

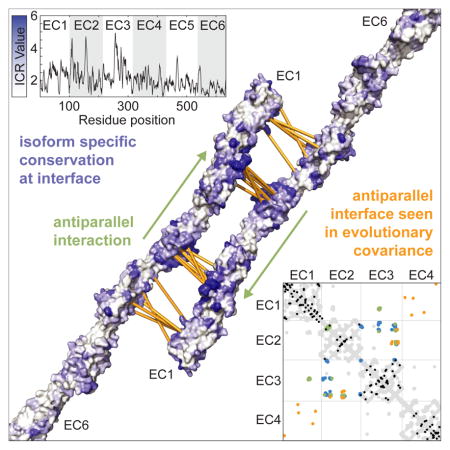
SUMMARY
Clustered protocadherin (Pcdh) proteins mediate dendritic self-avoidance in neurons via specific homophilic interactions in their extracellular cadherin (EC) domains. We determined crystal structures of EC1-EC3, containing the homophilic specificity-determining region, of two mouse clustered Pcdh isoforms (PcdhγA1 and PcdhγC3) to investigate the nature of the homophilic interaction. Within the crystal lattices, we observe antiparallel interfaces consistent with a role in trans cell-cell contact. Antiparallel dimerization is supported by evolutionary correlations. Two interfaces, located primarily on EC2-EC3, involve distinctive clustered Pcdh structure and sequence motifs, lack predicted glycosylation sites, and contain residues highly conserved in orthologs but not paralogs, pointing towards their biological significance as homophilic interaction interfaces. These two interfaces are similar yet distinct, reflecting a possible difference in interaction architecture between clustered Pcdh subfamilies. These structures initiate a molecular understanding of clustered Pcdh assemblies that are required to produce functional neuronal networks.
Graphical Abstract
INTRODUCTION
Clustered protocadherins (Pcdhs) have an important role in cell-cell interactions in neurons (Weiner and Jontes, 2013; Yagi, 2012). The Pcdh gene cluster comprises three groups, Pcdhα, Pcdhβ and Pcdhγ, producing 53 variable isoforms in humans. Various studies have associated clustered Pcdhs with synapse maintenance and formation (Fernández-Monreal et al., 2009; Li et al., 2012; Weiner et al., 2005), promotion of synapse development by astrocytes (Garrett and Weiner, 2009), connectivity between terminals of type Ia afferent neurons and ventral interneurons (Prasad and Weiner, 2011), and arborization of cortical pyramidal neurons (Garrett et al., 2012). Loss of the Pcdhγ cluster has been linked to apoptosis, neurodegeneration and synapse loss in different neuron populations (Chen et al., 2012; Emond and Jontes, 2008; Lefebvre et al., 2008; Prasad et al., 2008; Su et al., 2010; Wang et al., 2002), indicating that the role of clustered Pcdhs is complex and multifaceted.
The clustered Pcdhs have also been associated with self-avoidance in Purkinje and starburst amacrine cells (SACs), which results in dendritic self/non-self discrimination during synaptogenesis (Kostadinov and Sanes, 2015; Lefebvre et al., 2012). Each neuron stochastically expresses 5–10 isoforms (Esumi et al., 2005; Kaneko et al., 2006; Yagi, 2012; Yokota et al., 2011). In addition, the five C-type isoforms are constitutively expressed (Kaneko et al., 2006). Part of the cadherin superfamily of Ca2+-dependent adhesion proteins, clustered Pcdhs are thought to use specific homophilic interactions to signal self-avoidance (Lefebvre et al., 2012). In insects, the Dscam1 gene plays a role analogous to that of the clustered Pcdhs (Zipursky and Sanes, 2010). Unlike Dscam1, where the homophilic interfaces are well characterized (Meijers et al., 2007; Sawaya et al., 2008), the structural determinants of clustered Pcdhs interactions are largely unknown.
Each clustered Pcdh isoform contains six extracellular cadherin (EC) repeats followed by a transmembrane helix and a C-terminal intracellular domain. Although there are many known classical cadherin structures, few structures of the protocadherin subfamily are available, with the only known interface being the tip-link Pcdh15-Cdh23 complex involved in mammalian hearing (Sotomayor et al., 2012). Understanding the putative homophilic interactions of clustered protocadherins requires structures of these complexes.
Specific homophilic interactions are common in the cadherin superfamily. In classical cadherins, the first repeat, EC1, mediates homophilic interactions, either through an ‘X-dimer’ complex or a tryptophan-containing strand-swapping mechanism (Brasch et al., 2012; Sotomayor et al., 2014). In contrast, while the clustered Pcdh EC1 is required for complex formation, EC2 and EC3 determine interaction specificity (Schreiner and Weiner, 2010; Thu et al., 2014). Thus the nature of clustered Pcdh homophilic interactions is distinct from those of classical cadherins, prompting investigation into the difference in homophilic interaction specificity between these two cadherin subfamilies.
In cell aggregation assays, chimeric Pcdhγ isoforms were used to demonstrate that isoform concordance in both EC2 and EC3 is necessary for homophilic interactions (Schreiner and Weiner, 2010), implying the formation of parallel complexes, in which EC2 interacts with EC2 and EC3 with EC3. Concomitantly, immunoprecipitation experiments suggested that Pcdhγ isoforms form promiscuous cis complexes within a cell (Biswas et al., 2012; Han et al., 2010; Schalm et al., 2010; Schreiner and Weiner, 2010). These data led to a proposed model of a cis-bundle promoting parallel homophilic trans interactions, i.e. across two cells. Cis interactions have a combinatorial behavior such that expression of combinations of isoforms leads to novel specificity (Thu et al., 2014) – equivalent to an ‘AND’ logic gate. This is strong evidence that combinatorial interactions provide sufficient diversification potential to support the clustered Pcdhs as cell-identity markers in neurons. Cell aggregation assays cannot easily distinguish homophilic interactions that occur in trans from promiscuous cis interactions, especially if they are interdependent, indicating that we need other methods to determine the architecture of the specificity complex.
We know little about the structures of either the cis or trans interactions in clustered Pcdhs that lead to cellular self/non-self discrimination. We thus sought to identify possible interfaces present in these complexes as a way to explore the overall architecture. Using a combination of x-ray crystallography and bioinformatics we identified and analyzed possible interfaces. Here we describe a set of antiparallel interfaces that are similar to the trans Pcdh15-Cdh23 interactions (Sotomayor et al., 2012). These structures and analyses initiate a molecular understanding of the complicated interactions in the clustered Pcdhs.
RESULTS
Structures of protocadherin γA1 EC1-3 and γC3 EC1-3
We obtained structures of mouse protocadherin γA1 EC1-3 (A1) and γC3 EC1-3 (C3) (Table 1). The A1 and C3 protomers form elongated structures containing three Greek-key β-sandwich motif EC domains (Figure 1A), consistent with other available cadherin structures and NMR structures of EC1 from several other clustered Pcdh isoforms (Figure S1) (Morishita et al., 2006). The three repeats are arranged in tandem with two inter-repeat linker regions each containing three Ca2+-binding sites comprising canonical Ca2+-binding residues (Figure 1B).
Table 1.
Statistics for A1 and C3 structures
| PcdhγA1 EC1-3 | PcdhγC3 EC1-3 | |
|---|---|---|
| PDB ID | 4ZI9 | 4ZI8 |
| SBGrid Data Bank ID | 173 | 174 |
| Data Collection | ||
| Beam source | APS 24-ID-E | APS 24-ID-C |
| Wavelength (Å) | 0.97918 | 0.97918 |
| Space group | P21 | P212121 |
| Unit cell | ||
| a, b, c (Å) | 67.751, 63.687, 107.486 | 44.123, 82.286, 237.69 |
| α, β, γ (°) | 90, 99.693, 90 | 90, 90, 90 |
| Resolution (Å) | 50 – 1.77 (1.833 – 1.77) | 50 – 1.70 (1.73 – 1.70) |
| Total reflections | 269651 (13875) | 281547 (8193) |
| Unique reflections | 77361 (5120) | 84132 (3718) |
| Multiplicity | 3.5 (2.7) | 3.3 (2.2) |
| Completeness (%) | 87 (58) | 86 (39) |
| Mean I/σ(I) | 5.70 (0.46) | 5.38 (0.53) |
| Resolution shell at I/σ(I) = 2 (Å) | 2.14 – 2.07 | 2.07 – 2.02 |
| Wilson B-factor | 22.96 | 25 |
| Rmerge | 0.125 (1.499) | 0.112 (1.079) |
| Rmeas | 0.1469 (1.804) | 0.1316 (1.342) |
| CC1/2 | 0.995 (0.513) | 0.995 (0.292) |
| CC* | 0.999 (0.824) | 0.999 (0.672) |
| Refinement | ||
| Refinement resolution range | 33.51 – 1.77 (1.833 – 1.77) | 48.17 – 1.698 (1.759 – 1.698) |
| Reflections used in refinement | 76754 (4773) | 83396 (3379) |
| Reflections used for R-free | 2031 (130) | 1983 (79) |
| Rwork | 0.2274 (0.4453) | 0.2043 (0.3800) |
| Rfree | 0.2649 (0.4659) | 0.2394 (0.4085) |
| CCwork | 0.964 (0.647) | 0.961 (0.529) |
| CCfree | 0.946 (0.550) | 0.948 (0.640) |
| Number of non-hydrogen atoms | 5424 | 5655 |
| Macromolecules | 4935 | 5001 |
| Ligands | 12 | 48 |
| Protein residues | 618 | 636 |
| RMS | ||
| Bonds (Å) | 0.008 | 0.011 |
| Angles (°) | 1.10 | 1.23 |
| Clashscore | 2.03 | 2.4 |
| Average B-factor | 46.60 | 36.15 |
| Macromolecules | 46.78 | 35.03 |
| Ligands | 31.22 | 52.37 |
| Solvent | 45.16 | 44.12 |
| Ramachandran plot regions | ||
| Favored (%) | 96 | 100 |
| Allowed (%) | 3.5 | 0.31 |
| Outliers (%) | 0 | 0 |
| Rotamer outliers (%) | 0 | 0.18 (H256) |
Figure 1.

Sequence and structural properties of clustered Pcdhs. (A) Superposition of A1 (blue with cyan Ca2+) and C3 (green with yellow Ca2+) structures showing the Cys-X5-Cys, Phe-X10-Phe loop, and EC2 β4-β5 loops in purple. (B) Alignment of the six EC repeats of mouse PcdhγA1 and PcdhγC3. Ca2+-binding motifs are highlighted in yellow, and the A1 EC1-3 secondary structure is shown below the sequence. See also Figure S1.
Comparing A1 and C3, each of the three EC repeat structures are very similar (rmsd of 1.05, 0.824, and 0.593 Å for EC1, EC2, and EC3, respectively). However, superposition of A1 and C3 shows that A1 EC3 and C3 EC3 vary in tilt by 15° relative to EC2 (Figure 1A). There is some flexibility in fully Ca2+-bound linkers between cadherin repeats (Sotomayor and Schulten, 2008), which can explain the difference in relative EC3 orientation in A1 and C3. Since the overall A1 and C3 structures are similar, for simplicity figures refer to the A1 structure and sequence unless otherwise indicated.
The clustered Pcdh structures reveal distinctive features compared to classical cadherins (Figure 1). The most distinguishing feature is a conserved disulfide-clamped Cys-X5-Cys loop in EC1. This loop, elongated by the X5 sequence compared to classical cadherins, is required for cell-surface expression of Pcdh isoforms (Schreiner and Weiner, 2010), but its role in Pcdh interactions is unknown. Another feature of clustered Pcdhs is the Phe-X10-Phe loop-helix motif in EC3. Lastly, the loop between β4 and β5 in EC2 is elongated when compared to classical cadherins. All three of these regions have low conservation between human paralogs (15–25% average overall identity), suggesting that they may be sources of isoform diversity.
Crystallographic interfaces suggest possible biological interfaces
The purified A1 construct behaved as a monomer (37.7 kDa) in multiangle laser light scattering (MALLS) measurements at concentrations of up to 137 μM (Figure S2). Furthermore, C3 and A1 behaved similarly in size exclusion chromatography, indicating that C3 also behaves as a monomer in solution. This suggests that interactions between EC1-3 in solution are weak, precluding the use of solution-based experiments to investigate dimerization interfaces with these constructs. In the absence of in-solution interactions, we analyzed the physicochemical properties of the crystallographic interfaces to determine their biological relevance.
To select potential interfaces, we calculated the difference in accessible surface area (ΔASA) for all observed crystal packing interfaces. We further examined all interfaces of at least 600 Å2, except one C3 interface with ΔASA = 650 Å2 because it was highly solvated and the affinity-tag contributed more than 80 Å2. We thus selected four potential biological interfaces to evaluate (Figure 2). Interestingly, all four are antiparallel dimerization interfaces (EC1-EC3 directions in the protomers are antiparallel to each other) with two-fold rotational symmetry approximately perpendicular to the long axis of the protomers. We found two mutually compatible A1 interfaces: one where EC2 interacts with EC3 (from now on called the A1 EC23 interface; ΔASA=1229 Å2) and the other where EC1 and EC2 interact (A1 EC12 interface; ΔASA=1381 Å2) (Figure 2). The combination of these two A1 interfaces yields crystal packing along one axis. We found two more potential interfaces in the C3 structure: an extended interface over the length of EC1-3 (C3 extended; ΔASA=1156 Å2), and an EC1-EC1 interface (C3 EC1 interface; ΔASA=614 Å2) (Figure 2). These four interfaces may represent some of the interactions present in the cis or trans complexes of clustered Pcdhs.
Figure 2.

Observed antiparallel dimeric interfaces in the A1 and C3 crystal lattices. Side view of the four interfaces with ΔASA greater than 600 Å2: A1 EC12, A1 EC23, C3 EC1, and C3 extended. Residues with BSA > 0 Å2 are colored. See also Figure S2.
The largest determinant, by far, of biological significance of a protein complex interface is the ΔASA (Janin and Rodier, 1995). In a survey of PDB structures, a 856 Å2 cut-off distinguished a biological interface from a crystal contact with 85% accuracy (Ponstingl et al., 2000). All but the C3 EC1 interface are significantly above the 856 Å2 threshold (Table 2), indicating that they are likely biological in nature.
Table 2.
Physicochemical properties of clustered Pcdh interactions
| Interface | ΔASA (Å2) | #HBa | #SBb | Scc | NOXclass
|
||||
|---|---|---|---|---|---|---|---|---|---|
| Interface area ratio | Interface/surface correlation | GAPd | Biological (%) | Obligate (%) | |||||
| A1 EC12 | 1381 | 26 | 6 | 0.701 | 0.0845 | 0.5773 | 8.9 | 98.73 | 66.89 |
| A1 EC23 | 1229 | 11 | 4 | 0.662 | 0.0718 | 0.6007 | 12.2 | 90.03 | 85.64 |
| C3 extended | 1156 | 17 | 2 | 0.648 | 0.0672 | 0.6941 | 15.1 | 68.14 | 91.73 |
| C3 EC1 | 614 | 16 | 13 | 0.666 | 0.0356 | 0.7195 | 21.7 | 0 | 91.81 |
Number of hydrogen-bonds at interface
Number of salt bridges at interfaces
Shape correlation statistic
Gap volume index
The number of hydrogen bonds is indicative of specific protein interactions versus crystallization artifacts. Crystal contacts tend to have few hydrogen bonds (~5) while homodimers and protein complexes have ~10–20 (Bahadur et al., 2004). The interfaces have relatively large number of hydrogen bonds (Table 2), suggesting they are not crystallization artifacts, and that they are, if valid, non-obligate in nature, as obligate interactions rely more heavily on nonpolar interactions (Jones and Thornton, 1996).
Strong and specific interactions typically have highly complementary surfaces. In our interfaces, the A1 EC12 interface has the highest shape correlation (Sc = 0.70), while the C3 extended interface has the lowest (Sc = 0.65) (Table 2). These are all near the range reported for antibody/antigen complexes (Sc = 0.64–0.68), suggesting the interfaces are specific, yet non-obligate (Lawrence and Colman, 1993), which would agree with the expected properties of the clustered Pcdh interactions.
NOXclass is a protein-protein interaction classifier that distinguishes crystal packing from biological interactions and predicts whether an interface is likely obligate or non-obligate (Zhu et al., 2006). This prediction is useful for the non-obligate clustered Pcdhs interactions. According to NOXclass (Table 2), the C3 EC1 interface has a zero probability of being biological. A1 EC12 and A1 EC23 are both highly likely biological with 98.7% and 90% probability, respectively, while the C3 extended interface is 68% probable. All three are predicted to be obligate, but the A1 EC12 interface the least so with a 67% probability. This provides further support for the biological significance of all but the C3 EC1 interface.
The A1 EC23 and C3 extended interfaces include some of the structural features particular to the clustered Pcdhs described above (Figures 1 and 3). Polar interactions distribute towards the edge of the interface, while hydrophobic interactions are found towards the center of the interface, a property of biological interfaces (Chakrabarti and Janin, 2002). In A1 EC23 (Figure 3A), the Phe-X10-Phe loop of EC3 interacts with EC2 (Figure 3B). In the C3 extended interface (Figure 3C), the elongated β4-β5 loop in EC2 interacts with EC3 (Figure 3D). There is also extensive hydrogen bonding between the Cys-X5-Cys loop of EC1 and the Phe-X10-Phe loop of EC3 (Figure 3E). In this network T70 forms hydrogen bonds with S255 and H256 of the other protomer, which holds H256 as a rotamer outlier in both protomers (Figure S3), suggesting a strong and specific interaction.
Figure 3.

Structural details of the A1 EC23 (A, B) and C3 extended (C–E) interfaces. A1 EC23 (A) and C3 extended (C) interfaces are shown in an “open-book” orientation. Hydrogen-bonding (black) and salt-bridge (red) interactions are labeled on the top half of the interfaces, and residues with BSA > 20 Å2 on the bottom half. (B) In A1 EC23, the Phe-X10-Phe loop of EC3 (dark blue) makes specific interactions with β1 and β2 of EC2 (light blue). (D) In the C3 extended interface, the β4–β5 loop in EC2 of one protomer (dark green) interacts with β7 of EC3 in the other protomer (light green) and (E) the Cys-X5-Cys loop of EC1 (light green) forms a specific hydrogen-bonding network with Phe-X10-Phe loop of EC3 (dark green). (F) The A1 EC23 (blue) and C3 extended (green) interfaces overlap extensively (orange). See also Figure S3.
The A1 EC23 and C3 extended interfaces overlap, as they both include the β1-β2 loop and β2 of EC2, and the β3-β4 and β6-β7 loops of EC3. The two interfaces are related by a slide of the proteins by 22 Å and a slight rotation about their long axis (Figure 3F). The highly dissimilar sequences in this region may explain the difference in geometry between the A1 EC23 and C3 extended interfaces.
Homology modeling of EC1-6 clustered protocadherin predicts a linear structure
To gain further insights into the complete structure of a clustered protocadherin extracellular domain, we used the A1 and C3 EC1-3 structures along with sequence and structural analysis of other cadherin structures to construct a homology model of EC1-6.
The Ca2+-binding sites rigidify the linkers between EC repeats (Cailliez and Lavery, 2005; Sotomayor and Schulten, 2008). We predict that all clustered Pcdh EC linkers are fully occupied with three Ca2+ ions because all Ca2+-binding motifs are strictly conserved in each of the five EC linker regions (96% conservation of canonical Ca2+-binding residues in Figure 1B). To determine how to orient EC4-6 relative to each other and EC3, we surveyed cadherin structures with at least two ECs and canonical three-Ca2+ linker regions (Table S1). We defined the tilt angle as the angle between the long axes of two adjacent ovoid EC repeats (Figure 4A). Rotation of each EC around this primary axis determines the azimuthal rotation of adjacent ECs, which reflects the orientation of the cadherin fold for consecutive repeats. The average tilt and azimuthal angles for 17 canonical classical cadherin EC repeat pairs are 153±6° and 239±9°, respectively, versus 165±6° and 217±7° for the 13 non-classical cadherins and protocadherins (protocadherin-15, cadherin-23, A1 and C3). Thus adjacent EC pairs of non-classical and protocadherins are more linear than classical cadherins. The origin of this difference is unknown, but could denote an important structural difference between cadherin subfamilies.
Figure 4.

Clustered Pcdh EC1-6 homology models as elongated molecules. (A) The orientation of adjacent EC repeats can be defined by the tilt and azimuthal rotation of the EC domain principal axes. (B) Distribution of tilt and azimuthal angles from adjacent EC repeat pairs of classical (black), Pcdh15 and Cdh23 (blue) and clustered Pcdh (red). Gray and pink open circles represent averages of classical and non-classical/clustered Pcdh tilt and azimuthal angles. Crosses show the EC repeat pairs closest to the average values; these pairs were used to generate the homology models. (C) Homology models of A1 using the C3 EC2-3 (blue) and cadherin-6 EC1-2 (3lnd_a; green) model pairs. Classical EP-cadherin (1l3w; magenta; five EC repeats) is shown for comparison. (D) Models of trans-interacting antiparallel full-length dimers corresponding to the A1 EC12 (lilac) A1 EC23 (blue), C3 EC1 (yellow) and C3 extended (green) interfaces based on the full-length A1 homology model. See also Table S1.
We used two EC pairs as a starting model to position clustered Pcdh EC4-6: one model using the C3 EC2-EC3 pair, closest to the non-classical/protocadherin average (tilt = 164°, azimuthal = 212°), and one using mouse cadherin-6 EC1-EC2 (3lnd_a), closest to the classical cadherin average (tilt = 154°, azimuthal = 239°). We then used these models to construct homology models of the full mouse Pcdhγ A1 extracellular domain (Figure 4C). The two overall linear structures vary slightly in their overall curvature, but neither is incompatible with any of the potential interfaces in the A1 or C3 structures (Figure 4D).
The resulting clustered Pcdh model differs significantly from the five-EC classical cadherin structures, due to the relative orientations of the repeats (Figure 4C). Non-canonical Ca2+-binding sites in classical cadherins cause a significantly smaller EC3-EC4 tilt angle (~135°), yielding a nearly perpendicular orientation of EC1 and EC5. The clustered Pcdh models are more linear, reflecting the fact that all canonical Ca2+-binding motifs are completely conserved across all linker regions in clustered Pcdhs.
The A1 EC23 and C3 extended interfaces lack potential glycosylation sites
Posttranslationally-modified N- and O-glycosylation sites in classical cadherins expressed in Xenopus laevis or in human embryonic kidney 293 cells are distributed away from the homophilic binding interface (Boggon et al., 2002; Harrison et al., 2011), indicating that glycosylation is disfavored at cadherin interfaces. Since our recombinantly-expressed Pcdh constructs are not glycosylated, we mapped predicted glycosylation sites on the Pcdh structures. We used the NetNGlyc v1.0 and NetOGlyc v4.0 servers to predict N- and O- glycosylation sites, respectively, in all human clustered Pcdhs (Blom et al., 2004; Steentoft et al., 2013). Fourteen predicted N-glycosylation sites distributed across EC2-6 are well conserved across similar isoforms, and none overlap with the interfaces (Figure 5). Surprisingly, only two O-glycosylation sites were predicted: T325 in PcdhγA1 and position 357 (mouse PcdhγA1 numbering) in Pcdhα2, Pcdhα9 and Pcdhα10 (Figure 5). This contrasts with the high O-glycosylation seen in C-, N- and E-cadherin structures (Boggon et al., 2002; Harrison et al., 2011). The NetOGlyc server predicts no O-glycosylation sites for mouse C-, N-, or E-cadherin, indicating that it is not well trained for predicting O-glycosylation sites in cadherins.
Figure 5.

Predicted clustered Pcdh glycosylation sites largely do not overlap with the identified antiparallel interfaces. N- (magenta) or O- (black) glycosylation sites predicted in at least five human clustered Pcdh isoforms or a single C-type isoform and O-mannosylation sites (purple) from the SimpleCell strategy (Vester-Christensen et al., 2013) in clustered Pcdh EC repeats are overlaid with the observed interfaces (A1 EC12, lilac; A1 EC23, blue; C3 EC1, yellow; C3 extended, green; and A1 EC23 and C3 extended overlapping, orange). The left and right panels are related by a 180° rotation; numbering corresponds to mouse PcdhγA1.
O-mannosylation sites in clustered Pcdhs were identified by the SimpleCell strategy (Vester-Christensen et al., 2013). Many O-mannosylation sites were found in EC2, EC3, EC5, and EC6 of various isoforms. Several O-mannosylated residues overlap the A1 EC12 interface (T185, S195, and T197) (Figure 5). A site at position 300, found in PcdhγB1, overlaps the A1 EC23 and C3 extended interfaces, but a methionine in γA1 (M300) or arginine in γC3 indicates this glycosylation is absent in these isoforms. While glycosylation site prediction could not provide striking support for a particular interface, it does suggest that glycosylation may prevent the A1 EC12 interaction.
A1 EC23 and C3 extended interfaces correlate with isoform specific conservation
Sequence conservation analysis confirmed the homophilic interaction epitopes in the Drosophila Dscam1 variable exons (Meijers et al., 2007). We searched for regions of high isoform-specific conservation, which may signify regions that impart homophilic specificity, by comparing the conservation of Pcdh paralogs in humans to orthologs of each isoform in mammalian species. Conservation in mammalian orthologs is higher (average overall identity for all orthologs is 78±6%) than in human paralogs (50% overall identity). Thus, we defined an isoform conservation ratio (ICR), the ratio of ortholog conservation of an isoform to paralog conservation of all isoforms in a species (in our case, humans), as an index for residue-by-residue sequence diversification. We then calculated residue-by-residue average ICR for all 60 mammalian isoforms (Figure 6A).
Figure 6.

Isoform-specific conservation in clustered Pcdhs localizes to EC2 and EC3 surfaces. (A) ICR average of all clustered Pcdh isoforms compared to human clustered Pcdhs, as a sliding average with a 5-residue window size. (B) Residual of the ICR value between the overall and subfamily averages (α = black, β = red, γ = blue, and C-type = magenta). (C) ICR values mapped on the EC2 and EC3 surfaces with a high density of high ICR values, numbered according to mouse PcdhγA1. (D) ICR values mapped on full-length homology models of the A1 EC23 and C3 extended interfaces. (E) Comparison of the average ICR values of non-interface (white) and interface (gray) residues. Interface residues are defined as those having a BSA of at least 0 Å2 (light gray) or 55 Å2 (dark gray). See also File S3 and S4.
Average ICR values for EC1-3 are significantly higher than in EC4-6, and highest for EC2 and EC3 (Figure 6A). This preponderance of isoform-specific positions corroborates clustered Pcdh-driven cell aggregation assays that demonstrated the importance of EC23 in homophilic specificity (Schreiner and Weiner, 2010). These data also correlate with the presence of positively selected sites in EC2 and EC3, where the rate of nonsynonymous substitution at some positions is higher than the rate of synonymous mutations, implying that isoforms are under selective diversification (Wu, 2005). The average ICR of these previously identified positively selected sites (4.5±0.5) is significantly higher than the overall average ICR (2.09±0.05), supporting the ICR as a metric for selective diversification. We also compared the ICR values of clustered Pcdh subfamilies (α, β, γ, C-type) to the entire cluster by calculating the residual of the subfamily ICR from the entire cluster ICR (Figure 6B). No region has significantly different ICR values in the α, β, and γ subfamilies, supporting the notion that the homophilic specificity regions are common to all isoforms. In contrast, the C-type isoforms contain some regions with significantly decreased ICR values, specifically in EC2 and EC3. These regions could suggest a difference in C-type subfamily homophilic interactions and/or could reflect the fact that C-types may be under stronger conservation selection due to their essentiality in neuronal development (Chen et al., 2012).
The highest concentration of large ICRs is on a small portion of EC2 and a face on EC3 (Figure 6C), two regions proximal to both the A1 EC23 and C3 extended interfaces (Figure 6D). For example, the previously noted hydrogen-bond network in the C3 extended interface (Figure 3D) includes residues with high ICR values (T70 = 5.48, S255 = 6.18 and H256 = 5.49), indicating that this interaction is highly specific to the γC3 isoform.
To quantify the correlation between the observed interfaces and the ICR values we calculated the average ICR value for non-interface and interface residues (judged as residues with a buried surface area (BSA) of more than 0 Å2). The average ICR of non-interface residues is 2.36±0.08, while the average ICR of interface residues of the A1 EC23 and C3 extended complexes are 3.1±0.3 and 2.7±0.2, respectively (Figure 6E). In contrast the average interface ICR for the A1 EC12 and C3 EC1 complexes are both 2.3±0.2. To determine whether the correlation between ICR and our interfaces was amplified in residues that contribute most significantly to the interface, we reduced our selection of interface residues to those with a BSA of at least 55 Å2 per residue (Figure 6E). At this BSA cut-off, the average ICR of interface residues increases to 3.8±0.7 for A1 EC23 and 3.7±0.5 for C3 extended, whereas the A1 EC12 and C3 EC1 interface ICR values at this BSA cut-off are indistinguishable from non-interface ICR values. This result indicates that residues in the A1 EC23 and C3 extended complexes contributing appreciably to the interface surface area are more likely to have a high ICR value, which supports the biological significance of these interfaces.
Evolutionary covariance in clustered Pcdhs largely corresponds to intramolecular structural features
Pairs of residues near each other in the structure can be identified via evolutionary covariance caused by compensatory mutations. These evolutionary correlations can indicate structural contacts within a given protein (Aurell and Ekeberg, 2012; Hopf et al., 2012; Kamisetty et al., 2013; Marks et al., 2011) or between proteins that interact (Hopf et al., 2014; Ovchinnikov et al., 2014). We used EVCouplings (adapted from (Hopf et al., 2014; Marks et al., 2011)) to search for evolutionary correlations arising from homotypic interactions from an alignment generated with mouse PcdhγC3 as a query. The top 320 (~alignment length/2) evolutionary correlations (Figure S4A) predominantly reflect structural features of the cadherin fold (Figure 7A). Eleven pairs in EC1-4 are not close to each other in the monomer, but nevertheless higher scoring than many true positives, suggesting they represent oligomeric contacts (Figure 7A). These pairs – between EC2 and EC3, or EC1 and EC4 – support an antiparallel complex. Our structures corroborate the EC2-EC3 pairs, and although we did not crystallize EC4, our full-length homology model suggests EC1 and EC4 interact (Figure 4D).
Figure 7.

Evolutionary covariance supports antiparallel oligomeric contacts. (A) The top 320 correlated pairs (sequence length/2) are shown with the contact map of the EC1-6 clustered Pcdh homology model (gray) for reference. Correlated pairs matching structural constraints are shown in black while predicted oligomeric contact pairs are in orange. The observed interface contact residues are also mapped (A1 EC23, blue and C3 extended, green). The predicted oligomeric contact correlated pairs are mapped (orange spheres) on the A1 EC23 (B) and C3 extended (C) complex models with a line between coupled residues. See also Figure S4, File S5 and S6.
The 11 non-structural correlated pairs in EC1-4 come in close proximity in the A1 EC23 and C3 extended interfaces (Figure 7B and 7C), corroborating our posited interfaces. A plot of coupling strength versus pair distance reveals that these pairs are outliers compared to the rest of the observed monomeric pairs (Figure S4B). Using the A1 EC23 and C3 extended full-length complex models to define the pair distance of these 11 correlated pairs as intermolecular contacts, we find that the pair distance-coupling strength distribution is better preserved, supporting their significance as oligomeric contacts instead of intramolecular structural ones. Interestingly, the A1 EC23 model yields shorter distances overall for the 11 correlated pairs which is evident from the sliding of A1 EC23 and C3 extended interfaces relative to each other along the long cadherin axis (Figure 7B and 7C). This could indicate that A1 EC23 better represents homodimeric clustered Pcdh interfaces. However, because the clustered Pcdh locus has relatively few C-type isoforms, our sequence alignment contains many more non-C-type isoform sequences. This may manifest as a bias of observed correlated pairs towards non-C-type versus C-type interfaces if, as biological data suggest (Chen et al., 2012), C-type and non-C-type have different modes of interaction.
DISCUSSION
There is strong evidence that EC1-3 mediate clustered Pcdh specificity of cell surface signaling from cell-aggregation assays and sequence analysis (Schreiner and Weiner, 2010; Wu, 2005). Our structures of mouse PcdhγA1 and PcdhγC3 EC1-3 and bioinformatics analyses provide further evidence for the importance of EC2 and EC3 and offer the first hypotheses for these interactions. We propose two biologically relevant interactions: A1 EC23 and C3 extended. The physicochemical properties of these interfaces are consistent with canonical biological interactions, and interface residues are not predicted glycosylation sites and are conserved in isoform orthologs. The antiparallel arrangement is consistent with evolutionary correlations observed between EC2 and EC3, and EC1 and EC4. Lastly, isoform-specific conservation suggests that all subfamilies have similar modes of homophilic recognition, with the possible exception of the C-type isoforms.
The combination of an antiparallel arrangement of the specificity-determining homodimer and the linearity of our EC1-6 homology model limits the geometry of a trans complex. To satisfy both constraints, the overall length of A1 EC23 or C3 extended complexes is approximately 40 nm (Figure 4D). This distance is consistent with intercellular distances of chemical synapses (20–40 nm), suggesting that this trans complex architecture would be effective in determining synaptic specificity at nascent synapses (Phillips et al., 2003).
The two interfaces we identified comprise similar sets of residues with high ICR values, but only partially overlap (Figures 2E and 5). Currently, we cannot offer any explanation for these similar yet distinct interfaces. However, the biological behaviors of the two isoforms we crystallized are different: C-type Pcdhγ isoforms are necessary for neonatal neuronal survival in mice, while A-type Pcdhγ isoforms show redundancy in behavior (Chen et al., 2012). Thus, A1 EC23 and C3 extended interfaces may be representative of γ-type and C-type Pcdh isoforms, respectively, and reflect structural adaptations to use overlapping surfaces to encode a large diversity of preferentially homotypic dimerization interfaces.
In our data, we find some indirect support for two potential cis architectures in relation to the identified antiparallel trans interactions (Figure 8). The lack of predicted glycosylation sites on a face of EC4-6 opposite the homophilic EC2-EC3 interface (Figure 5) and evolutionary correlations in EC5 and EC6 that are not part of intramolecular contacts (Figure 7A) suggest that EC5-6 could be part of promiscuous cis interactions (Figure 8A). Supporting this model is the finding that α isoforms require EC6 of a carrier isoform (β or γ) to be surface-expressed (Thu et al., 2014), suggesting cis EC6 interactions across paralogs. Furthermore, deletion of EC1-3 does not impair cis-multimers (Schreiner and Weiner, 2010), consistent with a model in which the C-terminal EC domains provide further multimerization interfaces. An alternative model derives from the A1 EC12 interface, which is statistically assessed as potentially biological (Table 2). The A1 EC12 interface is on the opposite side of the A1 EC23 interface, allowing a zippering of Pcdhs at an intercellular junction to form an oligomer (Figure 8B), implying the presence of trans-mediated promiscuous cis interactions. This model is supported by a relatively conserved interface across isoforms, but is challenged by predicted glycoslation sites on the A1 EC12 interface. While neither model fully clarifies the mechanism used to enforce combinatorial homophilic interactions or specifies the possible multimeric state of these complexes, they can serve as testable hypotheses. Future experiments will require the development of biochemical and biophysical methods for investigating weak adhesion at a molecular level.
Figure 8.

Model of clustered protocadherin antiparallel trans and possible cis interactions. (A) Lack of predicted glycosylation sites on one face of EC4-6 allows a model where EC4-6 engage in promiscuous cis interactions to promote oligomerization. (B) The A1 EC12 interaction, which does not show preponderance for high ICR value residues, could support a model for trans-mediated cis oligomerization through EC1-2. Isoform types are shown as either blue or green to indicate homophilic (same color) or promiscuous (different color) interactions.
The interactions we identified are novel for the cadherin superfamily. Unlike classical cadherins that interact through EC1 in an ‘X-dimer’ or strand-swapping mechanism (Brasch et al., 2012; Sotomayor et al., 2014), these interactions are mediated by EC2 and EC3. The cis interactions of classical cadherins are also distinct as they occur through EC1 and EC2 and are parallel in nature (Boggon et al., 2002; Nagar et al., 1996). The most similar interface described thus far is the antiparallel “hand-shake” Pcdh15-Cdh23 interaction in the tip link of hair cells (Sotomayor et al., 2012). The elongated antiparallel interaction spanning multiple EC repeats of the Pcdh15-Cdh23 complex and in the clustered Pcdh interfaces described here may be a general adhesion strategy for other cadherin superfamily interactions.
We have described homophilic interactions of two clustered Pcdh isoforms and presented physicochemical and bioinformatics evidence for their biological significance. Overall, we showed that the A1 EC23 and C3 extended interfaces are the most likely to be relevant in vivo. It is clear from these interfaces and the presence of both cis and trans interactions that the architecture of the entire clustered Pcdh signaling complex is multifaceted. In addition, clustered Pcdhs play many roles in neuronal development, suggesting that different complex architectures and interactions with other proteins provide further layers of complexity, which we need to better define before we reach a full structural understanding of these proteins. While cell-based aggregation assay have been indispensable for studying clustered Pcdh interactions, these assays have not yet been used to distinguish between cis and trans interactions (Schreiner and Weiner, 2010; Thu et al., 2014). Thus, better biochemical methods are needed to verify these and other interactions. The structures, homology models, and interactions we describe provide a glimpse into the details of this complex system and constraints to better study clustered Pcdh signaling assemblies.
EXPERIMENTAL PROCEDURES
Protein expression and purification
Mouse PcdhγA1 EC1-3 (residues 1-311 referred to as A1) and PcdhγC3 EC1-3 (residues 1-314, referred to as C3) with C- and N-terminal hexahistidine tags, respectively, were expressed in BL21 Gold (DE3) E. coli cells in Terrific Broth, induced at OD600 = 0.8 with 0.5 mM IPTG at 37°C for 4 h. Cells were lysed by sonication in 8 M guanidine hydrochloride (GuHCl), 20 mM imidazole, 50 mM HEPES pH 7.5, 2 mM CaCl2, 1 mM benzamidine and 1 mM phenylmethanesulfonylfluoride. Cleared lysates were diluted to 5 M GuHCl, loaded onto Ni-sepharose, washed with 5 M GuHCl, 250 mM NaCl, 25 mM imidazole pH 8, 50 mM Tris pH 8.5, and eluted with 200 mM imidazole. A1 was refolded overnight at room temperature by drop dilution from 2 mg/mL protein in elution buffer to a final concentration of 0.2 M GuHCl with refolding buffer (100 mM Tris pH 8.5, 10 mM CaCl2, 1 mM EDTA, 3 mM GSH, 0.3 mM GSSG). C3 was refolded at 1 mg/mL by 12-hour dialysis steps into 3, 1.5 and 0 M GuHCl in refolding buffer with 0.2 M sucrose and 0.5 M L-arginine monohydrochloride. Concentrated refolded protein was purified by size-exclusion chromatography on a Superdex200 (GE Healthcare) in 20 mM Tris pH 8.5, 200 mM NaCl, 2 mM CaCl2. C3 was further purified on Resource Q (GE Healthcare) with a gradient of 50 to 500 mM NaCl in 20 mM Tris pH 8.5, 2 mM CaCl2. The A1 protein molecular mass was determined in runs at 5 mg/mL (137 μM) over a Superdex S200 10/300 column with an in-line MALLS instrument (Wyatt) in 20 mM Tris pH 8.5, 200 mM NaCl, 2 mM CaCl2.
Crystallization, data collection, and structure determination
A1 crystals were grown by vapor diffusion at room temperature in 100 mM Tris pH 8.5, 7.5% PEG-8000 and 200 mM NDSB-221 (Hampton Research, Aliso Viejo, CA). Crystals were cryo-protected in reservoir plus 20% glycerol. C3 crystals were grown by vapor diffusion at 4°C. Initial crystals, obtained with a drop ratio of 0.5 μL 4.3 mg/mL C3 to 0.3 μL reservoir (100 mM HEPES pH 7, 60% MPD, 100 mM CaCl2), were used as seeds to grow larger crystals in 100 mM HEPES pH 7, 20% MPD with a 0.5 μL protein : 0.2 μL reservoir : 0.1 μL seeds ratio. Crystals were cryo-protected in reservoir plus 15% MPD. Crystals were cryocooled in liquid N2. X-ray data were collected according to Table 1 and processed in HKL2000 (Otwinowski and Minor, 1997). The A1 structure was determined by molecular replacement in PHENIX (Adams et al., 2010), using cadherin-23 EC12 as a starting model (Sotomayor et al., 2010). The C3 structure was solved by molecular replacement using a C3 homology model based on A1. Model building was done in COOT (Emsley and Cowtan, 2004) and refinement in PHENIX (Adams et al., 2010). Both structures contain two protomers per asymmetric unit. In addition to canonical inter-EC linker Ca2+ ions, the C3 structure has three Ca2+ ions bridging a EC3-EC3, tail-end-to-tail-end, crystal packing interface between the two chains, chelated by EC3-EC4 linker residues in a non-physiological way. Validation reports for the A1 (File S1) and C3 (File S2) are provided in Supplemental Information.
Structural analysis of interface significance
PISA webserver was used to analyze the observed Pcdh interfaces (Krissinel and Henrick, 2007). ΔASA is (ASAA+ASAB−ASAAB)/2, where AB is a complex of A and B. BSA is the solvent-accessible surface area buried upon complex formation for a given residue. The SC program in CCP4 was used to calculate surface complementarity according to the following parameters (Lawrence and Colman, 1993): dot density = 15 Å−2, interface separation = 8 Å, trim width = 1.5 Å, probe radius = 1.7 Å, weight factor 0.5 Å−2. NOXclass was used to evaluate the observed interfaces using interface area, interface area ratio, correlation between interface and protein surface, and gap volume index parameters (Zhu et al., 2006).
Homology model construction
Structures of cadherin proteins containing at least two EC repeats were collected from the Protein Data Bank (Table S1). EC N was aligned to EC N+1 for each pair and Singular Value Decomposition was used to calculate the principal components of the EC N repeat and the original and aligned positions. The angles between the respective components were calculated to determine the tilt (between the first components) and azimuthal (average of the second and third components) angles. Models were constructed in PyMOL (Schrodinger) and then Modeller (Šali and Blundell, 1993) was used to generate the EC1-6 homology model.
Sequence analyses
For ICR analysis, Pcdh isoforms sequences were collected from the UCSC Genome Browser (Kent et al., 2002) to create a multiple sequence alignment. Overall %identity of the most common residue at each position was used to calculate ICR values. The alignment (File S3) and ICR data (File S4) are provided in Supplemental Information.
Evolutionary couplings were predicted using EVcouplings (Aurell and Ekeberg, 2012; Marks et al., 2011; Morcos et al., 2011), applying a pseudolikelihood maximization (PLM) approximation (Balakrishnan et al., 2011; Ekeberg et al., 2013; Kamisetty et al., 2013). The multiple sequence alignment used was generated starting with EC1-EC6 of mouse Pcdhγc3 (Q91XX1_MOUSE, residues 30–670) as a query sequence for Jackhmmer 3.1 (Johnson et al., 2010). The alignment was created in five iterations against the UniProt Reference Cluster UniRef100 (release 02/2015 (Suzek et al., 2007)) then filtered to exclude sequences that are not part of the clustered protocadherin family. To build the global maximum entropy model, sequences with at least 90% identity were clustered and weighted according to their cluster size (theta 0.1). Alignment columns that contained gaps in 50% or more of the members were excluded from the model and later predictions. The effect number of sequences in the final alignment was 3525.48 (sequences/alignment length = 5.56). Correlated pairs were normalized by the magnitude of the minimum coupling score to suppress background noise (Hopf et al., 2014). Pairs with normalized scores above 1.6 (~alignment length/2 = 320 couplings) are considered statistically significant (>80% predictive of contact; true positive rate (<8 Å distance) = 66% in EC1-EC3, 84% in EC2-EC3). The alignment (File S5) and raw coupling scores (File S6) for all pairs are included in Supplemental Information.
Supplementary Material
Highlights.
Clustered protocadherin EC1-3 fragments form antiparallel complexes in crystals
A full extracellular cadherin repeat region model yields an extended molecule
Isoform-specific conservation of interfaces explains strict homophilic specificity
Evolutionary correlations provide support for antiparallel arrangement
Acknowledgments
We thank Dr. Marcos Sotomayor and Dr. Josh Sanes for helpful discussions. We thank Sriram Srikant for help with phylogenetic analysis. Financial support (to J.M.N.) was provided by the National Defense Science and Engineering Graduate Fellowship. We thank the beamline staff of NE-CAT at the Advanced Photon Source (Argonne, IL, USA) for help with data collection. NE-CAT is funded by NIH (P41 GM103403 and S10 RR029205), and the Advanced Photon Source by the U.S. Department of Energy (DE-AC02-06CH11357).
Footnotes
AUTHOR CONTRIBUTIONS
W.A.W. and R.G. initiated the project; J.M.N., W.A.W. and S.L. screened constructs for purification and crystallization; J.M.N. and W.A.W. crystallized and solved structures; S.L. performed MALLS measurements; J.M.N constructed homology model and completed ICR analysis; J.M.N., C.P.I.S, D.S.M and R.G. completed evolutionary correlation analysis; R.G. directed the research; J.M.N. and R.G. wrote the manuscript with input from all authors.
We declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurell E, Ekeberg M. Inverse ising inference using all the data. Phys Rev Lett. 2012;108:1–5. doi: 10.1103/PhysRevLett.108.090201. [DOI] [PubMed] [Google Scholar]
- Bahadur RP, Chakrabarti P, Rodier F, Janin J. A Dissection of Specific and Non-specific Protein – Protein Interfaces. J Mol Biol. 2004;336:943–955. doi: 10.1016/j.jmb.2003.12.073. [DOI] [PubMed] [Google Scholar]
- Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79:1061–1078. doi: 10.1002/prot.22934. [DOI] [PubMed] [Google Scholar]
- Biswas S, Emond MR, Jontes JD. The clustered protocadherins Pcdhα and Pcdhγ form a heteromeric complex in zebrafish. Neuroscience. 2012;219:280–289. doi: 10.1016/j.neuroscience.2012.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Murray J, Chappuis-flament S, Wong E, Gumbiner BM, Shapiro L. C-Cadherin Ectodomain Structure and Implications for Cell Adhesion Mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- Brasch J, Harrison OJ, Honig B, Shapiro L. Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol. 2012;22:299–310. doi: 10.1016/j.tcb.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailliez F, Lavery R. Cadherin mechanics and complexation: the importance of calcium binding. Biophys J. 2005;89:3895–3903. doi: 10.1529/biophysj.105.067322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti P, Janin J. Dissecting protein-protein recognition sites. Proteins Struct Funct Genet. 2002;47:334–343. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- Chen WV, Alvarez FJ, Lefebvre JL, Friedman B, Nwakeze C, Geiman E, Smith C, Thu CA, Tapia JC, Tasic B, et al. Functional Significance of Isoform Diversification in the Protocadherin Gamma Gene Cluster. Neuron. 2012;75:402–409. doi: 10.1016/j.neuron.2012.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekeberg M, Lövkvist C, Lan Y, Weigt M, Aurell E. Improved contact prediction in proteins: Using pseudolikelihoods to infer Potts models. Phys Rev E - Stat Nonlinear, Soft Matter Phys. 2013;87:1–16. doi: 10.1103/PhysRevE.87.012707. [DOI] [PubMed] [Google Scholar]
- Emond MR, Jontes JD. Inhibition of protocadherin-α function results in neuronal death in the developing zebrafish. Dev Biol. 2008;321:175–187. doi: 10.1016/j.ydbio.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Esumi S, Kakazu N, Taguchi Y, Hirayama T, Sasaki A, Hirabayashi T, Koide T, Kitsukawa T, Hamada S, Yagi T. Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nat Genet. 2005;37:171–176. doi: 10.1038/ng1500. [DOI] [PubMed] [Google Scholar]
- Fernández-Monreal M, Kang S, Phillips GR. Gamma-protocadherin homophilic interaction and intracellular trafficking is controlled by the cytoplasmic domain in neurons. Mol Cell Neurosci. 2009;40:344–353. doi: 10.1016/j.mcn.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett AM, Weiner Ja. Control of CNS synapse development by {gamma}-protocadherin-mediated astrocyte-neuron contact. J Neurosci. 2009;29:11723–11731. doi: 10.1523/JNEUROSCI.2818-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett AM, Schreiner D, Lobas Ma, Weiner Ja. Gamma-Protocadherins Control Cortical Dendrite Arborization by Regulating the Activity of a FAK/PKC/MARCKS Signaling Pathway. Neuron. 2012;74:269–276. doi: 10.1016/j.neuron.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Lin C, Meng S, Wang X. Proteomics analysis reveals overlapping functions of clustered protocadherins. Mol Cell Proteomics. 2010;9:71–83. doi: 10.1074/mcp.M900343-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, et al. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure. 2011;19:244–256. doi: 10.1016/j.str.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf Ta, Colwell LJ, Sheridan R, Rost B, Sander C, Marks DS. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf TA, Schärfe CPI, Rodrigues JPGLM, Green AG, Sander C, Bonvin AMJJ, Marks DS. Sequence co-evolution gives 3D contacts and structures of protein complexes. eLife. 2014;3:e03430. doi: 10.7554/eLife.03430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J, Rodier F. Protein-protein interaction at crystal contacts. Proteins. 1995;23:580–587. doi: 10.1002/prot.340230413. [DOI] [PubMed] [Google Scholar]
- Johnson LS, Eddy SR, Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 2010;11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc Natl Acad Sci U S A. 2013;110:1–6. doi: 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko R, Kato H, Kawamura Y, Esumi S, Hirayama T, Hirabayashi T, Yagi T. Allelic gene regulation of Pcdh-α and Pcdh-γ clusters involving both monoallelic and biallelic expression in single Purkinje cells. J Biol Chem. 2006;281:30551–30560. doi: 10.1074/jbc.M605677200. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostadinov D, Sanes JR. Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit formation. eLife. 2015 Jul 3; doi: 10.7554/eLife.08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Colman PM. Shape complementarity at protein-protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Lefebvre JL, Zhang Y, Meister M, Wang X, Sanes JR. gamma-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development. 2008;135:4141–4151. doi: 10.1242/dev.027912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Kostadinov D, Chen WV, Maniatis T, Sanes JR. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 2012;488:517–521. doi: 10.1038/nature11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xiao H, Chiou TT, Jin H, Bonhomme B, Miralles CP, Pinal N, Ali R, Chen WV, Maniatis T, et al. Molecular and Functional Interaction between Protocadherin- C5 and GABAA Receptors. J Neurosci. 2012;32:11780–11797. doi: 10.1523/JNEUROSCI.0969-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, Sander C. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijers R, Puettmann-Holgado R, Skiniotis G, Liu J, Walz T, Wang J, Schmucker D. Structural basis of Dscam isoform specificity. Nature. 2007;449:487–491. doi: 10.1038/nature06147. [DOI] [PubMed] [Google Scholar]
- Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci. 2011;108:E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, Umitsu M, Murata Y, Shibata N, Udaka K, Higuchi Y, Akutsu H, Yamaguchi T, Yagi T, Ikegami T. Structure of the cadherin-related neuronal receptor/protocadherin-alpha first extracellular cadherin domain reveals diversity across cadherin families. J Biol Chem. 2006;281:33650–33663. doi: 10.1074/jbc.M603298200. [DOI] [PubMed] [Google Scholar]
- Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology, Volume 276: Macromolecular Crystallography, Part A. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. eLife. 2014;3:e02030. doi: 10.7554/eLife.02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GR, Tanaka H, Frank M, Elste A, Fidler L, Benson DL, Colman DR. Gamma-protocadherins are targeted to subsets of synapses and intracellular organelles in neurons. J Neurosci. 2003;23:5096–5104. doi: 10.1523/JNEUROSCI.23-12-05096.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponstingl H, Henrick K, Thornton JM. Discriminating Between Homodimeric and Monomeric Proteins in the Crystalline State. 2000;57:47–57. doi: 10.1002/1097-0134(20001001)41:1<47::aid-prot80>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- Prasad T, Weiner Ja. Direct and Indirect Regulation of Spinal Cord Ia Afferent Terminal Formation by the γ-Protocadherins. Front Mol Neurosci. 2011;4:1–12. doi: 10.3389/fnmol.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad T, Wang X, Gray Pa, Weiner Ja. A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin-gamma gene cluster. Development. 2008;135:4153–4164. doi: 10.1242/dev.026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Wojtowicz WM, Andre I, Qian B, Wu W, Baker D, Eisenberg D, Zipursky SL. A double S shape provides the structural basis for the extraordinary binding specificity of Dscam isoforms. Cell. 2008;134:1007–1018. doi: 10.1016/j.cell.2008.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, Ballif BA, Buchanan SM, Phillips GR, Maniatis T. Phosphorylation of protocadherin proteins by the receptor tyrosine kinase Ret. Proc Natl Acad Sci U S A. 2010;107:13894–13899. doi: 10.1073/pnas.1007182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner D, Weiner JA. Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proc Natl Acad Sci U S A. 2010;107:14893–14898. doi: 10.1073/pnas.1004526107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M, Schulten K. The allosteric role of the Ca2+ switch in adhesion and elasticity of C-cadherin. Biophys J. 2008;94:4621–4633. doi: 10.1529/biophysj.107.125591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M, Weihofen WA, Gaudet R, Corey DP. Structural determinants of cadherin-23 function in hearing and deafness. Neuron. 2010;66:85–100. doi: 10.1016/j.neuron.2010.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M, Weihofen WA, Gaudet R, Corey DP. Structure of a force-conveying cadherin bond essential for inner-ear mechanotransduction. Nature. 2012;492:128–132. doi: 10.1038/nature11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M, Gaudet R, Corey DP. Sorting out a promiscuous superfamily: towards cadherin connectomics. Trends Cell Biol. 2014;24:524–536. doi: 10.1016/j.tcb.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KTBG, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos-Silva L, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013;32:1478–1488. doi: 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Marcheva B, Meng S, Liang Fa, Kohsaka A, Kobayashi Y, Xu AW, Bass J, Wang X. Gamma-protocadherins regulate the functional integrity of hypothalamic feeding circuitry in mice. Dev Biol. 2010;339:38–50. doi: 10.1016/j.ydbio.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- Thu CA, Chen WV, Rubinstein R, Chevee M, Wolcott HN, Felsovalyi KO, Tapia JC, Shapiro L, Honig B, Maniatis T. Single-Cell Identity Generated by Combinatorial Homophilic Interactions between α, β, and γ Protocadherins. Cell. 2014;158:1045–1059. doi: 10.1016/j.cell.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester-Christensen MB, Halim A, Joshi HJ, Steentoft C, Bennett EP, Levery SB, Vakhrushev SY, Clausen H. Mining the O-mannose glycoproteome reveals cadherins as major O-mannosylated glycoproteins. Proc Natl Acad Sci. 2013;110:21018–21023. doi: 10.1073/pnas.1313446110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Weiner Ja, Levi S, Craig AM, Bradley A, Sanes JR. Gamma protocadherins are required for survival of spinal interneurons. Neuron. 2002;36:843–854. doi: 10.1016/s0896-6273(02)01090-5. [DOI] [PubMed] [Google Scholar]
- Weiner Ja, Jontes JD. Protocadherins, not prototypical: a complex tale of their interactions, expression, and functions. Front Mol Neurosci. 2013;6:1–10. doi: 10.3389/fnmol.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner Ja, Wang X, Tapia JC, Sanes JR. Gamma protocadherins are required for synaptic development in the spinal cord. Proc Natl Acad Sci U S A. 2005;102:8–14. doi: 10.1073/pnas.0407931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q. Comparative genomics and diversifying selection of the clustered vertebrate protocadherin genes. Genetics. 2005;169:2179–2188. doi: 10.1534/genetics.104.037606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T. Molecular codes for neuronal individuality and cell assembly in the brain. Front Mol Neurosci. 2012;5:1–11. doi: 10.3389/fnmol.2012.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota S, Hirayama T, Hirano K, Kaneko R, Toyoda S, Kawamura Y, Hirabayashi M, Hirabayashi T, Yagi T. Identification of the cluster control region for the protocadherin-beta genes located beyond the protocadherin-beta cluster. J Biol Chem. 2011;286:31885–31895. doi: 10.1074/jbc.M111.245605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Domingues FS, Sommer I, Lengauer T. NOXclass: prediction of protein-protein interaction types. BMC Bioinformatics. 2006;7:27. doi: 10.1186/1471-2105-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky SL, Sanes JR. Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell. 2010;143:343–353. doi: 10.1016/j.cell.2010.10.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.