

Summary
Exercise is essential in regulating energy metabolism and whole body insulin sensitivity. To explore the exercise signaling network we undertook a global analysis of protein phosphorylation in human skeletal muscle biopsies from untrained healthy males before and after a single high-intensity exercise bout revealing 1,004 unique exercise-regulated phosphosites on 562 proteins. These included substrates of known exercise-regulated kinases (AMPK, PKA, CaMK, MAPK, mTOR), yet the majority of kinases and substrate phosphosites have not previously been implicated in exercise signaling. Given the importance of AMPK in exercise-regulated metabolism we performed a targeted in vitro AMPK screen and employed machine learning to predict exercise-regulated AMPK substrates. We validated eight predicted AMPK substrates including AKAP1 using targeted phosphoproteomics. Functional characterization revealed an undescribed role for AMPK-dependent phosphorylation of AKAP1 in mitochondrial respiration. These data expose the unexplored complexity of acute exercise signaling and provide insights into the role of AMPK in mitochondrial biochemistry.
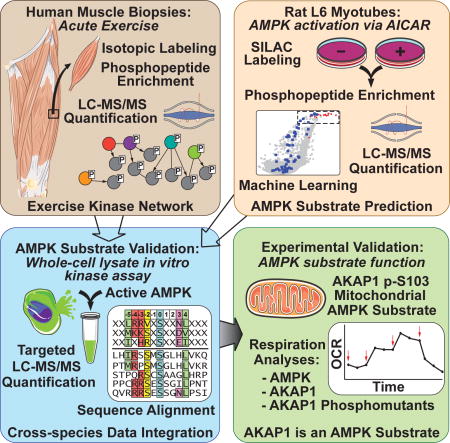
Graphical abstract
Introduction
Exercise plays an essential role in metabolic homeostasis and remains the most promising therapy for the prevention and treatment of obesity and its associated metabolic disorders. Physical inactivity causes insulin resistance in humans, while increased physical activity improves insulin sensitivity and whole body glucose and lipid metabolism (Hawley et al., 2014). Exercise regulates diverse biological functions throughout the body with a major perturbation of skeletal muscle cellular processes including fatty acid, glucose and protein metabolism via induction of a range of signal transduction pathways. Most studies of exercise signaling in muscle have focused on a limited subset of pathways, particularly those involving protein phosphorylation. The major protein kinases implicated in muscle exercise signaling include AMPK, PKA, CaMK, MAPK, PKC, FAK and mTOR (Egan and Zierath, 2013; Jeppesen et al., 2013; Sakamoto and Goodyear, 2002). The acute metabolic and mechanical demands in exercising muscle require a coordinated regulation of diverse signaling pathways that together form a complex signaling network that elicits a range of rapid cellular homeostatic adaptations. It is of major interest to define the key components within this network that confer the long term beneficial effects of exercise.
The energy sensing kinase AMPK (Hardie, 2014) plays an important role in the beneficial effects of exercise on whole body metabolic homeostasis (Richter and Ruderman, 2009; Steinberg and Kemp, 2009). Muscle-specific AMPK knockout mouse models have demonstrated a crucial role for AMPK in the metabolic adaptation of muscle during exercise (Fentz et al., 2015; O’Neill et al., 2011). However, several reports have suggested the previously characterized metabolic perturbations induced with exercise are not dependent upon AMPK (Jeppesen et al., 2013; Maarbjerg et al., 2009). Despite this, the beneficial effects of the antidiabetic drug metformin are at least partially mediated via AMPK (Foretz et al., 2014; Fullerton et al., 2013; Rena et al., 2013). Therefore, AMPK is a major therapeutic target for metabolic disease. Although AMPK regulates diverse metabolic processes, surprisingly few studies have interrogated the full repertoire of AMPK biological targets and pinpointed their specific phosphorylation sites. One such study used a chemical genetics approach and confirmed six AMPK substrates (Banko et al., 2011). However, more global approaches focusing on exercise are yet to be undertaken.
We report a global phosphoproteomic analysis of acute exercise signaling in human skeletal muscle revealing the complexity of the exercise signaling network. We quantified 8,511 unique phosphorylation sites of which ~12% were regulated with exercise, including more than 900 sites not previously annotated as exercise-responsive. By combining the human exercise phosphoproteome, two additional targeted AMPK assays, and bioinformatics analyses including machine learning, we have identified eight highly predicted AMPK substrates that are involved in mitochondrial function, vesicle transport and excitation-contraction coupling. One substrate, A kinase anchor protein 1 (AKAP1), was shown to regulate mitochondrial respiration via AMPK-dependent phosphorylation highlighting the utility of the exercise-regulated phosphoproteome. These data serve as a rich resource for future studies into how acute exercise orchestrates skeletal muscle mechanics and metabolism.
Results
Acute exercise-regulated phosphorylation in human skeletal muscle
Four healthy male volunteers (age: 24 – 27; BMI: 24.2 – 25.9 kg/m2, maximal oxygen uptake (VO2 max): 48–59 ml/kg/min, maximal watts (W) produced (Wmax): 320–375 W) underwent a single bout of high-intensity cycle exercise at 85% increasing to 92% Wmax for 9–11 min (Fig. S1). Quantitative phosphoproteomic analysis of muscle biopsies pre- and post-exercise was performed with multiplexed isobaric labeling and phosphopeptide enrichment coupled to tandem mass spectrometry (MS) (Fig. 1A). The analysis also included quantification of non-phosphorylated peptides to investigate changes in protein abundance. A total of 11,903 unique phosphopeptides (8,511 phosphosites with >90% localization probability) and 4,317 proteins were quantified in all four subjects (Fig. 1B; Tables S1, S2). The phosphorylation profile in each subject was highly reproducible with an average Pearson’s correlation coefficient r = 0.72 (Fig. 1C). We identified 1,322 phosphopeptides (1,004 phosphosites with >90% localization probability) significantly regulated with acute exercise (Fig. 1D; Table S1; adjusted P < 0.05, moderated t-test). Only 5 proteins were quantified as having altered abundance following acute exercise indicating that changes in phosphopeptide abundance are a direct result of phosphorylation. Of the regulated phosphosites, 592 were annotated in PhosphoSitePlus (Hornbeck et al., 2012) while 412 have not been annotated.
Fig. 1. Acute exercise-regulated phosphoproteome in human skeletal muscle.

(A) Experimental design of the phosphoproteomic analysis of exercise in human muscle is shown. Muscle biopsies pre- and post-exercise from four healthy males were collected. Protein was extracted, digested with Lys-C/trypsin and peptides were isobarically labeled with iTRAQ or TMT tags. Phosphopeptides were enriched by titanium dioxide chromatography and sequential elution from immobilized metal ion affinity chromatography (SIMAC). The unbound non-phosphorylated fraction and phosphorylated fraction was further separated by hydrophilic interaction liquid chromatography (HILIC) into 12–16 fractions. Each fraction was analyzed by nano-ultra high pressure liquid chromatography coupled to tandem MS (nanoUHPLC-MS/MS) on a Q-Exactive MS operated in DDA. (B) Volcano plot showing the median phosphopeptide Log2 fold-change (post- / pre-exercise) plotted against the −Log10 P-value highlighting significantly regulated phosphopeptides (blue; P < 0.05, n=4, moderated t-test). Dotted lines indicate (+/−) 1.5-fold change (Log2 = 0.58). (C) Pearson’s correlation analysis of phosphopeptide fold-change quantification (post- / pre-exercise) between the 4 subjects is shown. (D) Summary of the quantified and regulated proteome and phosphoproteome is shown.
Kinase regulation in response to acute exercise
Pathway over-representation analysis of the phosphoproteins containing exercise-regulated phosphosites revealed an enrichment of signaling pathways regulating a broad range of cellular functions, underpinning the pervasive role of exercise in human biology. This included well characterized exercise-regulated signaling pathways such as AMPK, MAPK, PKA, mTOR, S6 kinase, and Ca2+ signaling as well as pathways with a less defined role in exercise including CDK and ILK signaling (Table S3). Phosphorylation of proteins involved in insulin receptor, cell-junctional and cytoskeletal signaling including Rho and actin signaling was also significantly enriched. In addition, since kinases themselves are often modulated by phosphorylation we determined which kinases were phosphorylated in response to exercise. A total of 45 protein kinases contained at least one regulated phosphorylation site, including kinases known to be activated during exercise such as AMPK, MAPK and CAMK2 (Table S1).
We next retrieved site-specific information for experimentally annotated kinase-substrate relationships from PhosphoSitePlus. Significantly regulated phosphosites were first assigned to their upstream kinase. The relative changes in substrate phosphorylation were then used to infer kinase activity. Of the 592 identified phosphosites in PhosphoSitePlus, experimental evidence for the upstream kinase(s) was reported for only 79 sites on 66 proteins (Table S4). This analysis revealed the extensive breadth of kinases regulated by exercise comprising 15 kinases that had ≥2 substrates: AMPK, MAPKAPK2, p38 MAPK, ERK, RSK, PKA, Akt, CaMK, mTOR, ROCK, PKC, CDK, EEF2K, Src kinase family members SRC/FYN/LCK/YES and PDHK (Fig. 2A). Phosphorylation of many of these substrates was confirmed by immunoblotting using site specific antibodies (Fig. 2B). Next, we generated an integrative network of the exercise-regulated kinase interactome (Fig. 2C). Experimentally validated human protein-protein interactions were retrieved from the Human Protein Reference Database (HPRD) and STRING databases, and kinase-substrate relationships were retrieved from PhosphoSitePlus. This kinome network highlights the complexity and interconnectivity of the exercise signaling landscape that are not apparent from analyzing kinase-substrate relationships alone.
Fig. 2. Site-specific kinase-substrate regulation in response to acute exercise.

(A) Significantly regulated phosphopeptides (+/− 1.5-fold change, P < 0.05, n=4, moderated t-test) were clustered according to PhosphoSitePlus-annotated upstream kinases. Co-regulation of substrate phosphorylation sites (inner dotted circle = decreased phosphorylation; outer dotted circle = increased phosphorylation) provides insights into kinase activity in response to exercise. (B) Muscle biopsy lysates from four subjects pre- and post-exercise were immunoblotted for kinases and substrates in the phosphoproteomics data. (C) An integrative network of the exercise-regulated kinase interactome was generated using experimentally validated human protein-protein interactions and annotated kinase-substrate relationships. Direct increases (green arrows) and decreases (red arrows) in substrate phosphorylation are shown, and dotted lines represent protein-protein interactions. Colors represent kinases with exercise-regulated activity as shown in (A). Additional kinases (grey) and substrates (white) contain regulated phosphorylation sites.
Predicting AMPK substrates
The acute exercise kinome network highlighted multiple interconnected nodes involving diverse families of kinases. However, one of the most functionally relevant kinases associated with exercise, AMPK, did not feature prominently. Despite the well-established activation of AMPK in exercise, only six bona fide substrates were quantified and regulated. Since hundreds of the exercise-regulated phosphosites identified have unknown upstream kinases, we sought to expand the list of validated AMPK substrates. A total of 516 exercise-regulated phosphosites reported in PhosphoSitePlus had no annotated upstream kinase. A further 412 exercise-regulated phosphosites were absent from the database amounting to 928 regulated phosphosites with no associated upstream kinase. Identification of kinases for these substrates will likely yield insights into mechanisms of exercise action. To begin to identify kinases for these substrates we performed bioinformatic analysis using NetworKIN and NetPhorest, which combines consensus substrate motif analysis with protein-protein interaction databases (Linding et al., 2008).
Following prediction thresholding with PhosphoSiteAnalyzer (Bennetzen et al., 2012), 21 kinase groups were predicted for 575 of these phosphosites. Using phosphosite fold changes, a kinase-substrate set enrichment analysis revealed 11 kinases were associated with greater activity and two kinases were associated with decreased activity following exercise (P < 0.05; Kolmogorov-Smirnov test; Table S5). Kinase groups with greater activity included two well-characterized kinases associated with the exercise response, PKA and CaMKII. A large fraction of phosphorylation sites that were increased with exercise contained basophilic amino acids in the −2 and −3 positions and included predictions for kinases with a less defined role in exercise including DMPK, PIM, PAK and NEK. MAPK and CDK were associated with decreased activity following exercise with predicted substrates containing proline in the +1 position of the phosphorylation site. Surprisingly, we identified only five predicted AMPK substrates by NetworKIN. These included four known substrates; ACC1, ACC2, Raptor, TBC1D1, and a previously predicted substrate GPHN (Banko et al., 2011).
To extract additional AMPK substrates from the human exercise phosphoproteome we integrated these data with data from additional, more AMPK focused assays. In our first assay, SILAC labeled L6 myotubes were treated with or without the AMPK activator 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR; 2 mM; 30 min; n=4). Quantitative phosphoproteomic analysis was performed with phosphopeptide enrichment coupled to tandem MS (Fig. 3A). The analysis included quantification of non-phosphorylated peptides to investigate changes in protein abundance (n=3). A total of 17,212 unique phosphopeptides were identified (12,695 phosphosites with >90% localisation probability), and 7,464 phosphopeptides were quantified in two out of four biological replicates (7,421 phosphosites with >90% localisation probability) (Fig. 3B, 3C; Table S6). At least 6,844 proteins were identified with 3,938 quantified in two out of three biological replicates (Table S7). No significant changes in protein abundance were detected. AICAR significantly modulated 132 unique phosphopeptides (107 phosphosites with >90% localisation probability) relative to control, of which 65 and 42 phosphosites were up- and down-regulated, respectively (adjusted P < 0.05, moderated t-test; Fig. 3C; Table S6). Among AICAR up-regulated sites were known AMPK substrates including S231 on TBC1D1, S79 on ACC1, S637 on ULK1 and S108 on AMPKβ1 (Fig. 3D). To obtain an overview of the signaling pathways regulated by AICAR, phosphoproteins containing significantly regulated phosphosites were subjected to pathway analysis (Table S8). Significantly enriched pathways included actin cytoskeleton signaling, PPAR activation, ILK, cAMP signaling and AMPK signaling.
Fig. 3. AMPK substrate prediction using AMPK-activated phosphoproteomics.

(A) Experimental design of the phosphoproteomic analysis of AICAR stimulation is depicted. 2-plex SILAC labeled L6 myotubes were treated with or without AICAR (n=4 with 2 label switching experiments). Proteins were extracted and digested with trypsin. Peptides were fractionated by strong cation exchange (SCX) chromatography and phosphopeptides enriched using titanium dioxide chromatography. An aliquot of the non-phosphorylated peptides was fractionated by HILIC. Phosphopeptide and non-phosphopeptide fractions were analysed by nanoUHPLC-MS/MS on a Q-Exactive MS operated in data-dependent acquisition followed by MaxQuant analysis. (B) Volcano plot showing median phosphopeptide Log2 fold-change (AICAR/basal) plotted against −Log10 P-value highlighting significantly regulated phosphopeptides (blue; *P < 0.05, n=4, moderated t-test). Dotted lines indicate (+/−) 1.5-fold change (Log2 = 0.58). (C) Summary of the quantified and regulated proteome and phosphoproteome is shown. (D) AMPK substrate prediction using the AICAR-regulated L6 myotube phosphoproteome. The model was trained based on the primary amino acid motif surrounding the phosphosites and magnitude of up-regulation by AICAR (AICAR-regulated sites in blue) using known AMPK substrates quantified (red).
We adapted a machine learning-based model previously developed by our group for predicting kinase-substrate relationships (Humphrey et al., 2013) to predict AMPK substrates from the L6 phosphoproteomics data. A curated positive training set was obtained, and model prediction scores were calculated based on; i) a motif score where the amino acid sequence surrounding the phosphorylated residue matches the AMPK consensus motifs (Gwinn et al., 2008); and ii) the fold-change of a specific phosphorylation site in response to AICAR (Fig. 3D; Table S9). Multiple candidate AMPK substrates were identified and cross-referenced with the human exercise-regulated phosphoproteome. To integrate phosphoproteomics data across species we developed a web application, PhosphOrtholog, capable of mapping both annotated and non-annotated orthologous phosphorylation sites between human and rat proteins based on pairwise sequence alignment (Chaudhuri et al., 2015). Of the 1,004 human exercise-regulated phosphosites, only 55 orthologous rat phosphosites annotated in PhosphoSitePlus were also identified in rat L6 myotubes. PhosphOrtholog performed considerably better and mapped an additional 103 phosphosites identified in both the human muscle and rat L6 myotubes which were not annotated in PhosphoSitePlus (Table S10). Six candidate AMPK substrates regulated by exercise and AICAR were identified based on the prediction model. These comprised 2 known AMPK substrates TBC1D1 S237 (rat S231), ACC1 S80 (rat S79) and candidate substrates AKAP1 S107 (rat S103), stromal interaction molecule 1 (STIM1) S257 and S521, and VAMP-associated protein A (VAPA) S164.
To validate that these candidate phosphorylation sites were bona fide AMPK substrates we performed a whole cell lysate AMPK in vitro kinase analysis, based on (Knight et al., 2012), and combined this with targeted phosphopeptide quantification using data-independent acquisition MS (DIA-MS). This strategy consisted of three steps (Fig. S2): i) spectral library construction, ii) purification of active AMPK, and iii) global AMPK in vitro kinase analysis. A spectral library containing 35,897 phosphopeptides (22,772 phosphosites with >90% localization probability) was generated using phosphopeptide enrichment coupled to tandem MS from HEK293 cells treated with the AMPK agonists AICAR and A-769662 (Table S11). This spectral library contained 368 of the 1,004 exercise-regulated phosphopeptides identified in human muscle. A separate batch of HEK293 cells were lysed in kinase buffer and treated with a phosphatase and pan-kinase inhibitor cocktail. The lysate was treated with; i) buffer alone, ii) active AMPK, and iii) active AMPK in the presence of the AMPK inhibitor, Compound C. Lysates were used for either immunoblotting or phosphopeptide enrichment coupled to DIA-MS. Immunoblotting confirmed AMPK phosphorylation of S80 on ACC and S792 on Raptor, which were partially attenuated by Compound C despite the reduced efficacy of this drug in the presence of elevated AMP concentrations likely to be present in the cell lysate (Zhou et al., 2001) (Fig. 4A). Targeted data extraction (Gillet et al., 2012) on the single-shot DIA-MS data using the 368 exercise-regulated phosphopeptides resulted in quantification of 69 phosphopeptides (mProphet q-value <0.01 and CV <20%) (Table S12). Ten phosphopeptides were significantly increased by AMPK addition relative to control and the majority were attenuated by Compound C (Table S12). These included known AMPK substrates ACC1 S80 (ACACA; Fig. 4B), an additional AMPK phosphorylation site on Raptor, S722 (RPTOR; Fig. 4C), and several exercise-regulated sites (Fig. 4D – I). We performed multiple sequence alignment of exercise-regulated phosphopeptides that were significantly increased by AMPK in the DIA-MS data or AICAR in L6 cells (Fig. 5) with previously characterized AMPK consensus motifs (Gwinn et al., 2008). Seven known AMPK substrates regulated by exercise (black circles) aligned with the consensus motif as expected. Eight substrates regulated with exercise were confirmed with AICAR stimulation and/or DIA-MS (green circles). Of these, six were confirmed by DIA-MS (black diamonds ◆). An additional six phosphosites previously predicted as AMPK substrates (Banko et al., 2011) were regulated by exercise (yellow circles).
Fig. 4. AMPK substrate prediction using data-independent MS analysis of a global AMPK in vitro kinase assay.

(A) Immunoblot analysis of known AMPK substrates (ACC and Raptor) in HEK293 lysates subjected to a global AMPK (α1β1γ2) in vitro kinase assay ± active AMPK and the AMPK inhibitor Compound C. (B) – (I) Targeted quantification (mean ± standard deviation, one-way ANOVA corrected for multiple testing, *P < 0.05, **P < 0.01, ***P < 0.005, n=3) of phosphopeptides from HEK293 lysates subjected to a global AMPK (α1β1γ2) in vitro kinase assay using nanoUHPLC-MS/MS on a Q-Exactive MS operated in DIA.
Fig. 5. AMPK consensus motif and substrate sequence alignment.

AMPK consensus motif derived from (Gwinn et al., 2008) with multiple sequence alignment of exercise-regulated phosphosites in human muscle (Human Exercise) is displayed. The alignment shows: known AMPK substrates (black circles); internally predicted AMPK substrates with validation (blue circles) in either AICAR stimulated Rat L6 myotubes (Rat AICAR) or global AMPK in vitro kinase analysis in HEK lysates (black diamonds ◆); and externally predicted AMPK substrates from (Banko et al., 2011) (orange circles).
AKAP1 is a mitochondrial AMPK substrate
AKAP1 is a mitochondrial scaffold protein that binds mitochondrial-targeted PKA (Merrill and Strack, 2014) and other kinases such as PKC (Greenwald et al., 2014). To test the possibility that AKAP1 may play a role in mitochondrial AMPK signaling we generated a phospho-specific antibody to assess the predicted AMPK phosphorylation site S103 on AKAP1. This antibody recognized AKAP1 phosphorylated at S103 specifically in rodents. An increase in AKAP1 S103 phosphorylation induced by A-769662 was detected in HEK293 cells expressing FLAG-tagged mouse AKAP1 wild type (WT), but not empty vector (EV) FLAG or FLAG-tagged AKAP1 S103A phospho-dead mutant (Fig. 6A). To confirm AMPK as the AKAP1 S103 kinase, in vitro kinase assays were performed on immunoprecipitated AKAP1 in the absence or presence of active AMPK α1β1γ2. AMPK increased AKAP1 S103 phosphorylation similarly to ACC S79 and this was abolished by Compound C (Fig. 6B). Similarly to the characteristic increase in ACC S79 phosphorylation, AKAP1 S103 phosphorylation was increased following incubation of L6 myotubes with either AICAR, A-769662 or 2,4-dinitrophenol (DNP) (Fig. 6C). This A-769662 induced increase in AKAP1 S103 phosphorylation was ablated with siRNA knock down of the catalytic α1 and α2 subunits of AMPK (siAMPKα) compared to cells transfected with scrambled control siRNA (siScramble; Fig. 6D). Consistent with these findings, AICAR-dependent phosphorylation of AKAP1 S103 was absent in soleus muscle from mice expressing kinase dead AMPKα2 (Fig. 6E). It was notable that ACC phosphorylation was not inhibited to the same extent as AKAP1 in kinase dead AMPKα2 muscle, possibly reflecting an underlying subtlety to AMPK regulation that remains to be found. It was suggested that there may be an alternate ACC kinase, but so far attempts to identify this kinase have not been successful (Dzamko et al., 2008). AKAP1 S103 phosphorylation, in addition to AMPK T172 and ACC S79, was also increased in red quadricep muscles following treadmill exercise in mice (Fig. 6F).
Fig. 6. AKAP1 is a mitochondrial AMPK substrate.

(A) HEK293 cells expressing FLAG-tagged EV, AKAP1 WT or AKAP1 S103A were serum-starved for 2 h, followed by ± 100 μM A-769662 for 30 min. Cell lysates were immunoblotted with indicated antibodies. (B) Activated AMPK α1β1γ2 was added to immunoprecipitated AKAP1 or biotinylated ACC beads for 30 min at 33°C ± Compound C. In vitro kinase assay samples were immunoblotted. (C) L6 myotubes were serum-starved for 2 h, followed by incubation ± AMPK agonists AICAR (2 mM), A-769662 (100 μM) and DNP (200 μM) for 30 min. Lysates were immunoblotted. (D) L6 myoblasts were transfected with siScramble or siAMPKα. Cells were lysed after 72 h and immunoblotted. (E) Isolated soleus muscles from WT and AMPKα2 kinase dead (KD) mice were incubated in vitro ± 2 mM AICAR for 1 h and muscle lysates were immunoblotted. (F) WT mice were rested or subjected to treadmill exercise until exhaustion, red quadricep muscles were isolated and lysates were immunoblotted. (G) L6 myotubes were serum-starved for 2 h, followed by stimulation ± 100 μM A769662 for 30 min. Following subcellular fractionation, whole cell lysate (WCL), non-mitochondrial (Non-Mito) and mitochondrial (Mito) fractions were immunoblotted. (H) L6 myoblasts expressing pMito-LSSmOrange (Mito) and either FLAG-tagged AKAP1 WT or AKAP1 S103A were reseeded 24 h after transfection, fixed 24 h later and prepared for confocal microscopy (scale bar: 10 μm). Immunoblots are representative images from 3–6 independent experiments; quantitative AKAP1 pS103 data in panels E and F are normalized to total protein, and mean ± standard error of mean is shown from n=3–4 and n=5 mice, respectively, t-test, *P < 0.05 vs. basal WT muscle (E) or rest (F), #P < 0.05 vs. AICAR-stimulated WT muscle.
We next examined mitochondrial localization of AMPK and AKAP1. A-769662 increased AMPK T172 and ACC S79 phosphorylation in the whole cell lysate and non-mitochondrial cytoplasmic fractions isolated from L6 myotubes (Fig. 6G). As expected, AKAP1 and phospho-S103 AKAP1 were highly enriched in the mitochondrial fraction. Immunofluorescence confirmed that FLAG-tagged AKAP1 WT and the AKAP1 S103A mutant were primarily localized to mitochondria (Fig. 6H). Phospho-AMPK was also highly enriched in the mitochondrial fraction, while the majority of immunoreactive AMPKα was identified in the non-mitochondrial cytoplasmic fraction. Use of AMPK isoform-specific antibodies revealed that AMPKα2 was highly enriched in the mitochondrial fraction compared to AMPKα1. In addition, mitochondrial detection of AMPKα1 was increased upon A-769662 stimulation. Interestingly, both ACC1 and ACC2 were primarily localized to the non-mitochondrial cytoplasmic fraction. However, similarly to AMPK the minor pool of ACC localized at the mitochondria was robustly phosphorylated following AMPK activation (Fig. 6G).
AKAP1 S103 phosphorylation via AMPK facilitates mitochondrial respiration
Considering the mitochondrial localization of AKAP1 and regulation of mitochondrial energy utilization by AMPK, we next used siRNA knock down to explore their role in this process further. Knock down of AMPKα in L6 myoblasts significantly reduced mitochondrial respiration in the presence of both bovine serum albumin (BSA) and palmitate (Fig. 7A–7C). Similarly, knock down of AKAP1 (siAKAP1; Fig. 7D) significantly impaired mitochondrial respiration (JO2; Fig. 7E) and oxygen consumption (OCR; Fig. 7F) in the presence of BSA compared to myoblasts transfected with siScramble. Similar data were obtained using the oxygraph-2K respirometry systems (O2K; Fig. 7E) and extracellular flux analyzer (XF24; Fig. 7F). In addition, mitochondrial palmitate oxidation was blunted in myoblasts transfected with siAKAP1 compared to those transfected with siScramble (Fig. 7E, 7F).
Fig. 7. AKAP1 and AMPK mediated S103 phosphorylation facilitate mitochondrial respiration.

(A) L6 myoblasts were transfected with siScramble or siAMPKα. Cells were lysed after 72 h and immunoblotted with indicated antibodies. (B) Immunoblotting was performed in serum-starved L6 myoblasts incubated with BSA or BSA-conjugated palmitate (Palm.; 200 μM) ± 100 μM A-769662 for 30 min. Immunoblots are representative images from 3–4 independent experiments. (C) Oxygen consumption rate (OCR) was determined in serum-starved L6 myoblasts transfected with siScramble or siAMPKα and incubated with BSA or Palm. (200 μM) ± 100 μM A-769662. (D) L6 myoblasts were transfected with siScramble or AKAP1 siRNA (siAKAP1). Cells were lysed after 72 h and immunoblotted. Respiration (JO2 in E) and OCR (F) was determined in serum-starved L6 myoblasts transfected with siScramble or siAKAP1 and incubated with BSA or Palm. (200 μM) ± 100 μM A-769662. (G) L6 myoblasts were transfected with FLAG-tagged EV control, AKAP1 WT or AKAP1 S103A mutant, incubated with BSA or Palm., and immunoblotting was performed (G), respiration (H, I) and OCR (J) were measured. (I) Respiration is shown as % increase with A-769662 stimulation in cells incubated with Palm. Quantitative OCR and JO2 data represent mean ± standard error of mean, t-test, *P < 0.05 vs. siScramble BSA or EV/WT control BSA, #P < 0.05 vs. respective siScramble or EV/WT control, n=3–6.
To examine the role of AKAP1 S103 in mitochondrial respiration, we assessed whole cell respiration in L6 myoblasts expressing either FLAG-tagged AKAP1 WT or the AKAP1 S103A mutant (Fig. 7G). Using the O2K we observed the expected increases in respiration induced by palmitate and A-769662 in L6 myoblasts transfected with EV FLAG (Fig. 7H). Strikingly, L6 myoblasts expressing S103A displayed impaired A-769662-stimulated respiration compared to cells expressing either EV FLAG or WT AKAP1 (Fig. 7I). Similar to our siAKAP1 findings, L6 myoblasts expressing the S103A AKAP1 mutant displayed impaired OCR compared to those expressing WT AKAP1 in the presence of BSA and palmitate (Fig. 7J).
Discussion
This study represents a major advance in our understanding of how acute exercise signaling affects muscle biochemistry and physiology. Our use of unbiased, global phosphoproteomics has shed light on the complexity of exercise signaling. In this study, we have achieved three major goals. First, we provide a comprehensive map of the acute high-intensity exercise signaling network in human muscle and used this together with protein-protein interaction databases to generate an exercise kinome network. The entire network comprised over 900 exercise-regulated phosphorylation sites with no known upstream kinase. Second, we identified eight exercise-regulated AMPK substrates using a combination of global MS-based phosphoproteomics in muscle cells, machine learning AMPK substrate prediction, and integrated substrate validation using targeted DIA-MS analysis in HEK293 cells. Finally, we unveiled a function of AMPK in mitochondrial respiration via phosphorylation of AKAP1.
The presence of a small number of large abundant contractile proteins, which account for over 50% of tissue mass, presents a considerable challenge for muscle biochemistry (Geiger et al., 2013). By comparison to other tissues, a lower number (~3,500) of muscle phosphorylation sites have been reported using MS (Hojlund et al., 2009; Lundby et al., 2012; Zhao et al., 2011). In this study, we combined improved phosphopeptide enrichment strategies with multiplexed isobaric labeling and high resolution separation coupled to tandem MS to identify > 8,500 phosphorylation sites, ~12% of which were significantly regulated with exercise. To achieve this robust exercise response we utilized a high-intensity acute exercise regimen in humans, which likely engaged many muscle fibers and produced a robust signaling response. The paired experimental design combined with our optimized biopsy approach resulted in a highly reproducible exercise-induced phosphorylation response (Fig. 1).
Exercise imposes metabolic, oxidative and mechanical stress on muscle due to increased energy turnover. These stress pathways play a major role in the initiation of exercise signaling (reviewed in (Egan and Zierath, 2013)), culminating in the regulation of numerous kinases including AMPK, PKA, CaMK and MAPK. While we observed robust activation of these known kinases, this represented only a fraction of the regulated sites (~10%) identified in the present study. Thus, the vast majority of the exercise-regulated phosphosites reported here (~900 sites) have not previously been associated with the exercise response. This likely reflects the role of exercise to perturb multiple stimuli throughout the body to initiate signaling cascades in muscle. Our data further highlight exercise signaling pathways implicated in muscular dystrophy and associated muscle pathology including kinases such as DMPK, ILK and CDK. Interestingly, the exercise kinome network in skeletal muscle highlights many nodes involved in oxidative and/or intracellular stress including the p38 MAPK pathway, Wnk1/OSR1, DMPK, PKD, Aurora, MST3, PAK, ATM, PIM, Src kinase family members and DAPK. Consistent with this, exercise-induced reactive oxygen species purportedly plays a major role in the beneficial effects of exercise (Ristow et al., 2009). The effects of exercise on mTORC1/2 were of particular interest with altered phosphorylation observed in multiple components of this complex including RagC, DEPTOR, DAPK2, and the mTORC1 binding protein LARP1. Based on the direction of change in both Akt and mTORC1/2 substrates, our data are consistent with a model whereby exercise represses biosynthetic pathways to conserve substrates for energy production. However, there is no consensus concerning the effect of various exercise protocols on Akt activity (Thorell et al., 1999; Widegren et al., 1998; Wojtaszewski et al., 2001). We observed not only decreased Akt phosphorylation, but also reduced phosphorylation of a range of Akt substrates after exercise indicative of reduced Akt kinase activity. Therefore, we are confident that Akt kinase activity was reduced with our exercise protocol. The pathways described above, along with 45 protein kinases and 16 transcription factors that we observed to be phosphorylated during exercise, warrant further investigation (Table S1).
Given the known activation of AMPK during exercise and its suggested therapeutic potential for metabolic disease, there is considerable interest in identifying additional AMPK substrates. One of the most comprehensive studies was a chemical genetic screen that identified 28 potential AMPK substrates and validated six of these (Banko et al., 2011). In addition, a recent study applied antibody-based affinity proteomics to identify 57 candidate AMPK substrates in primary hepatocytes and validated two substrates (Ducommun et al., 2015). Another previous screen utilized a two-dimensional in vitro screening approach to identify proteins that interact with the AMPK α2β2γ1 isoform (Klaus et al., 2012). Here, we employed physiological activation of exercise pathways, combined with extensive interrogation of the AMPK signaling network using complementary MS techniques and bioinformatic approaches to identify eight high-confidence exercise-regulated AMPK substrates. We also identified six previously predicted AMPK substrates including GPHN, GOLGA4, IGF2R, TNIK, MINK1 and PPP1R12B (Banko et al., 2011).
Strikingly, five AMPK substrates are associated with the endoplasmic reticulum (ER) and Golgi apparatus (STIM1, VAPA, GPHN, GOLGA4 and IGF2/CIMPR), highlighting unidentified roles for AMPK in ER/Golgi function such as vesicle trafficking and Ca2+ regulation. STIM1 regulates store-operated Ca2+ entry and functions as an ER Ca2+ sensor. In response to depletion of intracellular Ca2+ stores STIM1 undergoes oligomerization and conformational changes triggering its interaction with the Ca2+ release activated channel (CRAC) Orai1 at the plasma membrane to facilitate Ca2+ entry into the cell (Soboloff et al., 2012). Interestingly, the AMPK-regulated S257 phosphosite on STIM1 is adjacent to the proposed hinge region suggesting that AMPK likely regulates STIM1 intra- and/or intermolecular dynamics, thereby affecting STIM1-Orai1 complex activation to regulate Ca2+ influx during acute exercise (Soboloff et al., 2012). AMPK-regulated phosphorylation of VAPA suggests additional unappreciated mechanisms of lipid and glucose transport regulation by AMPK. We also validated eight additional exercise-regulated AMPK substrates associated with other organelles (AKAP1, AGPAT9, SLC12A2, CTNND1, AKAP13, TNIK, MINK1 and PPP1R12B). Collectively, these studies expand the repertoire of AMPK biological functions to include: i) vesicle transport, ii) beta-catenin and Wnt signaling (TNIK, MINK1, CTNND1), iii) potassium and chloride transport (SLC12A2), which is crucial for sustaining exercise (Clausen, 2003), iv) calcium homeostasis and excitation contraction coupling (STIM1), v) myosin phosphorylation (PPP1R12B), vi) lipid signaling (AGPAT1), and vii) mitochondrial signaling (AKAP1).
The identification of AKAP1 as an AMPK substrate was of interest given its mitochondrial localization and the known role of AMPK in mitochondrial energetics. Currently the mitochondrial function of AMPK is thought to be primarily conveyed via AMPK-dependent phosphorylation of ACC to regulate mitochondrial fatty acid import and oxidation. AKAP1 is expressed at high levels in human muscle, heart and adipose tissue, and it has been implicated in fat metabolism (Bridges et al., 2006). Here, we demonstrate that AKAP1 is a bona fide AMPK substrate in muscle that plays an important role in AMPK regulated mitochondrial respiration. Consistent with this, knockdown of AKAP1 impairs mitochondrial membrane potential and ATP production (Livigni et al., 2006), and overexpression of AKAP1 increases cellular oxygen utilization (Merrill and Strack, 2014). Intriguingly, the expression of AKAP1 is reduced four-fold in adipose tissue from obese individuals (Marrades et al., 2010). However, we were unable to detect any difference between AKAP1 mRNA levels in human muscle from lean and obese individuals (data not shown).
The precise mechanism by which AKAP1 phosphorylation by AMPK regulates mitochondrial signaling and function is not understood. Phosphorylated AMPK is enriched in mitochondria together with AKAP1 in muscle cells (Fig. 6G). However, we were unable to detect a direct interaction between these proteins (data not shown). Additional exercise-regulated phosphosites were identified on AKAP1 including S128 and S592, which may contribute to the observed phenotype or have alternative functions. The present studies add to the growing body of evidence showing cross talk between AMPK and other kinases including PKA, potentially via AKAP1. For example, PKA phosphorylates and inhibits AMPK to promote efficiency of the lipolytic response in adipocytes (Djouder et al., 2010). Recent evidence highlights roles for AKAP1 in kinase regulation, including PKC (Greenwald et al., 2014) and Src (Livigni et al., 2006), that may impact mitochondrial function. Notably, exercise increased the activation loop phosphopeptide common to Src kinase family members Src, Fyn, Lck and Yes. An emerging concept for scaffold proteins such as AKAP1 is their role in scaffold state switching to allow signals and complexes to be insulated, amplified and/or accelerated within subcellular compartments such as mitochondria (Greenwald et al., 2014). Mitochondria are dynamic organelles that are continuously remodeled by fission and fusion. These reactions are sensitive to physiological stimuli such as energy stress and exercise to maintain mitochondrial integrity and survival (Bo et al., 2010). PKA bound to AKAP1 is known to inhibit mitochondrial fission (Merrill and Strack, 2014). The accessibility of PKA to AKAP1 is therefore expected to impact PKA’s ability to inhibit fission. Therefore, AMPK-stimulated AKAP1 phosphorylation in muscle during exercise and energy stress may limit PKA accessibility to AKAP1 to promote mitochondrial remodeling to meet metabolic demands. Additional mitochondrial phosphoproteins we found to be regulated by exercise, including mitochondrial fission factor (MFF) and mitochondrial fission regulator 1-like (MFR1L), may also contribute to this dynamic remodeling process. Notably, AKAP1 and MFR1L were regulated by both exercise and AICAR, and MFF was recently validated as an AMPK substrate (Ducommun et al., 2015). Therefore, AMPK may have several functions in mitochondrial remodeling and energy homeostasis.
It is well recognized that a better mechanistic understanding of exercise will provide a major step forward in harnessing the immense therapeutic potential of this lifestyle intervention (Neufer et al., 2015). It is our view that identifying the protein kinases triggered in various tissues in response to exercise may provide one tractable way of achieving this goal in view of the druggability of this class of molecules. A notable feature of the present study is that the exercise response is not governed by a single kinase, but rather it involves the synchronous activation of a series of kinases. The challenge is to delineate which of these is transmitting the beneficial effects of exercise with the long term goal of developing a combination kinase small molecule cocktail that can be used to recapitulate some of the beneficial effects of exercise in humans.
Taken together, the effort undertaken to map the exercise phosphorylation landscape in human muscle has filled a crucial gap in our understanding of muscle exercise signaling. For example, the breadth of signaling pathways and kinases modulated by exercise is far greater than previously appreciated. We hope that similar studies investigating the effects of other exercise protocols on other organs and other protein modifications in a temporal fashion will further expand our knowledge of cellular adaptation to exercise. Importantly, the combination of these data may provide a blueprint for future studies aimed at developing exercise mimetic therapies for metabolic disease treatment or prevention in the modern world. For example, testing the ability of these unappreciated exercise-regulated kinases to improve whole body insulin sensitivity in the absence of exercise, either through genetic or pharmacological studies, will undoubtedly increase our understanding of metabolic health. We anticipate that our unique, global approach to study exercise signaling in human muscle will help unleash the full potential of exercise in preventing obesity, type 2 diabetes and other major conditions that limit the health of our aging population.
Experimental Procedures
Brief experimental procedures are provided below. For complete details and MS data repository information, see Supplemental Experimental Procedures.
Human subjects and exercise
Four untrained healthy males (age: 24 – 27; BMI: 24.2 – 25.9 kg/m2, VO2 max: 48–59 ml/kg/min, Wmax: 320–375 W) underwent a single bout of high-intensity cycle exercise. Following warm up subjects exercised for 6 min at 85% of Wmax then to exhaustion at 92% of Wmax, which occurred after 9–11 min total exercise time. Blood was obtained before and during the last min of exercise, and 2 muscle biopsies were extracted from the vastus lateralis immediately before and upon exercise cessation.
Phosphopeptide enrichment and mass spectrometry
Human muscle peptides were labeled with isobaric tags for relative or absolute quantitation (iTRAQ) or tandem mass tags (TMT). L6 myotube SILAC labeling was performed as previously described (Humphrey et al., 2013). Phosphopeptides were enriched using titanium dioxide followed by sequential elution from immobilized metal ion affinity chromatography and fractionation by hydrophilic interaction liquid chromatography (TiSH) (Engholm-Keller et al., 2012). Labeled peptides were analyzed by MS using data-dependent acquisition (DDA) as previously described (Humphrey et al., 2013). Phosphopeptides enriched from the AMPK in vitro kinase assay were analyzed by data-independent acquisition (DIA) (Gillet et al., 2012).
Phosphoproteomic data analysis
DDA data from human muscle were processed using Proteome Discoverer v1.4 against the human UniProt database. DDA data from rat L6 myotubes were processed using MaxQuant v1.4.0.8 (Cox and Mann, 2008) against the rat UniProt database. All results were filtered to 1% FDRs. Median values of peptide spectral matches (PSMs) for each phosphosite were calculated using a script developed in Python. Bioinformatic analysis was performed primarily in the R programming environment [http://www.R-project.org, R Development Core Team (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0]. Protein quantification was performed using median values of all PSMs of the protein group. Significantly regulated phosphopeptides and proteins were determined using a moderated t-test from LIMMA package in R [Smyth, G.K, 2004, Statistical applications in genetic and molecular biology, 3, no.1, article 3]. Pathway analysis was performed with Ingenuity Pathway Analysis (www.ingenuity.com), and prediction of kinase-substrate relationships was performed with NetworKIN v2.0 (Linding et al., 2008) followed by processing and thresholding in PhosphoSiteAnalyzer (Bennetzen et al., 2012).
All DIA data were processed using Skyline v2.5.0.6157 (MacLean et al., 2010) and targeted data extraction was performed essentially as described (Gillet et al., 2012). Peak scoring models were trained based on mProphet (Reiter et al., 2011) using a combination of scores and filtered to 1% FDR. Significantly regulated phosphopeptides were determined by calculating the moderated t-statistic using LIMMA package in R where eBayes method was used for global variance shrinkage. Significance was defined by an adjusted P-value < 0.05.
Mitochondrial respiration assays
O2 consumption measurements were conducted using the OROBOROS Oxygraph-2K (OROBOROS Instruments) or the XF24 and XFp analyzers (Seahorse Biosciences).
Statistical Analysis
Error bars represent mean ± standard deviation (Fig. 4) or mean ± standard error of mean (Fig. 6 and 7). Statistical tests and experiment numbers are detailed in figure legends.
Supplementary Material
Acknowledgments
We thank John D. Scott for providing AKAP1 cDNA, Bruce E. Kemp and Jonathan S. Oakhill for AMPK α1, β1 and γ2 cDNA, Kei Sakamoto for A-769662, Merck for ACC1/2 antibodies, and Vladislav V. Verkhusha for pMito-LSSmOrange. We thank Dorte E. Kristensen, James R. Krycer and Xiao-Yi Zeng for help with enrolling human subjects, Seahorse experimental design, and mouse exercise experiments, respectively. This work was supported by National Health and Medical Research Council (NHMRC) project grants GNT1061122 and GNT1047067 (D.E.J.) and grants from the Diabetes Australia Research Trust (D.E.J., N.J.H., R.C.), Lundbeck Foundation, Novo Nordisk Foundation, University of Copenhagen Excellence Programme for Interdisciplinary Research (2016), and Council for Independent Research/Medicine (E.A.R.). D.E.J. is an NHMRC Senior Principal Research Fellow. B.L.P is a recipient of an NHMRC Early Career Fellowship, and S.J.H. is a recipient of a European Molecular Biology Organization Long-Term Fellowship. P.Y. and R.J. are supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Author Contributions
N.J.H. and B.L.P. designed experiments, performed MS sample preparation, data analysis, and wrote the manuscript. N.J.H. performed AKAP1 biological validation experiments. B.L.P. performed human muscle MS analysis and in vitro kinase assays. R.C. performed bioinformatics analyses and integrated AMPK substrate prediction. K.H.F-W. performed respirometry experiments. M.K. assisted with human MS sample preparation, and M.K. and T.E.J. performed AMPKα2 KD mouse muscle analyses. S.J.H. performed L6 myotube MS analysis, and S.J.H. and D.J.F. assisted with L6 myotube MS sample preparation. P.Y. and R.J. performed machine learning AMPK substrate prediction. M.H. and S.T. assisted with AKAP1 biological validation and mutagenesis, respectively. J.S. performed mouse exercise experiments. J.G.B. performed immunofluorescence experiments. J.S., B.K., J.F.P.W. and D.E.J. contributed to human exercise study design. B.K., J.F.P.W. and E.A.R. performed human exercise studies. E.A.R. and D.E.J. designed experiments, established collaborations, provided expert guidance and wrote the manuscript. All authors approved the final version of the manuscript.
The authors have no conflicts of interest.
The contents of the published material are solely the responsibility of the individual authors and the Administering Institution and do not reflect the views of the NHMRC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villen J, Wang B, Kim SR, Sakamoto K, et al. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Molecular cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen MV, Cox J, Mann M, Andersen JS. PhosphoSiteAnalyzer: a bioinformatic platform for deciphering phospho proteomes using kinase predictions retrieved from NetworKIN. Journal of proteome research. 2012;11:3480–3486. doi: 10.1021/pr300016e. [DOI] [PubMed] [Google Scholar]
- Bo H, Zhang Y, Ji LL. Redefining the role of mitochondria in exercise: a dynamic remodeling. Annals of the New York Academy of Sciences. 2010;1201:121–128. doi: 10.1111/j.1749-6632.2010.05618.x. [DOI] [PubMed] [Google Scholar]
- Bridges D, MacDonald JA, Wadzinski B, Moorhead GB. Identification and characterization of D-AKAP1 as a major adipocyte PKA and PP1 binding protein. Biochemical and biophysical research communications. 2006;346:351–357. doi: 10.1016/j.bbrc.2006.05.138. [DOI] [PubMed] [Google Scholar]
- Chaudhuri R, Sadrieh A, Hoffman NJ, Parker BL, Humphrey SJ, Stöckli JS, Hill A, James DE, Yang JY. PhosphOrtholog: A web-based tool for cross-species mapping of orthologous protein post-translational modifications. BMC Genomics. 2015 doi: 10.1186/s12864-015-1820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T. Na+-K+ pump regulation and skeletal muscle contractility. Physiological reviews. 2003;83:1269–1324. doi: 10.1152/physrev.00011.2003. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. The EMBO journal. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducommun S, Deak M, Sumpton D, Ford RJ, Galindo AN, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cellular signalling. 2015 doi: 10.1016/j.cellsig.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Dzamko N, Schertzer JD, Ryall JG, Steel R, Macaulay SL, Wee S, Chen ZP, Michell BJ, Oakhill JS, Watt MJ, et al. AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. The Journal of physiology. 2008;586:5819–5831. doi: 10.1113/jphysiol.2008.159814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell metabolism. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Engholm–Keller K, Birck P, Storling J, Pociot F, Mandrup-Poulsen T, Larsen MR. TiSH--a robust and sensitive global phosphoproteomics strategy employing a combination of TiO2, SIMAC, and HILIC. Journal of proteomics. 2012;75:5749–5761. doi: 10.1016/j.jprot.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Fentz J, Kjobsted R, Birk JB, Jordy AB, Jeppesen J, Thorsen K, Schjerling P, Kiens B, Jessen N, Viollet B, et al. AMPKalpha is critical for enhancing skeletal muscle fatty acid utilization during in vivo exercise in mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2015;29:1725–1738. doi: 10.1096/fj.14-266650. [DOI] [PubMed] [Google Scholar]
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell metabolism. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nature medicine. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T, Velic A, Macek B, Lundberg E, Kampf C, Nagaraj N, Uhlen M, Cox J, Mann M. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Molecular & cellular proteomics : MCP. 2013;12:1709–1722. doi: 10.1074/mcp.M112.024919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Molecular & cellular proteomics : MCP. 2012;11:O111016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald EC, Redden JM, Dodge-Kafka KL, Saucerman JJ. Scaffold state switching amplifies, accelerates, and insulates protein kinase C signaling. The Journal of biological chemistry. 2014;289:2353–2360. doi: 10.1074/jbc.M113.497941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMPK-Sensing Energy while Talking to Other Signaling Pathways. Cell metabolism. 2014;20:939–952. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ, Zierath JR. Integrative Biology of Exercise. Cell. 2014;159:738–749. doi: 10.1016/j.cell.2014.10.029. [DOI] [PubMed] [Google Scholar]
- Hojlund K, Bowen BP, Hwang H, Flynn CR, Madireddy L, Geetha T, Langlais P, Meyer C, Mandarino LJ, Yi Z. In vivo phosphoproteome of human skeletal muscle revealed by phosphopeptide enrichment and HPLC-ESI-MS/MS. Journal of proteome research. 2009;8:4954–4965. doi: 10.1021/pr9007267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic acids research. 2012;40:D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stockli J, Yang JY, James DE. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell metabolism. 2013;17:1009–1020. doi: 10.1016/j.cmet.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen J, Maarbjerg SJ, Jordy AB, Fritzen AM, Pehmoller C, Sylow L, Serup AK, Jessen N, Thorsen K, Prats C, et al. LKB1 regulates lipid oxidation during exercise independently of AMPK. Diabetes. 2013;62:1490–1499. doi: 10.2337/db12-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Polge C, Zorman S, Auchli Y, Brunisholz R, Schlattner U. A two-dimensional screen for AMPK substrates identifies tumor suppressor fumarate hydratase as a preferential AMPKalpha2 substrate. Journal of proteomics. 2012;75:3304–3313. doi: 10.1016/j.jprot.2012.03.040. [DOI] [PubMed] [Google Scholar]
- Knight JD, Tian R, Lee RE, Wang F, Beauvais A, Zou H, Megeney LA, Gingras AC, Pawson T, Figeys D, et al. A novel whole-cell lysate kinase assay identifies substrates of the p38 MAPK in differentiating myoblasts. Skeletal muscle. 2012;2:5. doi: 10.1186/2044-5040-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Pasculescu A, Olhovsky M, Colwill K, Bork P, Yaffe MB, Pawson T. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic acids research. 2008;36:D695–699. doi: 10.1093/nar/gkm902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livigni A, Scorziello A, Agnese S, Adornetto A, Carlucci A, Garbi C, Castaldo I, Annunziato L, Avvedimento EV, Feliciello A. Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Molecular biology of the cell. 2006;17:263–271. doi: 10.1091/mbc.E05-09-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nature communications. 2012;3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarbjerg SJ, Jorgensen SB, Rose AJ, Jeppesen J, Jensen TE, Treebak JT, Birk JB, Schjerling P, Wojtaszewski JF, Richter EA. Genetic impairment of AMPKalpha2 signaling does not reduce muscle glucose uptake during treadmill exercise in mice. American journal of physiology Endocrinology and metabolism. 2009;297:E924–934. doi: 10.1152/ajpendo.90653.2008. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrades MP, Gonzalez-Muniesa P, Martinez JA, Moreno-Aliaga MJ. A dysregulation in CES1, APOE and other lipid metabolism-related genes is associated to cardiovascular risk factors linked to obesity. Obesity facts. 2010;3:312–318. doi: 10.1159/000321451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill RA, Strack S. Mitochondria: a kinase anchoring protein 1, a signaling platform for mitochondrial form and function. The international journal of biochemistry & cell biology. 2014;48:92–96. doi: 10.1016/j.biocel.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufer PD, Bamman MM, Muoio DM, Bouchard C, Cooper DM, Goodpaster BH, Booth FW, Kohrt WM, Gerszten RE, Mattson MP, et al. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell metabolism. 2015 doi: 10.1016/j.cmet.2015.05.011. [DOI] [PubMed] [Google Scholar]
- O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L, Rinner O, Picotti P, Huttenhain R, Beck M, Brusniak MY, Hengartner MO, Aebersold R. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nature methods. 2011;8:430–435. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56:1898–1906. doi: 10.1007/s00125-013-2991-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. The Biochemical journal. 2009;418:261–275. doi: 10.1042/BJ20082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Goodyear LJ. Invited review: intracellular signaling in contracting skeletal muscle. Journal of applied physiology. 2002;93:369–383. doi: 10.1152/japplphysiol.00167.2002. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nature reviews Molecular cell biology. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiological reviews. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Thorell A, Hirshman MF, Nygren J, Jorfeldt L, Wojtaszewski JF, Dufresne SD, Horton ES, Ljungqvist O, Goodyear LJ. Exercise and insulin cause GLUT-4 translocation in human skeletal muscle. The American journal of physiology. 1999;277:E733–741. doi: 10.1152/ajpendo.1999.277.4.E733. [DOI] [PubMed] [Google Scholar]
- Widegren U, Jiang XJ, Krook A, Chibalin AV, Bjornholm M, Tally M, Roth RA, Henriksson J, Wallberg-henriksson H, Zierath JR. Divergent effects of exercise on metabolic and mitogenic signaling pathways in human skeletal muscle. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1998;12:1379–1389. doi: 10.1096/fasebj.12.13.1379. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen P, Kiens B, Richter EA. Regulation of glycogen synthase kinase-3 in human skeletal muscle: effects of food intake and bicycle exercise. Diabetes. 2001;50:265–269. doi: 10.2337/diabetes.50.2.265. [DOI] [PubMed] [Google Scholar]
- Zhao X, Leon IR, Bak S, Mogensen M, Wrzesinski K, Hojlund K, Jensen ON. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Molecular & cellular proteomics : MCP. 2011;10:M110000299. doi: 10.1074/mcp.M110.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. The Journal of clinical investigation. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.