]. The serotype, ST type, and PFGE bands of YM001 strain were consistent with those of HN016 [18]. However, the YM001 failed to cause disease or death in tilapia at the dose of 109 CFU/fish by intraperitoneal injection, while the HN016 was lethal to tilapia at the dose down to 103 CFU/fish [18]. Backpassage safety assay indicated that YM001 did not cause disease or death in tilapia after 11 generations of serial passage [9, 18]. The genomes of another 6 S. agalactiae strains were retrieved from the GenBank (Table 2). The strains GD201008-001, ZQ0910, and A909 all belonged to serotype Ia and ST7. The GD201008-001 and ZQ0910 strains were isolated from farmed tilapia in China, and the A909 was also proven closely related to GD201008-001 [5]. The serotypes of 138P, 138spar, and SA20-06 all belonged to Ib, and the 138spar was obtained by attenuation of 138P using sparfloxacin resistance strategy.
Genome sequencing and annotation
The draft genome sequences of S. agalactiae strain HN016 and YM001 were determined using Illumina Genome Analyzer II (GAII) at the Beijing Genomics Institute (BGI; Shenzhen, China). Draft assemblies were based on 454-Mb reads. All reads provided about 214-fold coverage of the genome. The GAII paired-end reads were assembled with the SOAPdenovo 2.04 program [40]. Gaps were closed by primer walking and sequencing of PCR products. Putative open reading frames (ORFs) with more than 30 amino acid residues were predicted using Glimmer 3.02 [41], while rRNAs and tRNAs were identified using RNAmmer 1.2 [42] and tRNAscan-SE 1.23 [43] respectively. The scaffolds were searched against the COG (Clusters of Orthologous Groups), GO (Gene Ontology), SwissProt, and KEGG (Kyoto Encyclopedia of Genes and Genomes) databases to annotate the gene descriptions.
Comparative genome analysis
Nucleotide comparisons and single nucleotide polymorphism (SNP) analysis for strains HN016 and YM001 were performed using the Artemis Comparison Tool (ACT) [44] and Mauve 2.3.1 genome alignment software [45]. ORF graphical visualization and manual annotation were carried out using Artemis, release 12 [46]. Screening for unusual coding differences between the HN016 and YM001 genomes (stops and frame shifts) was conducted using FASTA program packages [47, 48] and BLAST [49, 50]. The coding differences between the HN016 and YM001 genomes were checked manually.
Genome element prediction
CRISPRdb database, CRISPRs finder, and CRISPRcompar were used to display CRISPRs, generate dictionary of spacers and repeats, and compare CRISPRs (http://crispr.u-psud.fr/) [51–53]. PHAST (http://phast.wishartlab.com/index.html) was used to identify prophage sequences [54]. Amino acid sequences of the CDSs of 4 piscine strains and Ia strain were searched against the Virulence Factor of Pathogenic Bacteria database (VFDB, www.mgc.ac.cn/VFs/main.htm) using BLASTp [55–57]. An E value cut-off of 1e-5 was used to obtain the single best hit.
Accession numbers
The genome sequences of S. agalactiae strains HN016 and YM001 were deposited into the GenBank under the accession numbers of CP011325 and CP011326 respectively.
Acknowledgements
This work was supported by Guangxi Science and Technology Research Program (14121004-2-4), National Natural Science Foundation of China (31460695), Guangxi “bagui scholar” post special funds issue (BGXZ-LFY-04) and Guangxi Science Foundation (2014GXNSFBA118083).
Additional files
The large fragment deletion 1 detected in YM001 compared to HN016. (XLS 26 kb)
The large fragment deletion 2 detected in YM001 compared to HN016. (XLS 28 kb)
SNVs detected in YM001 compared to HN016. (XLS 34 kb)
Deletions detected in YM001 compared to HN016. (XLS 28 kb)
Footnotes
Rui Wang, Liping Li and Yan Huang contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: MC LFC RW LPL LWW. Performed the experiments/analyzed the sequence data: RW LPL LWW FGL TH. Contributed materials/analysis tools: XG YH AYL. All authors read and approved the final manuscript.
Contributor Information
Rui Wang, Email: raywongxx@163.com.
Liping Li, Email: pinglili2000@163.com.
Yan Huang, Email: huangyangxdx@163.com.
Fuguang Luo, Email: luofuguang3563@163.com.
Wanwen Liang, Email: nnlww@126.com.
Xi Gan, Email: ganxipaper@163.com.
Ting Huang, Email: htwish@163.com.
Aiying Lei, Email: nnleiaiying@sina.com.
Ming Chen, Email: cm990919@163.com.
Lianfu Chen, Email: chenllianfu@foxmail.com.
References
- 1.Brochet M, Couve E, Zouine M, Vallaeys T, Rusniok C, Lamy MC, et al. Genomic diversity and evolution within the species Streptococcus agalactiae. Microbes Infect. 2006;8(5):1227–1243. doi: 10.1016/j.micinf.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Chaiwarith R, Jullaket W, Bunchoo M, Nuntachit N, Sirisanthana T, Supparatpinyo K. Streptococcus agalactiae in adults at Chiang Mai University Hospital: a retrospective study. BMC Infect Dis. 2011;11:149. doi: 10.1186/1471-2334-11-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elliott JA, Facklam RR, Richter CB. Whole-cell protein patterns of nonhemolytic group B, type Ib, streptococci isolated from humans, mice, cattle, frogs, and fish. J Clin Microbiol. 1990;28(3):628–630. doi: 10.1128/jcm.28.3.628-630.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu G, Zhang W, Lu C. Comparative genomics analysis of Streptococcus agalactiae reveals that isolates from cultured tilapia in China are closely related to the human strain A909. BMC Genomics. 2013;14:775. doi: 10.1186/1471-2164-14-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosinski-Chupin I, Sauvage E, Mairey B, Mangenot S, Ma L, Da Cunha V, et al. Reductive evolution in Streptococcus agalactiae and the emergence of a host adapted lineage. BMC Genomics. 2013;14:252. doi: 10.1186/1471-2164-14-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pridgeon JW, Zhang D, Zhang L. Complete Genome of the Attenuated Sparfloxacin-Resistant Streptococcus agalactiae Strain 138spar. Genome Announc. 2014;2(3):e00431–14. doi: 10.1128/genomeA.00431-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, Su YL, Mai YZ, Li YW, Mo ZQ, Li AX. Comparative proteome analysis of two Streptococcus agalactiae strains from cultured tilapia with different virulence. Vet Microbiol. 2014;170(1-2):135–143. doi: 10.1016/j.vetmic.2014.01.033. [DOI] [PubMed] [Google Scholar]
- 9.Pridgeon JW, Klesius PH. Development of live attenuated Streptococcus agalactiae as potential vaccines by selecting for resistance to sparfloxacin. Vaccine. 2013;31(24):2705–2712. doi: 10.1016/j.vaccine.2013.03.066. [DOI] [PubMed] [Google Scholar]
- 10.Da Cunha V, Davies MR, Douarre PE, Rosinski-Chupin I, Margarit I, Spinali S, et al. Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline. Nat Commun. 2014;5:4544. doi: 10.1038/ncomms5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Wang R, Liang W, Gan X, Huang T, Huang Y, et al. Rare serotype occurrence and PFGE genotypic diversity of Streptococcus agalactiae isolated from tilapia in China. Vet Microbiol. 2013;167(3-4):719–724. doi: 10.1016/j.vetmic.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Dassa E, Bouige P. The ABC of ABCS: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol. 2001;152(3-4):211–229. doi: 10.1016/S0923-2508(01)01194-9. [DOI] [PubMed] [Google Scholar]
- 13.Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 14.Lee EW, Huda MN, Kuroda T, Mizushima T, Tsuchiya T. EfrAB, an ABC multidrug efflux pump in Enterococcus faecalis. Antimicrob Agents Chemother. 2003;47(12):3733–3738. doi: 10.1128/AAC.47.12.3733-3738.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavilla Lerma L, Benomar N, Valenzuela AS, Casado Munoz Mdel C, Galvez A, Abriouel H. Role of EfrAB efflux pump in biocide tolerance and antibiotic resistance of Enterococcus faecalis and Enterococcus faecium isolated from traditional fermented foods and the effect of EDTA as EfrAB inhibitor. Food Microbiol. 2014;44:249–257. doi: 10.1016/j.fm.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Murat D, Goncalves L, Dassa E. Deletion of the Escherichia coli uup gene encoding a protein of the ATP binding cassette superfamily affects bacterial competitiveness. Res Microbiol. 2008;159(9-10):671–677. doi: 10.1016/j.resmic.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Liu W, Xiao S, Li M, Guo S, Li S, Luo R, et al. Comparative genomic analyses of Mycoplasma hyopneumoniae pathogenic 168 strain and its high-passaged attenuated strain. BMC Genomics. 2013;14:80. doi: 10.1186/1471-2164-14-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li LP, Wang R, Liang WW, Huang T, Huang Y, Luo FG, et al. Development of live attenuated Streptococcus agalactiae vaccine for tilapia via continuous passage in vitro. Fish Shellfish Immunol. 2015;45(2):955–963. doi: 10.1016/j.fsi.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. 2004;2(10):820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 20.Johri AK, Paoletti LC, Glaser P, Dua M, Sharma PK, Grandi G, et al. Group B Streptococcus: global incidence and vaccine development. Nat Rev Microbiol. 2006;4(12):932–942. doi: 10.1038/nrmicro1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johri AK, Padilla J, Malin G, Paoletti LC. Oxygen regulates invasiveness and virulence of group B streptococcus. Infect Immun. 2003;71(12):6707–6711. doi: 10.1128/IAI.71.12.6707-6711.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gundogdu O, Mills DC, Elmi A, Martin MJ, Wren BW, Dorrell N. The Campylobacter jejuni transcriptional regulator Cj1556 plays a role in the oxidative and aerobic stress response and is important for bacterial survival in vivo. J Bacteriol. 2011;193(16):4238–4249. doi: 10.1128/JB.05189-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo CM, Chen RR, Kalhoro DH, Wang ZF, Liu GJ, Lu CP, et al. Identification of genes preferentially expressed by highly virulent piscine Streptococcus agalactiae upon interaction with macrophages. PLoS One. 2014;9(2):e87980.24. doi: 10.1371/journal.pone.0087980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajagopal L, Clancy A, Rubens CE. A eukaryotic type serine/threonine kinase and phosphatase in Streptococcus agalactiae reversibly phosphorylate an inorganic pyrophosphatase and affect growth, cell segregation, and virulence. J Biol Chem. 2003;278(16):14429–14441. doi: 10.1074/jbc.M212747200. [DOI] [PubMed] [Google Scholar]
- 25.Rajagopal L, Vo A, Silvestroni A, Rubens CE. Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol Microbiol. 2005;56(5):1329–1346. doi: 10.1111/j.1365-2958.2005.04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.deLos Reyes M, Burnside K, Lembo A, Harrell MI, Gurney M, Xue L, et al. Serine/threonine phosphatase Stp1 mediates post-transcriptional regulation of hemolysin, autolysis, and virulence of group B Streptococcus. J Biol Chem. 2011;286(51):44197–44210. doi: 10.1074/jbc.M111.313486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ball LA, Kaesberg P. Cleavage of the N-terminal formylmethionine residue from a bacteriophage coat protein in vitro. J Mol Biol. 1973;79(3):531–537. doi: 10.1016/0022-2836(73)90404-X. [DOI] [PubMed] [Google Scholar]
- 28.Margolis P, Hackbarth C, Lopez S, Maniar M, Wang W, Yuan Z, et al. Resistance of Streptococcus pneumoniae to deformylase inhibitors is due to mutations in defB. Antimicrob Agents Chemother. 2001;45(9):2432–2435. doi: 10.1128/AAC.45.9.2432-2435.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lofland D, Difuntorum S, Waller A, Clements JM, Weaver MK, Karlowsky JA, et al. In vitro antibacterial activity of the peptide deformylase inhibitor BB-83698. J Antimicrob Chemother. 2004;53(4):664–668. doi: 10.1093/jac/dkh129. [DOI] [PubMed] [Google Scholar]
- 30.Okwumabua O, Persaud JS, Reddy PG. Cloning and characterization of the gene encoding the glutamate dehydrogenase of Streptococcus suis serotype 2. Clin Diagn Lab Immunol. 2001;8(2):251–257. doi: 10.1128/CDLI.8.2.251-257.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol. 2010;64:475–493. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- 32.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 33.Lopez-Sanchez MJ, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, et al. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol Microbiol. 2012;85(6):1057–1071. doi: 10.1111/j.1365-2958.2012.08172.x. [DOI] [PubMed] [Google Scholar]
- 34.Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190(4):1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190(4):1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canchaya C, Fournous G, Brussow H. The impact of prophages on bacterial chromosomes. Mol Microbiol. 2004;53(1):9–18. doi: 10.1111/j.1365-2958.2004.04113.x. [DOI] [PubMed] [Google Scholar]
- 37.Porwollik S, Wong RM, McClelland M. Evolutionary genomics of Salmonella: gene acquisitions revealed by microarray analysis. Proc Natl Acad Sci U S A. 2002;99(13):8956–8961. doi: 10.1073/pnas.122153699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanlon GW. Bacteriophages: an appraisal of their role in the treatment of bacterial infections. Int J Antimicrob Agents. 2007;30(2):118–128. doi: 10.1016/j.ijantimicag.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Schmieger H, Schicklmaier P. Transduction of multiple drug resistance of Salmonella enterica serovar typhimurium DT104. FEMS Microbiol Lett. 1999;170(1):251–256. doi: 10.1111/j.1574-6968.1999.tb13381.x. [DOI] [PubMed] [Google Scholar]
- 40.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21(16):3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 45.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 47.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A. 1988;85(8):2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pearson WR, Wood T, Zhang Z, Miller W. Comparison of DNA sequences with protein sequences. Genomics. 1997;46(1):24–36. doi: 10.1006/geno.1997.4995. [DOI] [PubMed] [Google Scholar]
- 49.Mount DW. Using the Basic Local Alignment Search Tool (BLAST) CSH Protoc. 2007;2007:17. doi: 10.1101/pdb.top17. [DOI] [PubMed] [Google Scholar]
- 50.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 51.Grissa I, Vergnaud G, Pourcel C. CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2008;36(Web Server issue):W145–148. doi: 10.1093/nar/gkn228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35((Web Server issue):W52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinf. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39((Web Server issue):W347–352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(Database issue):D325–328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Chen L, Sun L, Yu J, Jin Q. VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res. 2008;36(Database issue):D539–542. doi: 10.1093/nar/gkm951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012;40(Database issue):D641–645. doi: 10.1093/nar/gkr989. [DOI] [PMC free article] [PubMed] [Google Scholar]