

Abstract
Type 1 diabetes mellitus (T1DM) patients have osteopenia and impaired fracture healing due to decreased osteoblast activity. Further, no adequate treatments are currently available that can restore impaired healing in T1DM; hence a significant need exists to investigate new therapeutics for treatment of orthopedic complications. Sclerostin (SOST), a WNT antagonist, negatively regulates bone formation, and SostAb is a potent bone anabolic agent. To determine whether SOST antibody (SostAb) treatment improves fracture healing in streptozotocin (STZ) induced T1DM mice, we administered SostAb twice weekly for up to 21 days post-fracture, and examined bone quality and callus outcomes at 21 days and 42 days post fracture (11 and 14 weeks of age, respectively). Here we show that SostAb treatment improves bone parameters; these improvements persist after cessation of antibody treatment. Markers of osteoblast differentiation such as RUNX2, collagen I, osteocalcin, and DMP1 were reduced, while an abundant number of SP7/osterix-positive early osteoblasts were observed on the bone surface of STZ calluses. These results suggest that STZ calluses have poor osteogenesis resulting from failure of osteoblasts to fully differentiate and produce mineralized matrix, which produces a less mineralized callus. SostAb treatment enhanced fracture healing in both normal and STZ groups, and in STZ+SostAb mice, also reversed the lower mineralization seen in STZ calluses. Micro-CT analysis of calluses revealed improved bone parameters with SostAb treatment, and the mineralized bone was comparable to Controls. Additionally, we found sclerostin levels to be elevated in STZ mice and β-catenin activity to be reduced. Consistent with its function as a WNT antagonist, SostAb treatment enhanced β-catenin activity, but also increased the levels of SOST in the callus and in circulation. Our results indicate that SostAb treatment rescues the impaired osteogenesis seen in the STZ induced T1DM fracture model by facilitating osteoblast differentiation and mineralization of bone.
Keywords: sclerostin, type I diabetes, fracture repair, streptozotocin, STZ, osteoblast differentiation, sclerostin antibody
Introduction
Type 1 diabetes mellitus (T1DM) patients develop osteopenia, which increases the risk of fractures; additionally, these fractures are more likely to result in delayed or impaired healing, caused by reduced bone formation(1-6). Fracture risk and the risk of osteoporosis increases with age, length of time after diagnosis, and with complications such as kidney disease(7). Recent studies have shown that the low bone mass in T1DM is mild, and caused by reduced osteoblast activity(3,8). Although some studies show association of T1DM and increased osteoclast activity(9,10), others fail to show any changes in resorption(2,11). In contrast, some type 2 diabetes mellitus (T2DM) patients have an increase in bone mineral density(12,13), however, similar to T1DM, T2DM patients are also more likely to develop fracture healing complications, suggesting a shared mechanism that contributes to the impaired fracture healing associated with diabetic bone repair(12,14).
Glucose control, although important for treatment, does not provide universal protection from the downstream effects of diabetes. The attachment of glucose moieties (glycation) that interfere with protein and tissue structure and function can be reduced but not prevented(15). Glycation of type I collagen in bone reduces the ability of osteoblasts to adhere to the extracellular matrix and diminishes alkaline phosphatase (bone-forming) activity. In T1DM rat models, closed fractures showed a delay in healing time in both insulin-controlled and uncontrolled groups(16), which is consistent with human patients(17). Although insulin control of blood glucose levels is an integral part of T1DM treatment, it cannot completely correct the bone loss(18), or the delayed fracture healing in T1DM; an additional therapeutic intervention is needed.
Canonical WNT signaling, which relies on β-catenin activity, in addition to its many roles during embryonic development(9-21), is also involved in fracture repair(22), and may provide a therapeutic path to treat diabetic fractures. In TCF reporter mice, a LacZ gene is expressed upon β-catenin activation and TCF binding, thus providing a way to assess canonical WNT signaling activity. After closed tibial fracture in mice, reporter activity indicated that elevated β-catenin signaling is sustained during the entire time course of fracture repair(22). Furthermore, β-catenin hyperactivity during fracture repair improved fracture healing even when increased in osteoblasts cells only(22). β-catenin activation is a requirement for mesenchymal stem cell differentiation into osteoblasts and osteoblast maturation; lack of β-catenin in mice results in decreased bone density(23). Type 1 diabetic rats showed a decrease in the WNT downstream effectors phosphorylated glycogen synthase kinase 3β (GSK3β) and activated β-catenin, along with an increase in SOST protein level, which resulted in decreased osteoblast activity(11). In addition, T2DM patients have elevated circulating SOST levels compared to non-diabetic patients(24), suggesting that WNT signaling is altered and may contribute to the observed osteopenia and delayed healing. Taken together, these data suggest that a high blood glucose environment alters osteoblast activity via WNT signaling.
To modulate WNT signaling, we have targeted sclerostin (SOST), a potent WNT antagonist secreted by osteocytes, which functions to inhibit bone formation(25). In animal models, overexpression of Sost causes osteopenia and limb defects(26,27), while lack of SOST causes 3-4 times more bone mass, consistent with human phenotypes(28,29). In humans, lack of sclerostin causes sclerosteosis, a generalized skeletal hyperostosis disorder that results from elevated WNT signaling/osteoblast activity(30,31), while non-coding deletions of gene regulatory regions that control SOST expression result in similar bone overgrowth(28,32). SOST antibodies (SostAb) have been shown to enhance bone healing in ovariectomized rats(33,34) by increasing bone formation and mass due to enhanced osteoblast function. SostAb treatment in T2DM rat models has also been shown to improve bone mass and strength(35).
In this study, we have administered SOST-neutralizing antibodies in vivo, in a pharmacological model of T1DM in mice during fracture repair. By enhancing canonical WNT signaling, we have shown improved fracture repair and rescued the osteopenia in T1DM mice. The improved bone quality persisted at least three weeks after treatment had been discontinued, suggesting an extended benefit to bone quality and fracture repair in the absence of glucose control. In addition, T1DM in our model induced enhanced bone marrow adipogenesis, which was rescued in healing fractures by SostAb treatment. Herein we demonstrate for the first time that sclerostin antibodies counteract effects of high glucose-driven elevation of SOST levels in uncontrolled diabetes, indicating a positive therapeutic effect of modulating WNT signaling in T1DM patients.
Methods
Animals and Fracture Model
Six week old C57BL6/J male mice were injected daily with Streptozotocin (STZ) (50mg/kg) or phosphate buffered saline (PBS) for 5 days; diabetic status was confirmed by a blood glucose readings of ≥300 mg/dL, one week after the last injection. At eight weeks of age, mid-femoral fractures were generated using a closed Einohorn model as previously described(36). SOST antibody, (SostAb; Regeneron) (25 mg/kg), or PBS was administered subcutaneously twice weekly up to 21 days post-fracture or 11 weeks of age, for a total of five injections, in Control, STZ, SostAb, and STZ+SostAb groups, Age-matched, uninjured cohorts (n=6-10 per group per time point) were also treated. At 21 days and 42 days post-fracture, bones were dissected and processed for microscale-computed tomography (μCT), histology and immunofluorescence (IF). All animal work was IACUC-approved and performed at Lawrence Livermore National Laboratory in an AAALAC-accredited facility.
Histology and Immunofluorescent Staining
Collected tissues were fixed, dehydrated, embedded and sectioned as described previously(28). For histology, slides were stained with Alcian Blue pH 1.5 and Nuclear Fast Red, or Masson's Trichrome. For immunofluorescence, Uni-trieve (Innovex) was used for the antigen retrieval for 30 minutes at 65°C, unless stated otherwise. Primary antibodies against RUNX2 (abcam, ab76956), collagen type 1 (calbiochem 234167), SP7/Osterix (ab22522), osteocalcin (abcam, ab10911), active caspase 3 (cellsig 9661), were used. Anti-SOST (R&D, AF1589) required Trypsin/EDTA at 37°C for 25 minutes for antigen retrieval. Anti-activated β-catenin (Millipore, 8E7, 05-665) required Uni-trieve, Proteinase K (15μg/ml) for 15 minutes, and Rodent Block. Secondary antibodies (Alexa Fluor 488 (green) or 594 (red), Molecular Probes) were used for detection. Negative control slides included secondary antibody-only, with the same antigen retrieval method used for the experimental samples (see also Supp. Fig.2). Stained slides were mounted with Prolong Gold with DAPI (Molecular Probes). ImagePro Plus V7.0 software and a QIClick CCD camera were used for imaging. Qualitative assessment of immunostains was performed by 2 blinded reviewers without knowledge of treatment group. For histological analysis of adipocytes and osteoclasts, cells were counted on complete bone sagittal sections (n=12 sections per animal) for n=3 animals per group by two blinded reviewers. Counts by each reviewer were averaged on a per-section basis for analysis. Cells were counted by hand for adipocytes, as they are represented by clear round empty spaces on the slide. Data are expressed as mean number of cells per section ± standard deviation. Cathepsin K immunostains were quantified using the Analyze Particles tool in ImageJ. Data are expressed as mean % stained area± standard deviation.
Micro-CT
Specimens were stored in 70% EtOH at 4°C. Specimens were processed without knowledge of sex, age or treatment, as these data were provided only after all the scanning and image analyses were complete. L4 vertebrae, mid femoral cortical bone, and fracture callus were measured and analyzed for bone parameters (n=6-10 per group) (μCT 40, SCANCO, Brüttisellen, Switzerland) according to the guidelines for μCT analysis of rodent bone structure(37): energy 55 kVp, intensity 114 mA, integration time 900 ms, 6 μm nominal voxel size. The threshold for “bone” was set at 375, which is approximately equal to 620 mg HA/cm3. The trabecular bone region was identified manually by tracing the region of interest. Images were thresholded using an adaptive-iterative algorithm and morphometric variables were computed from the binarized images using direct, 3D techniques that do not rely on any prior assumptions about the underlying structure. L4 vertebrae were scanned axially in a cranio-caudal orientation and the region of interest was placed 0.3mm beneath the cranial and above the caudal growth plates(38). Depending on the size of the vertebra, this ROI size ranged between 2220 and 2820 μm. 7 Images were segmented with the same algorithm and trabecular bone variables were assessed. To assess cortical bone parameters, 50 transverse μCT slices were obtained at the femoral mid-diaphysis using a 6 μm isotropic voxel size. Images were subjected to Gaussian filtration and segmented using a constant threshold. For fracture analysis, age-matched animals (11 or 14-weeks of age) were compared to 21 day or 42 day fractures for cortical measurements. Callus volume (TV) was measured including only callus tissue that excluded the native bone volume of the original cortical bone and marrow space. The whole callus was scanned, and we manually drew the contour line to define the region and volume of callus (TV). Then we used a unified threshold to segment mineralized callus from surrounding undermineralised callus, and only the callus that had similar BMD as native bone was counted into bone volume (BV).
Immunosorbent Assay
A mouse sclerostin ELISA kit (ALPCO, 41-SCLMS-E01) was used according to the manufacturer's instructions to measure serum sclerostin levels. Data is represented as mean +/- standard deviation (n=5 per group).
Statistics
All data were expressed as the mean ± standard deviation. For uCT and quantitative histological analysis, two-way ANOVA was performed with Sidak's correction for multiple comparisons using PRISM 6 software. Significant results are presented as a diabetic effect, antibody treatment effect, or diabetic x antibody treatment interaction effect; p<0.05 was considered significant. For statistical analysis of SOST ELISA results, Student's T-test with a two-tailed distribution was used, with two-sample equal variance (homoscedastic test), for significance. All groups were compared to Control; p<0.05 was considered significant.
Results
SostAb improves loss of bone in uninjured STZ mice
The experimental design included two collection time points at 11 weeks and 14 weeks of age, for uninjured age-matched cohorts; SostAb was discontinued after 3 weeks of treatment or 11 weeks of age (Fig. 1A). At 11 weeks of age, STZ-treated mice (STZ) showed abnormal growth plates, with reduced trabecular bone and less organized cartilage matrix deposition in the growth plate (Fig. 1D, yellow arrowheads), compared to Controls (Fig. 1B, D). We also noted an increase in adipocytes in the bone marrow of STZ-treated mice, (Fig. 1D,H, black arrowheads), as well as reduced trabecular bone (Fig. 1F, H), supporting previous reports of bone loss in STZ-treated animals(40,41). All groups showed increased trabecular bone in the metaphysis, in comparison to STZ, which showed less trabecular bone (Fig. 1B-E, and yellow arrowheads), STZ+SostAb also appeared to have a more organized growth plate (Fig. 1D,E). SostAb treatment dramatically increased trabecular bone formation in the epiphysis (Fig. 1F-H). Cortical bone was much thicker in SostAb treated groups (Fig. 1J-M, arrows), and while STZ mice showed residual cartilage matrix in the cortical bone (Fig. 1L, arrowhead) SostAb treatment of STZ mice resulted in less persistent cartilage (Fig. 1M).
Figure 1. SostAb treatment Improves Low Bone Mass in STZ mice.
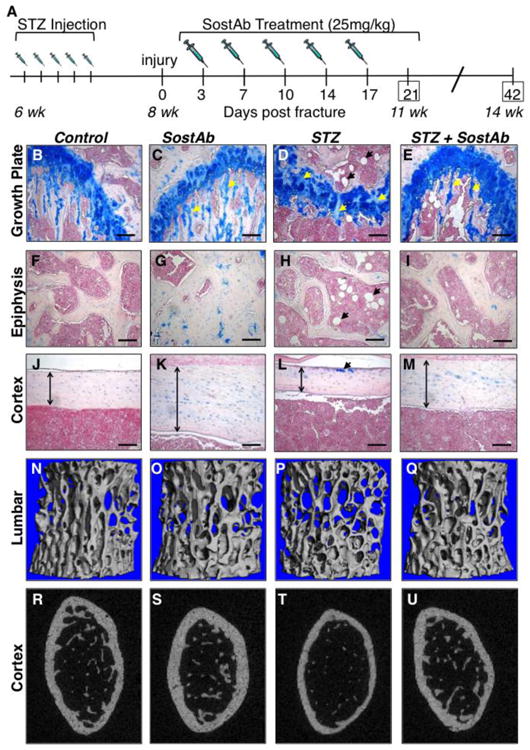
Experimental design (A). Histological sections and stain with Alcian Blue and Nuclear Fast Red (B-M). The growth plate (B-E), Epiphysis (F-l), and Cortex (J-M) represents the location of the cartilage, trabecular bone, and cortical bone respectively. Alcian Blue stains the cartilage in blue- see also yellow arrows (C, D, E). STZ mice showed some cartilage present also in the cortical bone (L, yellow arrow). White circles, indicated by black arrows (D, H, E) show adipocytes in the bone marrow. The cortical bone (J-M) shows degrees of cortical bone thickness (vertical arrows) in each group. Uninjured 11 week old mice were euthanized and the L4 vertebrae (N-Q) and distal femurs (R-U) were scanned for uCT. L4 vertebrae uCT scans reveals a visualized amount of trabecular bone is present within each group. A transverse plane on the distal uninjured femurs visually provides the differences of relative cortical bone thickness among each treated group. Calibration scale: bar = 100 μm.
μCT analysis reveals that uninjured STZ mice had significantly reduced bone volume to total bone volume ratio (BV/TV), trabecular thickness (Tb. Th.), and Structural Model Index (SMI) in lumbar vertebrae (L4) as a result of the diabetic state (Table 1, see also Fig. 1N,P). In cortical bone, bone area: total area (BA/TA) (p<0.0001), bone area (BA) (p<0.0001), and total area (TA) are reduced in uninjured STZ mice compared to Control (Table 1, see also Fig. 1R,T). No changes were identified in connectivity density (Conn. Dens.), trabecular number (Tb. N.), trabecular separation (Tb. Sp.) or bone mineral density (BMD). SostAb treated mice, both with and without diabetes, had a significant improvement in all bone parameters except L4 BMD in L4 vertebrae (Table 1, see also Fig.1N,O). In cortical bone, SostAb treated mice, with or without diabetes, had significantly increased BA/TA (p<0.0001) and BA (Table 1, see also Fig. 1R,S). In STZ+SostAb treated mice, BV/TV, SMI, and Tb. Th. (all p<0.0001), showed significant diabetes x antibody treatment interactions, suggesting that the effect of antibody treatment in diabetic animals may provide synergistic improvement for these parameters. Overall, our data from uninjured animals at 11 weeks of age indicates that SostAb improves diabetic osteopenia in the absence of fracture.
Table 1.
Bone Phenotyping Based on μCT Parameters in the Cancellous Bone Compartment of the L4 Vertebrae and Cortical Bone of uninjured 11-week old Male Mice Compared to Controls.
| Index | Control (n=6) | SostAb (n=6) | STZ (n=8) | STZ + SostAb (n=8) |
|---|---|---|---|---|
| BV/TV (%) | 0.235±0.038 | 0.299±0.006§ | 0.177± 0.010* | 0.333± 0.028§❖ |
| Conn. Dens. (1/mm3) | 326.203±22.476 | 294.848±23.435§ | 288.401±33.059 | 272.996±22.562§ |
| SMI (index [1]) | 0.395±0.219 | 0.32695±0.030§ | 0.8251±0.101* | -0.114875± 0.214§❖ |
| Tb.N (1/mm) | 5.490±0.339 | 5.572±0.165§ | 5.191±0.219 | 5.743±0.319§ |
| Tb.Th (mm) | 0.042±0.004 | 0.055±0.000§ | 0.0350±0.001* | 0.057± 0.003§❖ |
| Tb.Sp (mm) | 0.172±0.013 | 0.165±0.007§ | 0.183±0.009 | 0.160±0.011§ |
| BMD (μg HA/cm3) | 970.109±14.809 | 963.635±8.277 | 951.211±18.037 | 967.104±7.427 |
| BA/TA (%) | 0.393±0.023 | 0.464±0.020§ | 0.308±0.035* | 0.408±0.026§ |
| BA (mm2) | 0.927±0.131 | 1.143±0.107§ | 0.635±0.123* | 0.909±0.067§ |
| TA (mm2) | 2.349±0.235 | 2.463±0.184§ | 2.052±0.197* | 2.232±0.123§ |
| BMD (μgHA/cm3) | 1062.294±9.606 | 1067.389±12.429§ | 1049.428±11.972 | 1069.487±15.893§ |
Data represents mean ± standard deviation for parameters measured. BV=Bone Volume; TV= Total Volume; Conn. Dens.= Connectivity Density; SMI= Structural Model Index; Tb. N= Trabecular Number; Tb.Th= Trabecular Thickness; Tb.Sp= Trabecular Separation; BMD= Bone Mineral Density; TA=Total Area; BA= Bone Area.
p-values <0.05, antibody effect;
p-values < 0.05, diabetic effect;
p-values <0.05, diabetic x antibody interaction.
Improved bone parameters persist 3-weeks after discontinuation of SostAb treatment in uninjured STZ mice
To examine whether diabetic bone reverts to the osteopenic condition subsequent to SostAb treatment discontinuation, we examined bone parameters in 14-week old uninjured cohorts, or 3-weeks after cessation of SostAb treatment. We found no significant effect of diabetes on the variance in STZ mice, however, the means for all parameters in lumbar vertebrae were significantly different from Control mice (p<0.0001 for all parameters) (Table 2, see also Fig. 1N,P). This indicated that although the diabetic state had a significant negative impact on bone, most of the variance in the data is contributed by antibody treatment. Statistically, we find a significant antibody effect for all L4 bone parameters measured, with or without diabetes. At 14 weeks, different parameters show significant diabetes x antibody interaction effects (Conn. Dens., SMI, and Tb. N.) that at 11 weeks of age, suggesting that the synergistic benefits of antibody treatment on diabetic bone may change with age or growth status. In cortical bone, BA/TA, BA, and BMD did have a significant diabetic effect (Table 2). Antibody effects in SostAb STZ+SostAb mice increased BA/TA, BA, and BMD (all p<0.001), compared to Control and STZ groups (Table 2). Thus, at 14 weeks of age in uninjured animals, we saw continued bone quality improvement after discontinuation of SostAb treatment, in the absence of fracture.
Table 2.
Bone Phenotyping Based on μCT Parameters in the Cancellous Bone Compartment of the L4 Vertebrae and Cortical Bone of uninjured 14-week old Male Mice Compared to Controls.
| Index | Control (n=9) | SostAb (n=9) | STZ (n=9) | STZ + SostAb (n=8) |
|---|---|---|---|---|
| BV/TV (%) | 0.223±0.025 | 0.399±0.025§ | 0.191±0.027 | 0.403±0.053§ |
| Conn. Dens. (1/mm3) | 317.424±32.066 | 335.117±34.703§ | 282.650±23.429 | 362.147±55.315§❖ |
| SMI (index [1]) | 0.392±0.193 | -0.653±0.208§ | 0.601±0.149 | -0.844±0.424§❖ |
| Tb.N (1/mm) | 5.417±0.274 | 6.265±0.255§ | 5.073±0.263 | 6.350±0.372§❖ |
| Tb.Th (mm) | 0.041±0.003 | 0.067±0.004§ | 0.037±0.003 | 0.065±0.008§ |
| Tb.Sp (mm) | 0.177±0.009 | 0.148±0.006§ | 0.188±0.011 | 0.149±0.009§ |
| BMD (μg HA/cm3) | 969.230±11.598 | 977.497±12.198§ | 955.274±15.103 | 976.473±20.533§ |
| BA/TA (%) | 0.465±0.015 | 0.565±0.0127§ | 0.521±0.034* | 0.439±0.020§ |
| BA (mm2) | 0.945±0.094 | 1.084±0.067§ | 1.030±0.138* | 0.771±0.069§ |
| TA (mm2) | 2.030±0.190 | 1.920±0.133 | 1.971±0.155 | 1.757±0.117❖ |
| BMD (μgHA/cm3) | 1121.596±16.381 | 1146.834±11.915§ | 1123.519±16.748* | 1117.314±17.243§ |
Data represents mean ± standard deviation for parameters measured. BV=Bone Volume; TV= Total Volume; Conn. Dens.= Connectivity Density; SMI= Structural Model Index; Tb. N= Trabecular Number; Tb.Th= Trabecular Thickness; Tb.Sp= Trabecular Separation; BMD= Bone Mineral Density; TA=Total Area; BA= Bone Area.
p-values <0.05, antibody effect;
p-values < 0.05, diabetic effect;
p-values <0.05, diabetic x antibody interaction.
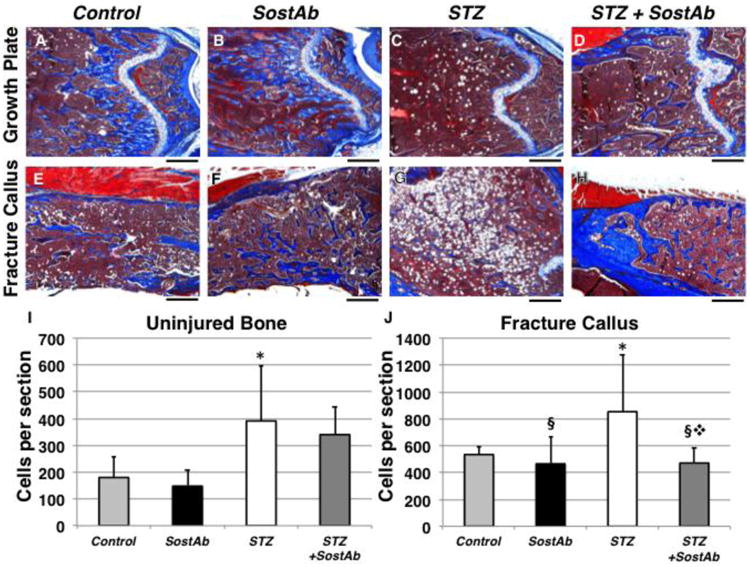
SostAb treatment improves bone parameters in fracture callus of injured STZ-treated mice
At 11 weeks of age, all groups had some amount of persistent cartilage in the mid-callus regions examined (Fig. 2A, 2B-E). Both the SostAb and STZ+SostAb groups appeared to have more mineralized bone within the callus compared to Control and STZ groups (Fig. 2B-E). At 14 weeks of age, bridging had occurred in all groups at the fracture site, and remodeling was evident by reduced trabecular bone in the callus region (Fig. 2F-I, compared to 2B-E). An enlarged marrow space at the bridging site was evident in STZ mice compared to Control, and abundant adipocytes persisted (Fig. 2F,H, and arrowheads), while both SostAb groups displayed more bone at the site of fracture compared to Control and STZ respectively (Fig. 2G,I).
Figure 2. Histology and MicroCT reveals SostAb Enhances Endochondral Healing.
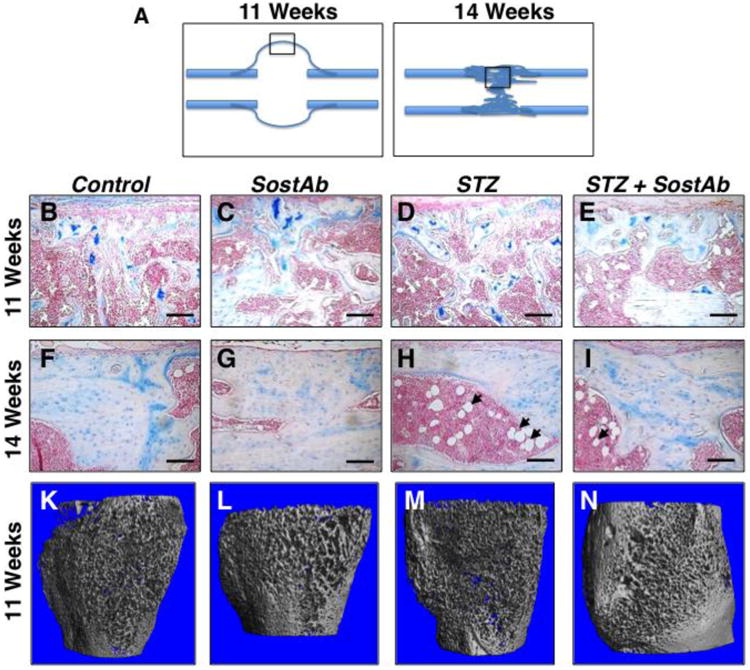
Histological sections on post fracture (dpf) callus at 11 and 14 weeks of age were analyzed and observed at the mid regions (diagrams, A). Calluses at 11 weeks of age (B-E) and 14 weeks of age (F-l) were sectioned and stained with Alcian Blue and Nuclear Fast Red. Alcian Blue stains the cartilage blue, easily observed in calluses of 11 week old mice (B-D). Adipocytes, indicated by black arrows (H, I) were observed in the bone marrow, easily seen in calluses of 14 week old mice. Calluses at 11 weeks of age were also scanned for uCT, providing a visual illustration of the amount of woven bone present in the callus (K-N). Calibration scale: bar = 100 μm.
Representative μCT reconstructions of calluses from 11 week old animals are shown in Fig. 2K-N. Reconstructions were consistent with histology, indicating a more porous bone structure in the calluses of STZ mice compared to all other groups (Fig. 2M). SostAb-treated groups (Fig. 2L,N) appeared to have thicker trabecular structure in calluses compared to Control and especially STZ mice (Fig. 2M). Quantitative callus analysis by μCT confirmed the histology (Table 3). Compared to Controls, STZ calluses at 11 weeks of age had decreased BV and TV, due to diabetic effects, indicating smaller fracture calluses, but this resulted in similar BV/TV between the two groups. Bone Mineral Content (BMC), the amount of mineralized bone per callus volume, had a significant diabetic effect, although the mean is unexpectedly higher than Controls; this may be due to the reduced volume of the callus in STZ animals. At 11 weeks of age, there was a significant antibody effect on all callus parameters except TV; and BMD had a significant diabetic x antibody interaction, which overall, reduced the BMD of the callus. Antibody effects on callus mineral suggested that SostAb treatment reduced BMD in the callus at relatively early healing stages.
Table 3.
Bone Phenotyping Based on μCT Parameters in the Cancellous Bone Compartment of Fracture Calluses of 11 Week Old and 14 Week Old Male Mice Compared to Controls.
| Index | Control (n=6) | SostAb (n=6) | STZ (n=6) | STZ + SostAb (n=6) |
|---|---|---|---|---|
| BV/TV (%) | 0.121±0.014 | 0.206±0.034§ | 0.132±0.0174 | 0.199±0.039§ |
| BV (mm2) | 2.814±0.496 | 4.368±1.543§ | 1.841±0.414* | 3.333±1.258§ |
| TV (mm2) | 23.447±4.642 | 21.473±7.424 | 14.429±5.038* | 16.458±3.638 |
| BMC (μgHA/cm3) | 172.312±5.433 | 253.603±25.708§ | 204.106±17.506* | 265.681±33.555§ |
| BMD (μgHA/cm3) | 945.376±15.143 | 905.870±23.073§ | 917.758±4.496 | 908.803±13.723§❖ |
|
| ||||
| Index | Control (n=6) | SostAb (n=8) | STZ (n=7) | STZ + SostAb (n=8) |
|
| ||||
| BV/TV (%) | 0.286±0.096 | 0.388±0.072§ | 0.234±0.037 | 0.366±0.056§ |
| BV (mm2) | 1.797±0.31 | 2.923±0.738 | 0.952±0.349 | 2.75±0.474 |
| TV (mm2) | 6.831±2.275 | 7.747±2.67 | 4.088±1.516 | 7.717±1.516 |
| BMC(μgHA/cm3) | 373.075±100.387 | 425.818±70.307§ | 279.438±66.801* | 412.253±53.769§ |
| BMD (μgHA/cm3) | 1033.059±32.891 | 1033.101±22.847 | 1004.713±31.445* | 1010.653±14.439 |
Data represents mean ± standard deviation for parameters measured. 11-week old mouse fracture callus data are presented in the top half of the table, 14 week-old mouse data are presented in the bottom half. BV=Bone Volume; TV= Total Volume; BMC= Bone Mineral Content; BMD= Bone Mineral Density.
p-values <0.05, antibody effect;
p-values < 0.05, diabetic effect;
p-values <0.05, diabetic x antibody interaction.
By 14 weeks of age, the fracture calluses have matured. Diabetic effects on the callus at this stage are only seen in BMC and BMD (Table 3, lower half), which reveals the long-term negative effects of diabetes on callus mineralization (p<0.0001 for both parameters). Antibody effects on calluses at 14 weeks of age show significant improvements in BV/TV and BMC, indicating that SostAb treatment, regardless of diabetic state, enhances mineralization or maturation of the callus. Significant gains in callus quality in STZ+SostAb treated animals are retained at 14 weeks of age, and the mineralization of the callus had reached or exceeded the level of Controls.
Injured STZ-treated mice have impaired osteogenesis during fracture repair
SP7/osterix, an early osteoblast marker (Fig. 3A-D), appeared dramatically elevated at 11 weeks of age, in STZ calluses (Fig. 3C, arrows), with large numbers of positive cells aligned on the bone surface, suggesting that lineage commitment and early osteoblast differentiation was not inhibited in STZ mice. However, RUNX2, a pro-osteogenic transcription factor that is increased early in the osteogenic differentiation program (Fig. 3E-H), appeared reduced in STZ mice compared to Controls (Fig. 3E,G), consistent with published results(42). In STZ mice, RUNX2 stain was less plentiful compared to Controls, (Fig. 3E,G); Treatment with SostAb did not increase the level of RUNX2(Fig. 3E,F); STZ+SostAb also did not appear to increase RUNX2 levels compared to STZ calluses (Fig. 3G,H), Collagen I, a matrix protein secreted by osteoblasts (Fig. 3I-L) was visibly reduced in STZ calluses (Fig. 3K), which indicated a decrease in mineralized matrix production consistent with delayed or impaired osteogenesis (see also Table 3), while SostAb and STZ+SostAb calluses showed increased levels (Fig. 3J,L), compared to Controls (Fig. 3I). Osteocalcin (Fig. 3M-P) and DMP1 (Fig. 3Q-T), markers of osteoblast maturation and osteocyte differentiation, appeared to have dramatically reduced signal in STZ calluses (Fig. 3O,S), which indicated delayed callus maturation in STZ calluses, while Control (Fig. 3M,Q), SostAb (Fig. 3N,R), and STZ+SostAb (Fig. 3P,T) groups revealed abundant signal along the surface of the bone. To determine if there is an alteration of cell apoptosis, activated caspase 3 antibody, an apoptosis marker (Fig. 3U-X), was stained on callus sections at 11 weeks of age, revealing no visible change in signal between any of the groups. Negative control stains for each antibody and antigen retrieval method are presented in Sup Fig. 2. This suggests that cell apoptosis was not contributing to poor bone in STZ calluses at 11 weeks of age. At this time point, STZ+SostAb calluses appeared to show patterns for all markers similar to Controls or SostAb calluses, rather than STZ calluses, which indicated a return to normal or better than normal healing, based on marker representation of the healing process.
Figure 3. SostAb Rescues Abnormal Osteoblast Maturation in STZ Observed by Immunofluorescence.
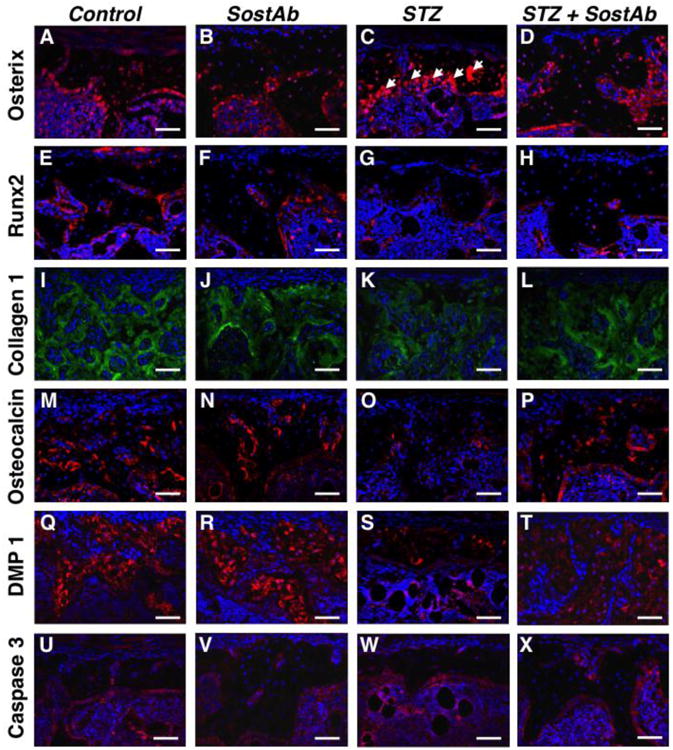
Fluorescent immunostains on callus sections of 11 week old mice were completed with markers of interest. Secondary antibodies of either green (Alexa Fluor 488) or red (Alexa Fluor 594) were used for detection and DAPI stains the nuclei of cells blue. An early osteoblast marker, osterix, showed osteoblast residing along the surface of the woven bone as indicated by white arrows (A-D). An early osteoblast transcription factor, RUNX2, is shown in panel E-H. Collagen type I, a protein secreted by osteoblasts, was shown in green within woven bone in panel I-L. A late osteoblast differentiation marker, osteocalcin, and osteocyte marker, DMP1, are represented in panels M-P and Q-T, respectively. Apoptosis marker, activated caspase 3, is shown along the woven bone and in the bone marrow in panel U-X. Negative control slides, see Supp. Fig. 2. Calibration scale: bar = 100 μm.
By 14 weeks of age, the callus had matured in injured Control animals, and SP7/osterix was still localized at the bone surface (Fig. 4A). Collagen I appeared more abundant compared to 11 weeks of age (Fig. 3I,4E), and DMP1 protein was plentiful in Control calluses (Fig. 4I). Compared to Controls, STZ calluses continued to have an abundant number of SP7/osterix –positive cells lining the bone surface, although not as plentiful as at 11 weeks of age (Fig. 3C,4C). STZ calluses continued to have visibly reduced signal for Collagen I compared to Controls (Fig. 4E,G), which suggested a continued delay in mineralization of the callus (see also Table 3). However, by 14 weeks of age, cells that became embedded in bone matrix of STZ calluses produced DMP1, at similar apparent levels compared to the other groups (Fig. 4I-L). Antibody-treated groups at this time point appeared to have more Collagen I in the callus relative to Controls or STZ calluses (Fig. 4E-H), which indicated possible enhanced mineralization. There appeared to have been little impact of antibody treatment on the abundance of DMP1 in the bony calluses of SostAb or STZ+ Sost Ab groups, as all groups seemed to have similar levels (Fig. 4I-L).
Figure 4. SostAb Anabolic Effect During Repair Continued After Treatment Termination.
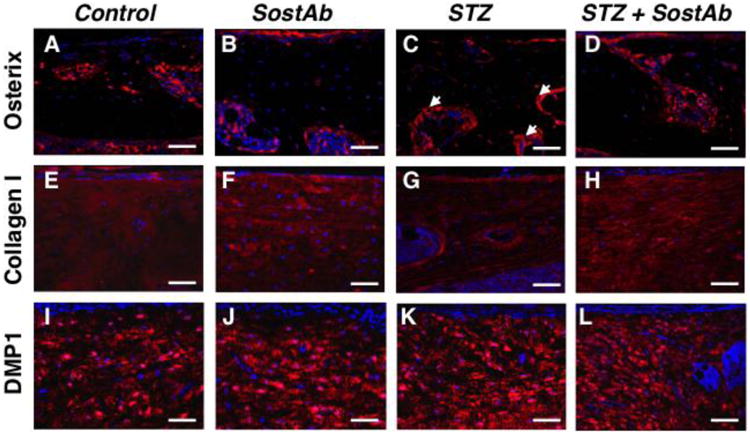
Calluses in mice at 14 weeks of age were stained with markers of interest by fluorescent immunostaining. Secondary antibodies of Alexa Fluor 488 (green) or Alexa Fluor 594 (red) were used for detection and DAPI stained the nuclei of cells blue. Osterix, an early osteoblast marker, signal was along the surface of the bone as indicated by the white arrows (A-D). Collagen 1, a protein secreted by osteoblasts, appeared within the bone in panels E-H. DMP1, an osteocyte marker, also appeared within the remodeling bone in panels I-L. Negative control slides, see Supp. Fig. 2. Calibration scale: bar = 100 μm.
While we did not see evidence of increased osteoblast apoptosis, we considered that enhanced osteoclast activity in STZ diabetic bones could contribute to fracture callus instability and poor bone structure. We performed osteoclast-specific tartrate-resistant acid phosphatase (TRAP) stain on uninjured bones and fracture calluses of 11 week old animals, and did not find any evidence of increased osteoclast activity with this marker in the growth plate (Sup. Fig. 1A-D), epiphyseal trabecular bone (Sup. Fig. 1E-H), or fracture callus (Sup. Fig. 1I-L) of STZ mice, nor did we see a change in TRAP activity with SostAb treatment. Additionally, we did not see a significant difference in cathepsin K when tested by two-way ANOVA, which was used to examine osteoclast maturity in epiphyseal bone and growth plate (Sup. Fig. 1M-Q). These data indicated that there is no difference in osteoclast activity of STZ diabetic mice, 5 weeks after onset of diabetes (at 11 weeks of age), and are consistent with other reports of normal osteoclast activity in T1DM(3,10,11). Our data indicated that osteoblast maturation markers were reduced in STZ-diabetes, while we found no evidence of osteoblast apoptosis or enhanced osteoclast activity to account for reduced bone mass; SostAb treatment of diabetic mice returned osteoblast marker levels to normal or enhanced levels.
WNT signaling is diminished during diabetic fracture healing, and reversed with SostAb administration
To examine WNT signaling activity we assessed activated β-catenin protein levels. In calluses of 11 week old mice, very low levels of activated β-catenin were observed in STZ treated samples (Fig. 5C). Control calluses showed modest levels of activated β-catenin, primarily localized to the bone-forming surface, with very few positive cells embedded in the bone matrix (Fig. 5A). However, both SostAb treated groups appeared to have higher levels of activated β-catenin at the bone surface, along with several positive cells in the bone matrix (Fig. 5B,D), suggesting enhanced β-catenin activity in osteocytes as well as in osteoblasts. By 14 weeks of age, in calluses, we found moderate levels of activated β-catenin in the bone of Control mice, and it was no longer confined to the bone-forming surface (Fig. 5I). STZ mice appeared to have small patches of activated β-catenin, but the signal was primarily absent from osteocytes in the bone matrix (Fig. 5K). In SostAb and STZ+SostAb treated mice, 42 day calluses showed robust activated β-catenin signal throughout the callus (Fig. 5J,L), which seemed to be increased over the levels seen in either Controls or STZ calluses (Fig. 5I,L). These data demonstrated that SostAb administration elevated β-catenin activity, consistent with SOST's role as a WNT antagonist(43).
Figure 5. Immunohistochemistry Reveals Altered Wnt Signaling in Treated Groups.
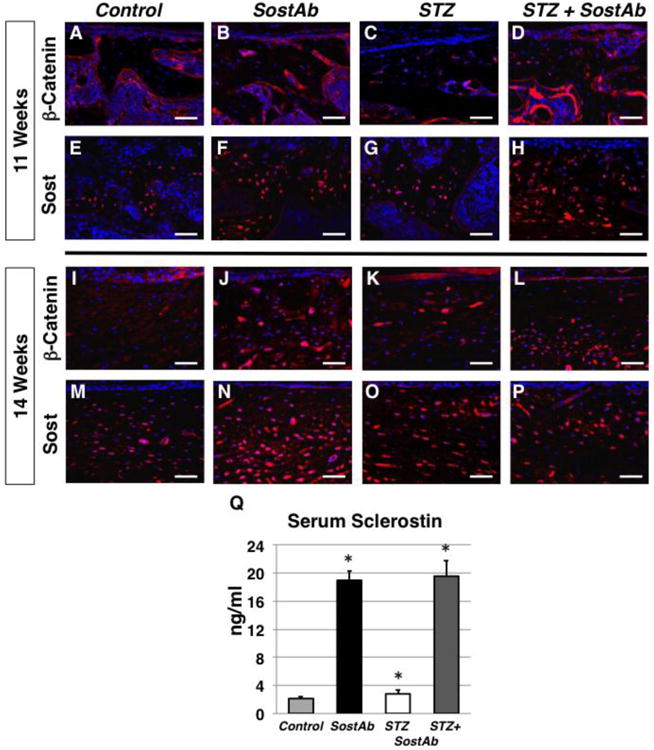
Fluorescent immunostains for β-catenin were stained on calluses at 11 (A-D) and 14 weeks of age (I-L). Alexa Fluor 594 (red) was used as a secondary antibody while the cells nuclei was stained with DAPI. Beta-catenin signal is initially expressed along the surface of the bone (A-D) and later within osteocytes during the remodeling stage (I-L). Sost antibody immunohistochemistry on calluses at 11 weeks of age (E-H) and 42 dpf calluses (M-P) is expressed with the woven bone and osteocytes during the later stages of healing. Sost serum levels in each treated groups was measured using an ELISA kit for mouse Sost (Q) and represented in ng/ml. p-values <0.05 are statistically significant (asterisks), compared to Controls. Negative control slides, see Supp. Fig. 2. Calibration scale: bar = 100 βm.
When examining sclerostin levels in the callus at 21 days, we saw modestly increased sclerostin in matrix-embedded cells of STZ mice, compared to Controls, consistent with expression in osteocytes (Fig. 5E,G). We did, however, observed dramatically increased sclerostin protein levels in the bone of SostAb treated groups (Fig. 5F,H). We also observed increased activated β-catenin activity in these same groups (Fig. 5B,D). By 42 days, we continued to find increased sclerostin signal in STZ calluses compared to Controls (Fig. 5M,O). We also found further increased sclerostin localization in SostAb and STZ+SostAb treatment groups (Fig. 5N,P). We questioned whether we may have detected the administered therapeutic SOST antibody, or whether we may have detected sclerostin that is inactivated, but still found in bone. Our negative control immunostains included secondary antibody stains against both the rabbit antibody that was used to examine SOST in tissues (Sup Fig. 2, A-D), and the mouse monoclonal therapeutic antibody that was administered (Sup Fig. 2, E-H), under the same antigen retrieval conditions. We found no evidence of non-specific signal, or the ability to detect the therapeutically administered antibody. Thus, we presume that the high SOST signal detected in the tissue of antibody-treated mice is the combined protein accumulation of biologically active SOST, and SOST that is inactivated and bound to SostAb but still in tissues or circulation. These findings were supported by the detection of elevated levels of SOST in the serum. STZ mice had a 30% increase in circulating sclerostin, relative to Controls (p<0.03) (Fig. 5Q). Circulating levels for sclerostin in Controls were within the reported range for the assay. However, we found 8 times the amount of circulating sclerostin in both antibody-treated groups compared to Controls (SostAb p<5×10-9 STZ+SostAb p<2×10-7). We find a pattern of elevated SOST and decreased β-catenin activity in the context of a high-glucose environment, while SostAb treatment enhances β-catenin activity despite high SOST levels.
Enhanced bone marrow adiposity in healing fractures of STZ mice is rescued by SostAb treatment
We saw some evidence that adipocytes are more abundant in the marrow compartment of STZ diabetic mice, as has been suggested by others, who have proposed that increased adipogenesis may be at the expense of precursor differentiation into osteoblasts, resulting in reduced bone formation(44-46). We additionally observed an up-regulation of the number of adipocytes in the marrow after fracture, compared to intact bone. To quantify the effects of diabetes on bone marrow adiposity, we examined Masson's Trichrome-stained sections from uninjured age-matched bones, and fracture calluses at 11 weeks of age (Figure 6). Total number of adipocytes per sagittal bone section were analyzed for both sets of samples and compared by two-way ANOVA. In uninjured bones we found a significant effect of diabetes on the adipocyte number (p<0.001) (Fig. 6I), with no effect of antibody treatment (Fig. 6A-D). However, in age-matched fracture calluses, we saw an increase in marrow adiposity in all groups, which was dramatically up-regulated in STZ diabetic mice (Fig. 6E-H). Quantification of the differences by two-way ANOVA revealed a significant diabetic effect in STZ mice, a significant antibody treatment effect, and a significant diabetes x antibody interaction effect, suggesting that SostAb treatment mitigates the enhanced adiposity seen in healing fracture callus of STZ diabetic mice. SostAb treatment may in this way enhance fracture healing by directly or indirectly reducing marrow adiposity, independent of its effects on osteoblast maturation or activity.
Figure 6. Histology Reveals increased adipogenesis in STZ mice that is reversed with SostAb tyreatment.
Masson Trichroms stain (collagen, red; nuclei, black; bone, blue; cartilage, white; adipocytes, empty circles) on uninjured bone sections and fracture callus at 11 weeks of age indicated increased marrow adiposity in STZ mice without injury (A-D, I). During fracture healing, adipogenesis increased in the marrow of all groups, but substantially in STZ calluses (E-J). Two-way ANOVA demostrated a diabetic effect in uninjured animals (I), and in fracture calluses, there was a diabetic effect, an antibody effect, and a diabetes x antibody interaction effect. *p<0.05, diabetic effect. § p<0.05, antibody effect. ❖ p<0.05, diabetes x antibody interaction. Calibration scale: bar = 500 μm.
Discussion
In this study, we treated fractured femurs in type I diabetic mice with SostAb to improve fracture healing outcomes. We utilized a chemically-induced STZ diabetic model, to cause early-onset diabetes in growing mice. Consistent with previous reports, STZ treated mice lose bone mass and quality within 3 weeks of becoming diabetic(47,48). The STZ mouse model we report here results in mild osteopenia, similar to T1DM patients(17); however, it does not represent a true model of T1DM, as there is no auto-immune trigger for the disease, so not all potential impacts of T1DM can be examined in this context. STZ-induced diabetes, at the dose we report here, has been reported as an appropriate model for T1DM effects on bone and are similar to spontaneous models of T1DM(10). The loss of bone we report here is primarily based on reduced bone formation, as we saw reduced mineral deposition, which was confirmed by immunofluorescence for matrix markers and μCT, and we did not find evidence of enhanced osteoclast activity in either uninjured bones or fracture calluses in our model.
We found that SostAb treatment of STZ mice dramatically enhanced bone formation, but despite the dramatic increases in bone volume, mineral density did not increase compared to Controls. This suggested that SostAb in the diabetic setting may function to promote osteoblast differentiation or osteoblast activity, and allows mineralization to catch up to normal levels. This is evidenced by the fact that neither SostAb nor STZ+SostAb had bone mineral densities at the end of the study that exceed the levels of Controls, while densities of both groups were less than Controls after 3 weeks of treatment. However, SostAb appears to enhance mineralization of fracture calluses tand thus potentially improve the strength of newly repaired bone, as SostAb treatment resulted in dramatically enhanced bone mineral content of callus at 14 weeks of age in both normal and diabetic mice. Similarly, recent reports have demonstrated reduced mineral content of bone after SOST antibody treatment, or due to genetic loss of Sost, despite abundant bone formation (49,50). It is likely that reduction of SOST via antibody treatment increases osteoblast activity to produce abundant osteoid, which then mineralizes at a normal rate. At later time points, this leads to higher mineral content in the abundantly formed new bone, thereby potentially increasing mechanical stability. Furthermore, early reports that successfully used SOST antibody treatment to promote fracture healing relied heavily on models that primarily healed by intramembranous bone formation(34,35,51,52). This suggested that SostAb may be beneficial in problematic fracture healing that results from poor osteogenesis; however, Li et al.,(53) reported fracture repair in an endochondral model in Sost-/- mice, and Feng et al.,(54) reported similar results with SOST antibody treatment, suggesting that fracture repair may benefit from early intervention by reducing sclerostin levels, rather than only during osteogenesis. While we found a significant benefit in callus BV and BV/TV of STZ+SostAb mice, we also saw an increase in callus size, suggesting a benefit in early repair events prior to osteogenesis.
The large number of osteoblasts and precursors at the bone surface that show SP7/osterix, in the absence of significant mineral, suggests that the osteopenia in STZ diabetic mice may be due to a defect in osteoblast maturation and matrix synthesis. In another report, STZ mice had the same amount of immature mesenchymal tissue but decreased osteoblast differentiation. This was associated with decreased collagen type 1, and Runx2 mRNA expression in an intramembranous bone repair model(55), similar to our findings. Song et al.,(56) demonstrated that loss of β-catenin in osteoblast precursors results in a switch from osteogenesis to adipogenesis that causes low bone mass. As our STZ mice displayed increased adiposity in the marrow, this is a plausible explanation for impaired osteogenesis in our STZ mice. SOST is not known to regulate the cell fate of precursor cells, however, our study shows evidence that adipogenesis dramatically increases during callus formation. This suggested that a pool of precursors have recently differentiated, and SostAb mitigated this effect, leaving open the possibility that elevated Wnt signaling influences the fate of precursor cells.
Furthermore, STZ mice appear to have much less osteocalcin at the bone surface, indicative of immature osteoblasts. However, reduced osteocalcin has also recently been implicated in metabolic activity of osteoblasts, in which reduced insulin signaling in osteoblasts and reduced Runx2 expression may fail to activate osteocalcin to promote local and systemic metabolic homeostasis in STZ diabetes(57). In STZ diabetic mice, osteoblasts are no longer able to depend on glucose as an energy source, but must rely on lipid metabolism to generate ATP(57). LRP5, a Wnt co-receptor, has been implicated in this process, and mice lacking Lrp5 develop metabolic imbalance with increased adiposity. We have shown that Sost primarily inhibits LRP5-dependent Wnt signaling(27), thus in the absence of insulin, SostAb may restore Wnt signaling through LRP5 to partially re-balance metabolic homeostasis, independent of glucose metabolism. In this way, metabolic stress on the osteoblasts is relieved, and differentiation and metabolism can proceed more normally.
Another STZ diabetic model showed that in T1DM, insulin and IGF-1 deficiency decreases osteogenesis due to negative regulation of Wnt signaling activity(11). It was demonstrated that the decreased bone formation was caused by reduced levels of activated β-catenin, precipitated by a loss of phosphorylated GSK3β, and phosphorylated AKT/PKB, targeting β-catenin for degradation. They also observed a dramatic increase in WNT inhibitor (Sost and Dkk1) levels. Loss of phosphorylated AKT/PKB can occur from a cycle of reduced WNT signaling, as in the presence of high SOST levels, or in the event of oxidative damage. As a result, cells (such as osteoblast precursors and early osteoblasts) in a low insulin, high SOST, high glucose (oxidative) environment may be subject to cell cycle arrest(58,59). Another study observed a decrease in cyclin D mRNA expression in osteoblasts cells treated in a high glucose environment(60), which correlated with increased apoptosis and reduced cell proliferation. Taken together, these findings may account for the reduced number of RUNX2-positive cells, and/or the apparent inhibition of differentiation of osterix-positive cells seen in our STZ mice. While we did not observe enhanced activated caspase-3-driven apoptosis in calluses at 21 days, it is possible that early osteoblast or precursor death at another time point may contribute to reduced osteogenesis in our T1DM model.
Reports are conflicting on whether T1DM induces changes in osteoclast activity, with some studies reporting enhanced, unchanged, or even reduced osteoclast activity in the context of T1DM. Reports of osteopenia compared to osteoporosis in T1DM are difficult to deconvolute, and are likely due to differences in age of animals or patients, time since diagnosis, diabetic complications (such as vascular or kidney disease), and body condition(3,4,7). We found no change in osteoclast activity, which is consistent with the recent time since diagnosis and young age of our study groups.
Oxidative stress in tissues leads to much of the destructive sequelae in diabetic patients, causing pancreatic beta-cell death, kidney disease, cardiovascular disease, and may contribute to the effects of diabetes seen in the skeleton(61). Reduced WNT signaling and increased WNT antagonism have been associated with increased oxidative stress, and reduced osteoblast activity(11,62,63). Oxidative stress activates FOXO signaling, which competes for β-catenin in the cell, to reduce activation of WNT target genes. Activation of WNT signaling activates the AKT/PKB pathway; PKB then phosphorylates and sequesters FOXO in the cytoplasm, and prevents it from competing with β-catenin(64). As SOST is an antagonist of β-catenin-mediated WNT signaling, reduction of bioactive SOST through SostAb treatment activates β-catenin signaling, potentially mitigating the effects of oxidative stress and competition by activated FOXO(62). Thus, WNT signaling may be suppressed by a mechanism that is independent of SOST, but can be mitigated by promoting WNT signaling through inhibition of SOST. In this way, SostAb can alleviate the effects on diabetic bone that cannot be overcome by treatment with insulin.
We examined sclerostin and β-catenin levels in our mice, to demonstrate that SostAb is effective at suppressing sclerostin activity and elevating WNT signaling activity through activation of β-catenin. We found elevated sclerostin in serum, and decreased activated β-catenin in STZ mice, consistent with other reports of diabetic animals and patients with elevated SOST levels(9,11,24,62). While we did find elevated β-catenin in antibody-treated groups, consistent with elevated WNT signaling and promotion of osteogenesis, we also saw an unexpected and very highly elevated level of sclerostin protein in the bone and blood of antibody-treated animals. This finding was unexpected, but there has been a recent report of sclerostin-antibody treatment associated with elevated expression of sclerostin(65). At these extreme levels of SOST protein we find in the serum and bone, however, it is likely that the majority of the protein is inactivated by antibody bound to it. SOST antibody has been reported to have a very long lifespan in circulation; Ominsky et al.,(66) reported SOST antibody levels in circulation of cynomolgus monkeys and found that a single dose can be detected in circulation up to nearly a month post-dose, while Padhi et al.,(67) reported a single dose detected in serum up to an amazing 85 days after a single dose. In addition, it has been reported that SostAb bound to sclerostin prevents internalization of sclerostin and Wnt inhibition by sclerostin, similar to our findings in vivo(68). These results pose some questions that remain unanswered: SostAb bound to sclerostin may have a very long lifespan in tissues; are high therapeutic doses of antibody needed to combat elevated expression and protein levels in response to treatment? Furthermore, sclerostin bound to SostAb may prevent degradation of either molecule, revealing a need to study downstream effects of this protein construct, which appears to be in circulation for extended periods of time. Future experiments are needed that examine the long-term effects of SostAb treatment on osteoblast metabolism; these experiments can be conducted in Ins2(Akita) mice, as they become diabetic by a nearly identical mechanism to STZ-induced diabetes, and have a much longer lifespan in the diabetic state. These studies have the potential to uncover more mechanistic insight into the interaction between the diabetic state and Lrp5-dependent/Sost-mediated osteoblast metabolism, and the contribution to whole body metabolic homeostasis. The phenotype of these mice is penetrant in both genders, which will facilitate much-needed studies in female animals.
Conclusions
Our study has revealed that anabolic doses of SostAb enhance bone formation and quality in an early-onset Type I diabetic fracture healing model. We provide evidence that SostAb alleviates inhibition on osteoblast differentiation caused by the diabetic state. We further demonstrated that the effects of SostAb treatment may extend well beyond the end of the dosing regimen, since our result show continued improvement of bone parameters in STZ diabetic mice treated with antibody, even three weeks after treatment had stopped. We observed SostAb-mediated reduction of marrow adiposity during callus formation, which may be of significant therapeutic benefit in diabetic fracture healing. We also uncovered an as yet undocumented in vivo phenomenon of apparent accumulation of sclerostin and/or sclerostin-antibody complexes as a result of treatment, which is worth further exploration. In summary, SostAb is an effective treatment to counteract the low osteogenesis that occurs in T1DM bone environments, and may provide extended benefits beyond the treatment span.
Supplementary Material
Supplementary Figure 1. Osteoclast Activity was Unchanged in Treated Groups. Histological sections on uninjured femora of 11 week old mice were stained with TRAP, staining (osteoclasts, red) and counterstained with Fast Green,(bone, blue-green); calibration scale: bar = 500 μm. (A-H). Analysis on osteoclast activity was analyzed at the growth plate (A-D) and epiphysis (E-H), representing the regions of natural osteogenesis and trabecular bone, respectively. No alterations in osteoclast activity was observed, which was confirmed from fluorescence immunohistochemistry on cathepsin K; calibration scale: bar=40 μm (I-L). Trabecular regions just below the growth plate were analyzed. Expression was shown by secondary antibody Alexa 594 (red) along the surface of trabecular bone. Two-way ANOVA was used to quantify Cathepsin K-positive area of growth plate and trabecular bone. Statistical analysis revealed no differences between groups (p>0.05).
Supplementary Figure 2. Negative antibody control stains show no evidence of cross-reactivity or non-specific signal. Histological sections on fracture calluses of 11 week old animals were stained with secondary antibodies corresponding to the primary antibodies used in this study. These stains utilized the antigen retrieval methods used for individual antibodies and are as follows: Donkey anti-goat 594 was used with trypsin/EDTA as a negative control for –Sost protein detection (A-D). Goat anti-mouse 594 was used with trypsin/EDTA as a negative control to detect administered mouse monoclonal SOST-neutralizing antibody (SostAb) (E-H). Goat anti-rabbit 594 was used with trypsin/EDTA as a negative control for active caspase 3 and osteocalcin (I-L). Goat anti-rabbit 488 was used with Unitrieve as a negative control for Collagen type I (M-P). Goat anti-rabbit 594 was used with Unitrieve as a negative control for SP7/Osterix, DMP1, and Cathepsin K (Q-T). Goat anti-mouse 594 was used with Unitrieve and Proteinase K as a negative control for activated b-catenin and Runx2. Calibration scale: bar = 100 μm.
Supplementary Methods: Histological sections of injured (21 days) and uninjured controls (11 weeks of age) were de-waxed and stained with TRAP working solution for 10 minutes at 37°C in the dark and counterstained with 0.02% Fast Green and mounted with 60% glycerol. Fluorescent immunostains using primary antibody against cathepsin K (abcam, ab19027) required Unitrieve (Innovex) for the antigen retrieval, for 30 minutes at 65°C. Secondary antibodies (Alexa Fluor 488 (green) or 594 (red), Molecular Probes) against mouse, rabbit, or goat antigens/primary antibodies were used for detection and negative control slides. Stained slides were mounted with Prolong Gold with DAPI (Molecular Probes). ImagePro Plus V7.0 software and a QIClick CCD camera were used for imaging. Cathepsin K quantification was performed using the Analyze Particles tool in Image J, where percent area of stain was quantified.
Highlights.
Diabetic effects on bone are rescued by SOST-neutralizing antibody treatment.
These effects persist even after discontinuation of treatment.
SOST-neutralizing antibody allows osteoblast differentiation inhibited by diabetes.
SOST-neutralizing antibody improves diabetic bone marrow during fracture repair.
Footnotes
Disclosures: CY performed the experiments, performed data analysis, and contributed to the manuscript, LX performed uCT analysis and interpretation, SH provided technical resources for SostAb usage, NH performed critical reading of the manuscript, DM contributed to animal experiments, ANE provided SostAb and contributed to the manuscript writing, GGL contributed to experimental design, mentorship, and manuscript writing, and NMC performed experiments, designed experiments, performed data analysis and wrote the manuscript. SH, LX, ANE are employees of Regeneron Pharmaceuticals, Inc. This study received funding from National Institutes of Health DK075730 and Laboratory-Directed Research and Development Grant 11-ERD-060. This work was conducted under the auspices of the United States Department of Energy by Lawrence Livermore National Laboratory (Contract #DE-AC52-07NA27344) (Manuscript release number LLNL-JRNL-666626).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Graves DT, Alblowi J, Paglia DN, O'Connor JP, Lin S. Impact of Diabetes on Fracture Healing. J Exp Clin Med. 2011;3:3–8. [Google Scholar]
- 2.Hamilton EJ, Rakic V, Davis WA, Chubb SA, Kamber N, Prince RL, Davis TM. Prevalence and predictors of osteopenia and osteoporosis in adults with Type 1 diabetes. Diabet Med. 2009;26:45–52. doi: 10.1111/j.1464-5491.2008.02608.x. [DOI] [PubMed] [Google Scholar]
- 3.McCabe LR. Understanding the Pathology and Mechanisms of Type I Diabetic Bone Loss. J Cell Biochem. 2007;102:1343–57. doi: 10.1002/jcb.21573. [DOI] [PubMed] [Google Scholar]
- 4.Hofbauer LC, Brueck CC, Singh SK, Bobnig H. Osteoporosis in Patients With Diabetes Mellitus. J Bone Miner Res. 2007 Sep;22(9):1317–28. doi: 10.1359/jbmr.070510. [DOI] [PubMed] [Google Scholar]
- 5.Nyman JS, Even JL, Jo CH, Herbert EG, Murry MR, Cockreil GE, Wahl EC, Bunn RC, Limpkin CK, Jr, Fowlkes JL, Thrailkill KM. Increasing duration of type 1 diabetes perturbs the strength-structure relationship and increases brittleness of bone. Bone. 2011;48:733–40. doi: 10.1016/j.bone.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz AV. Diabetes Mellitus: Does it Affect Bone? Calcif Tissue Int. 2003;73:515–9. doi: 10.1007/s00223-003-0023-7. [DOI] [PubMed] [Google Scholar]
- 7.Vestergaard P, Rejnmark L, Mosekilde L. Diabetes and Its Complications and Their Relationship with Risk of Fractures in Type 1 and 2 Diabetes. Calcif Tissue Int. 2009;84:45–55. doi: 10.1007/s00223-008-9195-5. [DOI] [PubMed] [Google Scholar]
- 8.Coe LM, Irwin R, Lippner D, McCabe LR. The Bone Marrow Microenvironment Contributes to Type 1 Diabetes Induced Osteoblast Death. J Cellular Physiology. 2010;226:477–83. doi: 10.1002/jcp.22357. [DOI] [PubMed] [Google Scholar]
- 9.Iitsuka N, Hie M, Tsukamoto I. Zinc supplementation inhibits the increase in osteoclastogenesis and decrease in osteoblastogenesis in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2013;714:41–7. doi: 10.1016/j.ejphar.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 10.Motyl K, McCabe LR. Streptozotocin, Type I Diabetes Severity and Bone. Biol Proced Online. 2009;11:296–315. doi: 10.1007/s12575-009-9000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hie M, Litsuka N, Otsuka T, Tsukamoto I. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int J Mol Med. 2011;28:455–62. doi: 10.3892/ijmm.2011.697. [DOI] [PubMed] [Google Scholar]
- 12.de Liefde II, van der Klift M, de Laet CE, vanDaele PL, Hofman A, Pols HA. Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam Study. Osteoporos Int. 2005;16:1713–20. doi: 10.1007/s00198-005-1909-1. [DOI] [PubMed] [Google Scholar]
- 13.Issa C, Zantout MS, Azar ST. Osteoporosis in men with diabetes mellitus. J Osteoporos. 2011;651867 doi: 10.4061/2011/651867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shu A, Win MT, Stein E, Cremers S, Dworakowski E, Ives R, Rubin MR. Bone Structure and turnover in type 2 diabetes mellitus. Osteoporosis Int. 2012;23:635–41. doi: 10.1007/s00198-011-1595-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ceriello A. The emerging challenge in diabetes: the “metabolic memory”. Vascul Pharmacol. 2012;57:133–8. doi: 10.1016/j.vph.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Beam HA, Parsons JR, Lin SS. The effects of blood glucose control upon fracture healing in the BB Wistar rat with diabetes mellitus. J Orthop Res. 2002;20:1210–6. doi: 10.1016/S0736-0266(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 17.Loder RT. The Influence of Diabetes Mellitus on the Healing of Closed Fractures. Clin Orthop Relat Res. 1988;232:210–6. [PubMed] [Google Scholar]
- 18.CamposPastor MM, Lopez-Ibarra PJ, Escobar-Jimenez F, SerranoPardo MD, Garcia-Cervigon AG. Intensive insulin therapy and bone mineral density in type 1 diabetes mellitus: a prospective study. Osteoporos Int. 2000;11:455–9. doi: 10.1007/s001980070114. [DOI] [PubMed] [Google Scholar]
- 19.Petersen CP, Reddien PW. Wnt Signaling and the Polarity of the Primary Body Axis. Cell. 2009;139:1056–68. doi: 10.1016/j.cell.2009.11.035. [DOI] [PubMed] [Google Scholar]
- 20.Halt K, Vainio S. Coordination of kidney organogenesis by Wnt signaling. Pediatr Nephrol. 2014;29:737–44. doi: 10.1007/s00467-013-2733-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Church VL, Francis-West P. Wnt signalling during limb development. Int J Dev Biol. 2002;46:927–36. [PubMed] [Google Scholar]
- 22.Chen Y, Whetstone HC, Lin AC, Nadesan P, Wei Q, Poon R, Alman BA. Beta-Catenin Signaling Plays a Disparate Role in Different Phases of Fracture Repair: Implications for Therapy to Improve Bone Healing. PLoS Medicine. 2007;4:e249. doi: 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/Beta-Catenin Signaling in Mesenchymal Progenitors Controls Osteoblast and Chondrocyte Differentiation during Vertebrate Skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Gennari L, Merlotti D, Valenti R, Ceccarelli E, Ruvio M, Pietrini MG, Capodarca C, Franci MB, Campagna MS, Calabro A, Cataldo D, Stolakis K, Dotta F, Nuti R. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J Clin Endocrino Metab. 2012;97:1737–44. doi: 10.1210/jc.2011-2958. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin Binds to LRP5/6 and Antagonizes Canonical Wnt Signaling. J Biol Chem. 2005;280:19883–7. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 26.Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, Ovcharenko D, Plajzer-Frick I, Rubin EM. Genomic deletion of a long-range bone enhancer mistregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–35. doi: 10.1101/gr.3437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collette NM, Genetos DC, Murugesh D, Harland RM, Loots GG. Genetic evidence that Sost inhibits Wnt signaling in the limb. Dev Biol. 2010;342:169–79. doi: 10.1016/j.ydbio.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, Lane NE, Harland RM, Loots GG. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci USA. 2012;109:14092–7. doi: 10.1073/pnas.1207188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X1, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–9. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 30.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balemans W, Van Den Ende J, Freire Paes-Alves A, Dikkers FG, Willems PJ, Vanhoenacker F, de Almeida-Melo N, Alves CF, Stratakis CA, Hill SC, Van Hul W. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am J Hum Genet. 1999;64:1661–9. doi: 10.1086/302416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suen PK, He Y, Chow DHK, Huang L, Li C, Ke HZ, Ominsky MS, Qin L. Sclerostin monoclonal antibody enhanced bone fracture healing in an open osteotomy model in rats. J Ortho Res. 2014;32:997–1005. doi: 10.1002/jor.22636. [DOI] [PubMed] [Google Scholar]
- 34.McDonald MM, Morse A, Mkulec K, Peacock L, Yu N, Baldock PA, Birke O, Liu M, Ke HZ, Little DG. Inhibition of sclerostin by systemic treatment with sclerostin antibody enhances healing of proximal tibial defects in ovariectomized rats. J Ortho Res. 2012;30:1542–8. doi: 10.1002/jor.22109. [DOI] [PubMed] [Google Scholar]
- 35.Hamann C, Rauner M, Höhna Y, Bernhardt R, Mettelsiefen J, Goettsch C, Günther KP, Stolina M, Han CY, Asuncion FJ, Ominsky MS, Hofbauer LC. Sclerostin antibody treatment improves bone mass, bone strength, and bone defect regeneration in rats with type 2 diabetes mellitus. J Bone Miner Res. 2013;28:627–38. doi: 10.1002/jbmr.1803. [DOI] [PubMed] [Google Scholar]
- 36.Bonnarens F, Einhorn TA. Production of a Standard Closed Fracture in Laboratory Animal Bone. J Orthop Res. 1984;2:97–101. doi: 10.1002/jor.1100020115. [DOI] [PubMed] [Google Scholar]
- 37.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro–computed tomography. J Bone Miner Res. 2010;25:1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 38.Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22:1197–207. doi: 10.1359/jbmr.070507. [DOI] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(– Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Hamada Y, Kitazawa S, Kitazawa R, Fujii H, Kasuga M, Fukagawa M. Histomorphometric analysis of diabetic osteopenia in streptozotocin-induced diabetic mice: a possible role of oxidative stress. Bone. 2007;40:1408–14. doi: 10.1016/j.bone.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 41.Fowlkes JL, Nyman JS, Bunn RC, Jo C, Wahl EC, Liu L, Cockrell GE, Morris LM, Lumpkin CK, Jr, Thrailkill KM. Osteo-promoting effects of insulin-like growth factor I (IGF-I) in a mouse model of type 1 diabetes. Bone. 2013;57:36–40. doi: 10.1016/j.bone.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.López-Herradón A, Portal-Núñez S, García-Martín A, Lozano D, Pérez-Martínez FC, Ceña V, Esbrit P. Inhibition of the Canonical Wnt Pathway by High Glucose Can Be Reversed by Parathyroid Hormone–Related Protein in Osteoblastic Cells. J Cell Biochem. 2013;114:1908–1916. doi: 10.1002/jcb.24535. [DOI] [PubMed] [Google Scholar]
- 43.Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S, Krumlauf R. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–49. doi: 10.1359/jbmr.060810. [DOI] [PubMed] [Google Scholar]
- 44.Chuang CC, Yang RS, Tsai KS, Ho FM, Liu SH. Hyperglycemia enhances adipogenic induction of lipid accumulation: involvement of extracellular signal-regulated protein kinase 1/2, phosphoinositide 3-kinase/Akt, and peroxisome proliferator-activated receptor gamma signaling. Endocrinology. 2007;148:4267–75. doi: 10.1210/en.2007-0179. [DOI] [PubMed] [Google Scholar]
- 45.Wang A, Midura RJ, Vasanji A, Wang AJ, Hascall VC. Hyperglycemia diverts dividing osteoblastic precursor cells to an adipogenic pathway and induces synthesis of a hyaluronan matrix that is adhesive for monocytes. J Biol Chem. 2014;289:11410–20. doi: 10.1074/jbc.M113.541458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148:198–205. doi: 10.1210/en.2006-1006. [DOI] [PubMed] [Google Scholar]
- 47.Musumeci G1, Loreto C, Clementi G, Fiore CE, Martinez G. An in vivo experimental study on osteopenia in diabetic rats. Acta Histochem. 2011;113:619–25. doi: 10.1016/j.acthis.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 48.Lozano D, Fernández-de-Castro L, Portal-Núñez S, López-Herradón A, Dapía S, Gómez-Barrena E, Esbrit P. The C-terminal fragment of parathyroid hormone-related peptide promotes bone formation in diabetic mice with low-turnover osteopaenia. Brit J Pharmacol. 2011;162:1424–1438. doi: 10.1111/j.1476-5381.2010.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross RD, Edwards LH, Acerbo AS, Ominsky MS, Virdi AS, Sena K, Miller LM, Sumner DR. Bone matrix quality after sclerostin antibody treatment. J Bone Miner Res. 2014;29:1597–607. doi: 10.1002/jbmr.2188. [DOI] [PubMed] [Google Scholar]
- 50.Hassler N1, Roschger A, Gamsjaeger S, Kramer I, Lueger S, van Lierop A, Roschger P, Klaushofer K, Paschalis EP, Kneissel M, Papapoulos S. Sclerostin deficiency is linked to altered bone composition. J Bone Miner Res. 2014;29:2144–51. doi: 10.1002/jbmr.2259. [DOI] [PubMed] [Google Scholar]
- 51.Ominsky MS, Li C, Li X, Tan HL, Lee E, Barrero M, Asuncion FJ, Dwyer D, Han CY, Vlasseros F, Samadfam R, Jolette J, Smith SY, Stolina M, Lacey DL, Simonet WS, Paszty C, Li G, Ke HZ. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res. 2011;26:1012–21. doi: 10.1002/jbmr.307. [DOI] [PubMed] [Google Scholar]
- 52.Jawad MU, Fritton KE, Ma T, Ren PG, Goodman SB, Ke HZ, Babij P, Genovese MC. Effects of sclerostin antibody on healing of a non-critical size femoral bone defect. J Orthop Res. 2013;31:155–63. doi: 10.1002/jor.22186. [DOI] [PubMed] [Google Scholar]
- 53.Li C, Ominsky MS, Tan HL, Barrero M, Niu QT, Asuncion FJ, Lee E, Liu M, Simonet WS, Paszty C, Ke HZ. Increased callus mass and enhanced strength during fracture healing in mice lacking the sclerostin gene. Bone. 2011;49:1178–85. doi: 10.1016/j.bone.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 54.Feng G, Chang-Qing Z, Yi-Min C, Xiao-Lin L. Systemic administration of sclerostin monoclonal antibody accelerates fracture healing in the femoral osteotomy model of young rats. Int Immunopharmacol. 2015;24:7–13. doi: 10.1016/j.intimp.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Lu H, Kraut D, Gerstenfeld LC, Graves DT. Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology. 2003;144:346–52. doi: 10.1210/en.2002-220072. [DOI] [PubMed] [Google Scholar]
- 56.Song L, Liu M, Ono N, Bringhurst FR, Kronenberg HM, Guo J. Loss of wnt/β-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J Bone Miner Res. 2012;27:2344–58. doi: 10.1002/jbmr.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q, Riddle RC, Clemens TL. Bone and the regulation of global energy balance. J Intern Med. 2015 Jan 16; doi: 10.1111/joim.12348. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang J, Slingerland JM. Multiple Roles of the PI3K/PKB(Akt) Pathyway in Cell Cycle Progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 59.Kim JY, Lee SK, Jo KJ, Song DY, Lim DM, Park KY, Bonewald LF, Kim BJ. Exendin-4 increases bone mineral density in type 2 diabetic OLETF rats potentially through the down-regulation of SOST/sclerostin in osteocytes. Life Sci. 2013;92:533–40. doi: 10.1016/j.lfs.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 60.Ma P, Gu B, Xiong W, Tan B, Geng W, Li J, Liu H. Glimepiride Promotes Osteogenic Differentiation in Rat Osteoblasts via the PI3K/Akt/eNOS Pathway in a High Glucose Microenvironment. PLoS ONE. 2014;9:e112243. doi: 10.1371/journal.pone.0112243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erejuwa OO. Management of diabetes mellitus: could simultaneous targeting of hyperglycemia and oxidative stress be a better panacea? Int J Mol Sci. 2012;13:2965–72. doi: 10.3390/ijms13032965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manolagas SC, Almeida M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol. 2007;21:2605–14. doi: 10.1210/me.2007-0259. [DOI] [PubMed] [Google Scholar]
- 63.Gaudio A, Privitera F, Battaglia K, Torrisi V, Sidoti MH, Pulvirenti I, Canzonieri E, Tringali G, Fiore CE. Sclerostin Levels Associated with Inhibition of the Wnt/β-Catenin Signaling and Reduced Bone Turnover in Type 2 Diabetes Mellitus. J Clin Endocrinol Metab. 2012;97:3744–50. doi: 10.1210/jc.2012-1901. [DOI] [PubMed] [Google Scholar]
- 64.Abiola M, Favier M, Christodoulou-Vafeiadou E, Pichard AL, Martellv I, Guillet-Deniau I. Activation of Wnt/beta-catenin signaling increases insulin sensitivity through a reciprocal regulation of Wnt10b and SREBP-1c in skeletal muscle cells. PLoS One. 2009;4:e8509. doi: 10.1371/journal.pone.0008509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stolina M, Dwyer D, Niu QT, Villasenor KS, Kurimoto P, Grisanti M, Han CY, Liu M, Li X, Ominsky MS, Ke HZ, Kostenuik PJ. Temporal changes in systemic and local expression of bone turnover markers during six months of sclerostin antibody administration to ovariectomized rats. Bone. 2014;67:305–13. doi: 10.1016/j.bone.2014.07.031. [DOI] [PubMed] [Google Scholar]
- 66.Ominsky MS1, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, Tipton B, Cai J, Deshpande R, Zhou L, Hale MD, Lightwood DJ, Henry AJ, Popplewell AG, Moore AR, Robinson MK, Lacey DL, Simonet WS, Paszty C. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25:948–59. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 67.Padhi D1, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 68.van Dinther M1, Zhang J, Weidauer SE, Boschert V, Muth EM, Knappik A, deGorter DJ, van Kasteren PB, Frisch C, Mueller TD, ten Dijke P. Anti-Sclerostin antibody inhibits internalization of Sclerostin and Sclerostin-mediated antagonism of Wnt/LRP6 signaling. PLoS One. 2013;8:e62295. doi: 10.1371/journal.pone.0062295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Osteoclast Activity was Unchanged in Treated Groups. Histological sections on uninjured femora of 11 week old mice were stained with TRAP, staining (osteoclasts, red) and counterstained with Fast Green,(bone, blue-green); calibration scale: bar = 500 μm. (A-H). Analysis on osteoclast activity was analyzed at the growth plate (A-D) and epiphysis (E-H), representing the regions of natural osteogenesis and trabecular bone, respectively. No alterations in osteoclast activity was observed, which was confirmed from fluorescence immunohistochemistry on cathepsin K; calibration scale: bar=40 μm (I-L). Trabecular regions just below the growth plate were analyzed. Expression was shown by secondary antibody Alexa 594 (red) along the surface of trabecular bone. Two-way ANOVA was used to quantify Cathepsin K-positive area of growth plate and trabecular bone. Statistical analysis revealed no differences between groups (p>0.05).
Supplementary Figure 2. Negative antibody control stains show no evidence of cross-reactivity or non-specific signal. Histological sections on fracture calluses of 11 week old animals were stained with secondary antibodies corresponding to the primary antibodies used in this study. These stains utilized the antigen retrieval methods used for individual antibodies and are as follows: Donkey anti-goat 594 was used with trypsin/EDTA as a negative control for –Sost protein detection (A-D). Goat anti-mouse 594 was used with trypsin/EDTA as a negative control to detect administered mouse monoclonal SOST-neutralizing antibody (SostAb) (E-H). Goat anti-rabbit 594 was used with trypsin/EDTA as a negative control for active caspase 3 and osteocalcin (I-L). Goat anti-rabbit 488 was used with Unitrieve as a negative control for Collagen type I (M-P). Goat anti-rabbit 594 was used with Unitrieve as a negative control for SP7/Osterix, DMP1, and Cathepsin K (Q-T). Goat anti-mouse 594 was used with Unitrieve and Proteinase K as a negative control for activated b-catenin and Runx2. Calibration scale: bar = 100 μm.
Supplementary Methods: Histological sections of injured (21 days) and uninjured controls (11 weeks of age) were de-waxed and stained with TRAP working solution for 10 minutes at 37°C in the dark and counterstained with 0.02% Fast Green and mounted with 60% glycerol. Fluorescent immunostains using primary antibody against cathepsin K (abcam, ab19027) required Unitrieve (Innovex) for the antigen retrieval, for 30 minutes at 65°C. Secondary antibodies (Alexa Fluor 488 (green) or 594 (red), Molecular Probes) against mouse, rabbit, or goat antigens/primary antibodies were used for detection and negative control slides. Stained slides were mounted with Prolong Gold with DAPI (Molecular Probes). ImagePro Plus V7.0 software and a QIClick CCD camera were used for imaging. Cathepsin K quantification was performed using the Analyze Particles tool in Image J, where percent area of stain was quantified.