

Abstract
There is no FDA approved treatment for acute respiratory distress syndrome (ARDS), in spite of the relatively large number of patients with the diagnosis. In this report, we provide an overview of preclinical studies as well as a description of completed and future clinical trials in humans with ARDS. Preclinical studies dealing with acute lung injury (ALI) have suggested roles for complement and complement receptors, as well as the evolving role of histones, but details of these pathways are inadequately understood. Anti-inflammatory interventions have not been convincingly effective. Various cell growth factors are being considered for clinical study. Interventions to block complement activation or its products are under consideration. Stem cell therapies have shown efficacy in preclinical studies, which have motivated phase I/II trials in humans with ARDS.
Epidemiology of ARDS
The Acute Respiratory Distress Syndrome (ARDS) is characterized by a severe, acute inflammatory responses within the lung, resulting in diffuse damage to the alveolar-capillary barrier, flooding the airspaces with protein-rich edema fluid, with resulting severe gas-exchange abnormalities (1, 2). This is a common syndrome, with an annual US incidence of greater 80 per 100,000 population, and especially common in the elderly (3). ARDS can be precipitated by either direct or indirect insult to the lung. Direct insults include pneumonia, aspiration of gastric contents, pulmonary contusion, or inhalation of injurious gases. Indirect injury can occur as a result of systemic processes such as sepsis, pancreatitis, multiple trauma, or massive transfusion of blood products. Sepsis is the most common cause of ARDS in humans. Sepsis due to a pulmonary cause carries with it the highest mortality as compared to other etiologies of ARDS (3). Early deaths in ARDS are due to hypoxic respiratory failure and development of multiple organ failure, whereas deaths after 2 weeks are usually attributable to progressive pulmonary dysfunction as a result of exuberant fibroproliferation and/or the development of nosocomial infection, most notably pneumonia (4, 5).
Pathogenesis of ARDS
In the initial phase of ARDS (referred to as the exudative phase), direct or indirect insults generally result in injury to both the capillary endothelium and the alveolar epithelium (6, 7). Type I alveolar epithelial cells (AEC) comprise >95% of the alveolar surface, and are particularly susceptible to injury. As a consequence of capillary endothelial and AEC injury, there is loss of alveolar-capillary barrier function, and accumulation of protein rich edema fluid within the pulmonary interstitium and alveolus (1). Denuded epithelium is replaced by the formation of proteinaceous hyaline membranes (Figure 1A). The exudative phase of ARDS is temporally associated with influx of neutrophils within pulmonary capillaries, margination and adherence to the activated endothelium, followed by exuberant accumulation of PMNs in both interstitial and alveolar spaces (8). Activated PMNs contribute to lung injury by releasing a variety of injurious molecules, including neutrophil elastase, metalloproteases, and other proteolytic enzymes, oxidants, and reactive nitrogen species (9, 10). In addition to PMNs, there is chemokine-dependent emigration of macrophages, which can amplify pulmonary injury by releasing inflammatory cytokines and apoptosis-inducing molecules (11).
Figure 1.
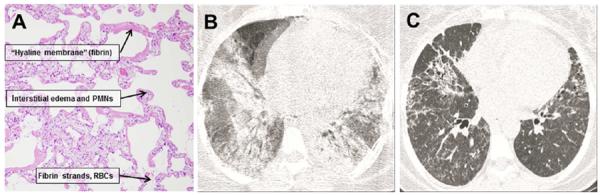
Histological and radiographic features of ARDS. A. Lung from a patient with ARDS, stained with hematoxylin and eosin. There are prominent “hyaline membranes” consisting of fibrin deposits along alveolar walls, widespread interstitial edema accompanied by neutrophils. Alveolar spaces also contain RBCs and fibrin strands. Panels B and C are radiographic appearance of exudative and fibroproliferative phases of ARDS. The exudative phase is characterized by diffuse ground glass and alveolar opacities, whereas the fibroproliferative phase is characterized by residual linear opacities, traction bronchiectasis and honeycombing.
The fibroproliferative phase of ARDS occurs early after injury (within initial 3 days) and temporally overlaps with inflammatory events that characterize the exudative phase. The alveolar space becomes engorged with proliferating mesenchymal cells, including fibroblasts, myofibroblasts, and locally generated pluripotential mesenchymal progenitor cells (12, 13). Type II AEC proliferate to replace necrotic and apoptotic type I cells, and new blood vessels form within the provisional matrix. There is also evidence of thrombogenesis and impaired fibrinolytic activity, as indicated by the accumulation of fibrin in the distal airspaces, together with microthrombi in small pulmonary vessels (14–16). In some patients, there is pronounced deposition of matrix components, including fibronectin and replacement of type III collagen by type 1 collagen (12). An exuberant fibroproliferative response in patients with ARDS is associated with a requirement for prolonged mechanical ventilation and increased mortality (5, 17). The CT appearance of the exudative and fibroproliferative phases of ARDS is shown in Figure 1B and C, respectively.
Animal Models of Acute Lung Injury (ALI)
No animal model recapitulates all of the key histopathological features of human ARDS. However, a number of models of ALI, mostly in rodents, have been described and are outlined in Table 1, along with the routes by which the various ALI-inducing agents are given. It has been suggested that agents in items 1a-d tend to be associated, at least early on, with damage of capillary endothelial cells in lungs (18–20). These agents include oleic acid which is known to be toxic to vascular endothelial cells (21). LPS, which is not directly toxic to cells, produces a sequence of events that can lead to both endothelial cell and alveolar epithelial damage. Live E. coli given via the airways produces a series of events in lung similar to the effects of LPS. Bleomycin causes injury to endothelial cells, usually with resultant fibrosis through a series of signaling pathways involving TGFβ and other signaling molecules. Agents cited in 2e-k (Table 1) are thought to chiefly target alveolar epithelial cells, at least in the early phases of lung injury. HCl given i.t. will cause reversible damage to the alveolar epithelium, although, in the case of aspiration pneumonia in humans, there is an abundance of particulates (from ingested food) that produces a much more pleomorphic and intense inflammatory response than that caused by HCl alone. A more recently described transgenic model involves selective diphtheria toxin-induced depletion of type II AEC, resulting in progressive fibroproliferation (22). This response is mediated in part by recruited macrophages, which elaborate both inflammatory and fibrogenic cytokines and produce collagen (23). The combination of damage to capillary endothelial cells and alveolar epithelial cells appears to be prominent in models indicated in Table 1 which includes ischemia-reperfusion (IR) injury directly involving the lung arterial system. It has been also established that IR injury involving distant sites, such as outflow tracts of the abdominal aorta (as in the case of a dissecting aneurysm or thrombotic IR of the femoral arterial system) can also lead to lung damage via systemic activation of the complement system and related events (24–26). It is clear that the designations in Table 1 are arbitrary and probably not completely accurate. For instance, airway delivery of LPS results in alveolar edema, PMN buildup, and fibrin deposits along with RBC presence in alveolar spaces (27). Based on the morphology, it is clear that both vascular endothelial cell and alveolar epithelial cell barriers have been damaged, leading to these morphologic changes. In the case of sepsis, whether due to infectious agents or related to damage or danger associated molecular patterns (DAMPs) as in “sterile sepsis” that occurs in hemorrhagic shock, the outcomes are often similar, resulting in severe sepsis or septic shock. Remarkably, most examples of ALI usually end up with “resolution” of inflammation (return to preinjury state) as well as repair or replacement of damaged cells. Usually, days or weeks later after onset of ALI, the morphology of the lung in rodents has returned to preinjury state, suggesting that the lung has remarkable regeneration and healing abilities, also involving regeneration of damaged or apoptotic or necrotic epithelial and endothelial cells. To what extent the “regenerative” outcome in ALI-damaged lung is linked to locally present “stem cells” or is due to bone marrow-derived stem cells is a matter of current dispute and is discussed below.
TABLE 1.
Models of Acute Lung Injury in Rodents*
| Chief Target | Injury-causing Agent | Usual Route of Delivery |
|---|---|---|
| 1. Capillary endothelium | a. Oleicacid | i.v. |
| b. LPS | i.t. | |
| c. Bleomycin | i.t. | |
| d. E. coli (live) | i.t. or aerosol | |
| 2. Alveolar epithelium | e. HCl | i.t. |
| f. Hyperoxia (100% O2) | airway | |
| g. Surfactant depletion | repetitive BAL | |
| h. Mechanical ventilation | airway intubation | |
| i. Bleomycin | i.t. | |
| j. FITC | i.t. | |
| k. Diphtheria toxin | i.p. | |
| 3. Both endothelial and alveolar epithelial cells | l. Ischemia-reperfusion (IR) injury | local (lung) pulmonary artery occlusion or distal IR injury |
| m. Sepsis (LPS, CLP, live bacteria) | i.t., i.v., i.p. | |
| n. Other conditions (hemorrhagic shock or “double-hit sepsis”, hemorrhagic shock followed by CLP) | variable |
Based in part on published reviews, including Bernard GR, et al. The American-European Consensus Conference on ARDS. Am J Respir Crit Care Med. 1994 Mar;149(3 Pt 1):818–24.
The propensity for development of fibroproliferation in ALI appears to be a function of the experimental agent employed (bleomycin), but at least experimentally it can also be linked to the extent to which there has already been extensive denuding of the alveolar epithelium and/or vascular endothelium, resulting in a fibrotic outcome before alveolar epithelial or endothelial cell regeneration can be completed. Earlier studies have also suggested that reactive oxygen species produced by both PMNs and macrophages, especially the hydroxyl radical and myeloperoxidase products (HOCl) from H2O2 also play important roles in the extent of alveolar and epithelial cell damage or destruction and a subsequent fibrotic response (28). Such events may result in strong activation of fibroblasts and rapid (within days) development of pulmonary fibrosis in rodents. Proteases (including matrix metalloproteases) and other inflammatory products also appear to contribute to such outcomes.
Molecular Events in Experimental ALI
Figure 2 summarizes experimental and clinical evidence related to molecular events leading to ALI and, often, to multiorgan failure. The latter occurs when the products generated in lung (complement activation products, chemokines and cytokines, proteases, etc.) become disseminated. Whether ALI is triggered by DAMPs (histones, HMGB1, etc.) or pathogen associated molecular patterns (PAMPs) such as LPS, lipoteichoic acid, etc., complement activation ensues as well as engagement of extracellular toll like receptors (TLRs) and perhaps intracellular NOD-like receptors (NLRs) (29, 30). The role of complement activation products and receptors are key in events leading to ALI (31). At the onset of ALI, generation of C5a ensues, followed by engagement of C5a with its two receptors (C5aR1, C5aR2) which are heavily expressed on PMNs but also at much lower levels on macrophages, endothelial cells and a variety of other cell types (31). C5a induces neutrophil extracellular traps (NET) formation in PMNs, resulting in local containment of bacteria and release of histones, myeloperoxidase, and other products of PMNs (32–34). Extracellular histones activate the NLRP3 inflammasome (35, 36) with release of IL-1β and IL-18, both of which are proinflammatory. Numerous other cytokines and chemokines are released, the bulk of which appear to be deriving from lung macrophages (32, 36), but also by structural cells that comprise the alveolus. These mediators have broad proinflammatory effects. Histones are also highly lung-damaging, resulting in severe disturbances in gas exchange between the capillary compartment and alveolar spaces. In addition, histones are highly prothrombotic and trigger venous thrombosis in lung (32). Use of a histone neutralizing mAb has been highly protective in the setting of ALI as well as in the setting of sepsis (35, 37). On the basis of analysis of BALF from humans and mice with ALI, the presence of histones has been verified by use of ELISA and Western blots. It appears that histones (H2A, H4) are present in BALF obtained from mice and humans with ALI (32). However, conclusions are incomplete, since quantitative detection of individual histones by ELISA has been difficult due to lack of suitable reagents.
Figure 2.
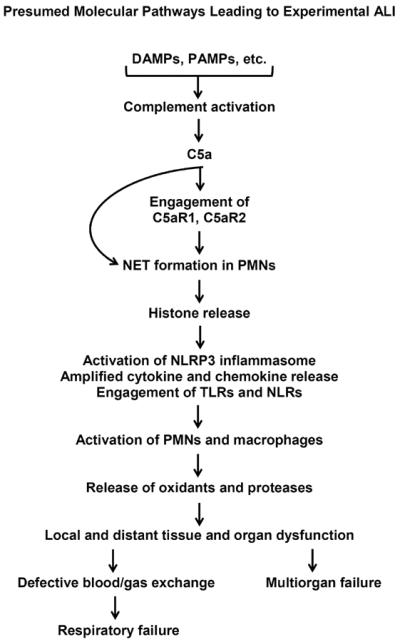
Pathophysiological pathways in experimental ALI. These involve component activation, engagement of the two C5a receptors, and engagement of TLRs and NLRs. PAMPs are pathogen-associated molecular patterns, such as LPS released from gram negative bacteria and LTA released from gram positive bacteria. DAMPs are danger-associated molecular patterns released from damaged tissues (HMGB1, heat shock proteins, DNA, RNA, etc.). Downstream events include release of cytokines and chemokines, formation of neutrophil extracellular traps (NETs), and release of histones which are highly cell and tissue damaging.
There are limited data suggesting that circulating stem cells as well as stem cells resident in the lung may attenuate ALI in mice induced by airway instillation of LPS (38). It may be that such outcomes are linked to paracrine release of protective products from marrow-derived stem cells resulting in enhanced function of residual type II epithelial cells or accelerate cell regeneration. In general, past studies have not convincingly established whether circulating or in situ stem cells in lung play an important role in recovery from defective lung function following onset of ALI. However, Vaughan and colleagues have recently shown for the first time that a lung-derived population of lineage-negative epithelial stem/progenitor cells proliferate and migrate to sites of alveolar epithelial injury to repopulated denuded epithelium in response to either influenza- or bleomycin-induced lung injury (39).
Protective Interventions in pre-clinical models of ALI
A large number of interventions has been described in the literature in order to block ALI. These include antioxidants, antiproteases, and signal transduction inhibitors. Most studies have been employed in rodents. Obviously, our discussion of protective interventions is very limited. Our knowledge regarding interventions that are protective in the setting of ALI has come from the use of experimental models of ALI. As indicated in Figure 2, ALI can be induced by airway instillation of C5a, IgG immune complexes or LPS, the latter two resulting in early complement activation and appearance of C5a, which is reactive with C5aR1 and C5aR2. These early events are keystones for events that will evolve into ALI. As shown in Table 2, antibody-induced neutralization of C5a or other antibodies that block C5aRs are highly protective in the setting of ALI and sepsis (31, 40–42). The appearance of histones soon after complement activation is PMN-dependent and requires availability of C5aRs (32), suggesting that the bulk of extracellular histones appearing in BALF are derived from NETs formed by PMNs. As indicated above, histones have strong proinflammatory (32), cell damaging and procoagulant activities for lungs. Targeting histones in lung with a neutralizing mAb provides high levels of protection against development of ALI (32). Another strategy to protect from ALI is to employ a heparin derivative that lacks anti-coagulant activity but has an acidic charge and binds to histones, resulting in inactivation of the biological activities. This strategy has been shown to be protective in the setting of sepsis (43). The heparin derivative is intriguing since it has already received FDA approval in humans (44). Because of the diversity of chemokines and cytokines and overlapping interactions with their receptors, it seems unlikely that in vivo neutralization of these mediators with mAbs would be effective in humans with ALI. The exception to this may be inflammasome-derived IL-1β and IL-18, using the IL-1β receptor antagonist (anakinra). This antagonist is clinically effective in rheumatoid arthritis (45) and also in mice reduces the intensity of collagen-induced arthritis, which is complement-dependent (46). Clearly, a more complete understanding of events coming into play in ALI is needed in order to identify the most effective candidates that may be considered for use to treat humans developing ALI.
TABLE 2.
Therapeutic Strategies that Ameliorate ALI in Mice
| 1. C5a blockade |
| 2. Inhibition of C5aR1, C5aR2 |
| 3. PMN or macrophage depletion |
| 4. Neutralization of extracellular histones |
| 5. Blockade of inflammasome [anakinra (IL-1βRa)] |
Human Clinical Trials Targeting Dysregulated Cellular and Molecular Pathways in ARDS
Translating discoveries made in pre-clinical animal models of ALI into effective therapies in patients with ARDS has been disappointing. These animal models have been informative in identifying cellular and molecular pathways involved but have been much less useful in predicting response to therapy. Temporal changes in pathophysiological events, the functional redundancy and pleotropic effects of cytokine and chemokine mediators and transcription factors, and the heterogeneity in host responses of syndromic diseases such as ARDS have made treatment especially challenging. To date, the only intervention proven effective in multi-center Phase III trials in ARDS is the use of lung-protective ventilation, which reduces alveolar stretch and systemic release of inflammatory mediators (e.g. IL-6) (47). Various cellular/molecular pathological pathways have been or are currently being targeted (Table 3) in ARDS patients or at-risk patients.
Table 3.
Ongoing Immunomodulatory Clinical Trials in ARDS
| Molecular Target | Therapeutic | Trial Design | Outcome Measures |
|---|---|---|---|
| 1. Anti-inflammatory | a. Low dose hydrocortisone | Multi-center Phase II/III RCT | Mortality, ARDS severity |
| b. Vitamin C | Multi-center Phase II RCT | ARDS severity | |
| c. Vitamin D | Phase I/II RCT | Prevention of ARDS | |
| 2. Growth factors | d. GM-CSF | Multi-center Phase III RCT (planned) | Mortality, prevention and severity of ARDS |
| 3. Coagulation/complement cascade | e. Nebulized heparin | Multi-center Phase II RCT | Prevention and severity of ARDS |
| f. Aspirin (LIPS-A) | Multi-center Phase II RCT | ARDS prevention, mortality | |
| 4. Stem cell therapy | g. Allogeneic MSC infusion | Multi-center Phase I/II open label, dose escalation | Safety, ARDS severity |
Anti-inflammatory Strategies
Immune activation occurs early in ARDS in response to local or systemic insults, resulting in the intrapulmonary and in some instances systemic release of pro-inflammatory mediators. However, approaches to suppress deleterious inflammation have not consistently resulted in improvement in patient-oriented outcomes. Targeting of specific pro-inflammatory cytokines, including TNF-α and IL-1β, has not improved the outcome of patients with ARDS or altered susceptibility to ARDS development in at-risk patients. The early (first 48 hrs) or late (after 14 days) administration of high-dose corticosteroids has been ineffective in reducing the mortality or complications of ARDS (48, 49). However, there is some suggestion that corticosteroids given at more moderate doses administered during the earlier stages of fibroproliferative ARDS may be of benefit (50), and is currently being investigated in a multi-center phase II/III trial. Other failed strategies to broadly suppress inflammatory processes include the administration of prostaglandin E1, lysophylline, ketoconazole, statins, dietary oils and other anti-oxidants.
Neutrophils and/or neutrophil derived products are believed to play a major pathogenic role in ALI/ARDS. Despite promising pre-clinical and phase II results in patients with ARDS, administration of neutrophil elastase inhibitors (sivelestat) did not improve and in fact caused worsening outcomes in these patients (51). Attempts to inhibit CXC chemokine-dependent PMN recruitment and/or activation using CXCR1/2 blockade in humans have been contemplated but as yet not performed.
Recent data suggests that both vitamin C and vitamin D3 can exert anti-inflammatory properties, although the molecular mechanisms accounting for these effects are uncertain (52–54). There are ongoing phase II trials to delineate the effect of vitamin D supplementation on ARDS development in high risk patients and high dose vitamin C administration in patients with established ARDS.
Growth Factors
A recent paradigm in therapeutic strategies is to target factors that promote cytoprotective and mitogenic effects on lung epithelium. Examples include growth factors such as keratinocyte growth factor (KGF), granulocyte-macrophage colony stimulating factor (GM-CSF) and hepatocyte growth factor (HGF). Keratinocyte growth factor is a member of the fibroblast growth factor family (also known as FGF-7) which exerts both cytoprotective and mitogenic effects on lung epithelium (55). Importantly, KGF is an inducer of GM-CSF from alveolar epithelium, leading to enhanced GM-CSF-mediated alveolar macrophage antimicrobial function (56). Despite promising pre-clinical studies, a multicenter phase II trial of KGF administration in ARDS patients failed to demonstrated improvement in clinically relevant outcomes. GM-CSF is a pleotropic cytokine with potent activating and differentiation effects on myeloid cells (macrophages, dendritic cells and PMN) and promotes cytoprotection and mitogenic effects on epithelial surfaces, including alveolar and intestinal epithelium. A multicenter phase II trial of GM-CSF administration in patients with established ARDS did not change duration of mechanical ventilation, but did resultant in trends suggestive of reduced non-pulmonary organ failure and decreased mortality (6% absolute reduction, 26% relative reduction) as compared to placebo patients (57). These finding are consistent with improved clinical outcomes in other phase II trials involving non-neutropenic patients with sepsis (58–61), and raise the possibility that the salutary effects of GM-CSF observed may be attributable to reversal of leukocyte reprogramming of sepsis or mobilization of marrow derived progenitor cells, such as endothelial progenitor cells. A European multicenter trial of GM-CSF administration in patients with sepsis-induced immune reprogramming (as indicated by reduced monocyte HLA-DR expression) will begin this year. Moreover, a trial of early GM-CSF administration to prevent ARDS development and improve patient-oriented clinical outcomes in patients with septic shock or severe sepsis due to pneumonia is one of several trials being considered by the newly formed Prevention and Early Therapy in Acute Lung Injury (PETAL) Network. A final potential target is hepatocyte growth factor (HGF). HGF is produced primarily by fibroblasts which induces proliferation of alveolar type II epithelial cells (62–64) and human bronchial epithelial cells (65), raising the possibility that this molecule might play a role in lung repair post injury. Human trials evaluating effects of HGF administration are contemplated but not yet organized.
Coagulation/Complement Cascade
Sepsis and ARDS have in common robust systemic activation of both the coagulation and complement cascade, resulting in vascular injury, microthrombi formation, and complement-mediated activation of leukocytes and platelets (66). The hypercoagulable state in sepsis/ARDS is mediated, in part, by endothelial damage and the systemic release of tissue factor, which has been shown to be secreted in membrane-bound vesicles (microparticles). Attempts to block factors that promote coagulation or administration of anticoagulants in sepsis and ARDS have been largely unsuccessful. However, a small single center phase II study suggested that nebulized heparin may reduce duration of mechanical ventilation in patients at risk for ARDS (67). This study has motivated a larger phase II trial to assess the impact of nebulized heparin on ARDS development in at-risk patients. Finally, observational data suggests that pre-admission cyclooxygenase inhibitor use was associated with reduced ARDS development (68). A multicenter phase II trial assessing the impact of aspirin administration on ARDS development in at risk patients (LIPS-A) has been completed and results of this trial have not yet been reported.
Immune activation triggers the rapid release of the anaphylatoxins C3a and C5a, dysregulated coagulation, and kallikrein/kinin system activation. Targeting of C3/C3a and/or C5/C5a has been limited by the inherent redundancy in anaphylatoxin biological effects and lack of available therapeutics. However, pre-clinical models indicate that these pathways can be effectively limited by the protein C1-esterase inhibitor (C1INH), a constitutively expressed and acute phase reactant plasma serine protease inhibitor (serpin). A recent multicenter phase II trial involving 61 patients with severe sepsis found that administration of purified human C1INH resulted in a substantial reduction in mortality (33% absolute reduction), with an even greater improvement in survival among patients with sepsis-induced ARDS (69). These intriguing findings have not yet motivated larger phase II/III trials.
Cell-based Therapies
Bone marrow-derived mesenchymal stem cells (MSCs) can modulate both local and systemic inflammatory and reparative responses by facilitating protective antimicrobial innate responses, elaborating anti-inflammatory molecules and growth factors, and differentiation into cells that can reconstitute epithelial and vascular surfaces (70, 71). Pre-clinical animal models suggest that exogenously administered MSCs can attenuate lung injury and reparative responses. In an ex-vivo perfused human lung model, infusion of clinical grade, cryopreserved human MSCs reduced alveolar permeability, suppressed inflammation and exerted antimicrobial effects after the i.t. administration of live E. coli (72). These preclinical observations have motivated phase I trials that indicate infusion of allogeneic adipose or bone marrow-derived MSCs is safe and may reduce circulating markers of alveolar epithelial injury in patients with moderate to severe ARDS (73–75). A larger multi-center phase II trial of bone marrow-derived human MSC administration involving up to 60 subjects with moderate-severe ARDS [Stem cells for ARDS Treatment (START)] is ongoing (76).
Future Directions
The results of recent and past clinical trials to disrupt dysregulated molecular pathways clearly indicate that dramatically new approaches to therapy are required. Candidate pathways for modulation include epigenetic machinery regulating gene expression, transcription factors (e.g. NF-kB, PPARs), pathways that regulate necrosis/apoptosis (e.g. FAS/FASL, TRAIL), angiogenic factors (e.g. VGEF, angiopoietin) and inhibitors of the renin-angiotensin system. Cell-based therapy is in its infancy but offers the advantage of modulating multiple disrupted cellular and molecular events in ARDS. Given the syndromic nature of ARDS, including the considerable heterogeneity in causes of ARDS, timing of presentation, and variability in host response, it is highly unlikely that a specific therapy will be identified that proves to be equally efficacious in all patients (i.e. the “magic bullet”). In addition, more broad-based therapies targeting fundamentally essential biological processes (e.g. transcription factors, epigenetic machinery, etc.) are not feasible due to far-reaching effects on complex compensatory and homeostatic pathways. For similar reasons, attempts to alter pathophysiological events in diseases that cause ARDS, most notably sepsis, have proven to be quite challenging. With the exception of low tidal volume ventilation, all approaches to modulate the local or systemic host response in sepsis have failed (77). Accordingly, a more personalized approach to therapy may ultimately be required, necessitating more comprehensive molecular phenotyping of ARDS patients or at-risk patients. Precision medicine in acute critical illness will be dependent upon a more coherent understanding of dysregulated molecular pathways involved and better biomarkers identifying anomalies in these pathways.
Acknowledgements
This work was supported by National Institutes of Health grants HL123515 and U01 HL123031 (TJS) and GM29507 and GM61656 (PAW). The authors declare no commercial, financial or personal relationship with organizations that could potentially be perceived as influencing the described research. The authors have read the journal's policy on disclosure of potential conflicts of interest. Both authors have read and participated in the writing of the manuscript. The authors thank Sue Scott and Melissa Rennells for their assistance in preparing the manuscript.
Abbreviations
- AEC
alveolar epithelial cells
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- BALF
bronchoalveolar lavage fluid
- DAMPs
danger-associated molecular patterns
- GM-CSF
granulocyte-macrophage colony stimulating factor
- IR
ischemia-reperfusion
- LPS
lipopolysaccharide
- NETs
neutrophils extracellular traps
- PAMPs
pathogen-associated molecular patterns
- PMNs
polymorphonuclear leukocytes (neutrophils)
- TLRs
toll-like receptors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. The New England journal of medicine. 2000 May 4;342(18):1334–49. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. The New England journal of medicine. 2005 Oct 20;353(16):1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 3.Hudson LD, Steinberg KP. Epidemiology of acute lung injury and ARDS. Chest. 1999 Jul;116(1 Suppl):74S–82S. doi: 10.1378/chest.116.suppl_1.74s-a. [DOI] [PubMed] [Google Scholar]
- 4.Dever LL, Johanson WG., Jr Pneumonia complicating adult respiratory distress syndrome. Clin Chest Med. 1995 Mar;16(1):147–53. [PubMed] [Google Scholar]
- 5.Marshall RP, Bellingan G, Webb S, et al. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med. 2000 Nov;162(5):1783–8. doi: 10.1164/ajrccm.162.5.2001061. [DOI] [PubMed] [Google Scholar]
- 6.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982 Jan;3(1):35–56. [PubMed] [Google Scholar]
- 7.Anderson WR, Thielen K. Correlative study of adult respiratory distress syndrome by light, scanning, and transmission electron microscopy. Ultrastruct Pathol. 1992 Nov-Dec;16(6):615–28. doi: 10.3109/01913129209023751. [DOI] [PubMed] [Google Scholar]
- 8.Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994 Jul;150(1):113–22. doi: 10.1164/ajrccm.150.1.8025736. [DOI] [PubMed] [Google Scholar]
- 9.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003 Apr;31(4 Suppl):S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 10.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001 Feb;7(1):1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Herold S, Steinmueller M, von Wulffen W, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. 2008 Dec 22;205(13):3065–77. doi: 10.1084/jem.20080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chesnutt AN, Matthay MA, Tibayan FA, Clark JG. Early detection of type III procollagen peptide in acute lung injury. Pathogenetic and prognostic significance. Am J Respir Crit Care Med. 1997 Sep;156(3 Pt 1):840–5. doi: 10.1164/ajrccm.156.3.9701124. [DOI] [PubMed] [Google Scholar]
- 13.Horowitz JC, Cui Z, Moore TA, et al. Constitutive activation of prosurvival signaling in alveolar mesenchymal cells isolated from patients with nonresolving acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2006 Mar;290(3):L415–25. doi: 10.1152/ajplung.00276.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abraham E. Coagulation abnormalities in acute lung injury and sepsis. Am J Respir Cell Mol Biol. 2000 Apr;22(4):401–4. doi: 10.1165/ajrcmb.22.4.f184. [DOI] [PubMed] [Google Scholar]
- 15.Idell S, James KK, Levin EG, et al. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest. 1989 Aug;84(2):695–705. doi: 10.1172/JCI114217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003 Jul;285(1):L20–8. doi: 10.1152/ajplung.00312.2002. [DOI] [PubMed] [Google Scholar]
- 17.Martin C, Papazian L, Payan MJ, Saux P, Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. 1995 Jan;107(1):196–200. doi: 10.1378/chest.107.1.196. [DOI] [PubMed] [Google Scholar]
- 18.Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011 May;44(5):725–38. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008 Sep;295(3):L379–99. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011 Mar-Apr;17(3–4):293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beilman G. Pathogenesis of oleic acid-induced lung injury in the rat: distribution of oleic acid during injury and early endothelial cell changes. Lipids. 1995 Sep;30(9):817–23. doi: 10.1007/BF02533957. [DOI] [PubMed] [Google Scholar]
- 22.Osterholzer JJ, Christensen PJ, Lama V, et al. PAI-1 promotes the accumulation of exudate macrophages and worsens pulmonary fibrosis following type II alveolar epithelial cell injury. J Pathol. 2012 Oct;228(2):170–80. doi: 10.1002/path.3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osterholzer JJ, Olszewski MA, Murdock BJ, et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J Immunol. 2013 Apr 1;190(7):3447–57. doi: 10.4049/jimmunol.1200604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harkin DW, Marron CD, Rother RP, Romaschin A, Rubin BB, Lindsay TF. C5 complement inhibition attenuates shock and acute lung injury in an experimental model of ruptured abdominal aortic aneurysm. Br J Surg. 2005 Oct;92(10):1227–34. doi: 10.1002/bjs.4938. [DOI] [PubMed] [Google Scholar]
- 25.Harkin DW, Romaschin A, Taylor SM, Rubin BB, Lindsay TF. Complement C5a receptor antagonist attenuates multiple organ injury in a model of ruptured abdominal aortic aneurysm. J Vasc Surg. 2004 Jan;39(1):196–206. doi: 10.1016/j.jvs.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Groeneveld AB, Raijmakers PG, Rauwerda JA, Hack CE. The inflammatory response to vascular surgery-associated ischaemia and reperfusion in man: effect on postoperative pulmonary function. Eur J Vasc Endovasc Surg. 1997 Nov;14(5):351–9. doi: 10.1016/s1078-5884(97)80284-5. [DOI] [PubMed] [Google Scholar]
- 27.Bosmann M, Grailer JJ, Zhu K, et al. Anti-inflammatory effects of beta2 adrenergic receptor agonists in experimental acute lung injury. FASEB J. 2012 May;26(5):2137–44. doi: 10.1096/fj.11-201640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal. 2007 Nov;9(11):1991–2002. doi: 10.1089/ars.2007.1785. [DOI] [PubMed] [Google Scholar]
- 29.Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012 Mar;4(3) doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010 Mar 4;464(7285):104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol. 2012;946:147–59. doi: 10.1007/978-1-4614-0106-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bosmann M, Grailer JJ, Ruemmler R, et al. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J. 2013 Dec;27(12):5010–21. doi: 10.1096/fj.13-236380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fattahi F, Grailer JJ, Jajou L, Zetoune FS, Andjelkovic AV, Ward PA. Organ distribution of histones after intravenous infusion of FITC histones or after sepsis. Immunol Res. 2015 Mar;61(3):177–86. doi: 10.1007/s12026-015-8628-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010 Mar;10(3):210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 35.Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol. 2013 Dec;43(12):3336–42. doi: 10.1002/eji.201243224. [DOI] [PubMed] [Google Scholar]
- 36.Grailer JJ, Canning BA, Kalbitz M, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014 Jun 15;192(12):5974–83. doi: 10.4049/jimmunol.1400368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009 Nov;15(11):1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L, He H, Liu A, et al. Therapeutic effects of bone marrow-derived mesenchymal stem cells in models of pulmonary and extrapulmonary acute lung injury. Cell Transplant. 2015 Feb 18; doi: 10.3727/096368915X687499. [DOI] [PubMed] [Google Scholar]
- 39.Vaughan AE, Brumwell AN, Xi Y, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015 Jan 29;517(7536):621–5. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laudes IJ, Chu JC, Sikranth S, et al. Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. Am J Pathol. 2002 May;160(5):1867–75. doi: 10.1016/S0002-9440(10)61133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones HD, Crother TR, Gonzalez-Villalobos RA, et al. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol. 2014 Feb;50(2):270–80. doi: 10.1165/rcmb.2013-0087OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uriarte SM, Rane MJ, Merchant ML, et al. Inhibition of neutrophil exocytosis ameliorates acute lung injury in rats. Shock. 2013 Mar;39(3):286–92. doi: 10.1097/SHK.0b013e318282c9a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wildhagen KC, Garcia de Frutos P, Reutelingsperger CP, et al. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood. 2014 Feb 13;123(7):1098–101. doi: 10.1182/blood-2013-07-514984. [DOI] [PubMed] [Google Scholar]
- 44.The identity problem. Nat Biotechnol. 2010 Sep;28(9):877. doi: 10.1038/nbt0910-877. [DOI] [PubMed] [Google Scholar]
- 45.Kalliolias GD, Liossis SN. The future of the IL-1 receptor antagonist anakinra: from rheumatoid arthritis to adult-onset Still's disease and systemic-onset juvenile idiopathic arthritis. Expert Opin Investig Drugs. 2008 Mar;17(3):349–59. doi: 10.1517/13543784.17.3.349. [DOI] [PubMed] [Google Scholar]
- 46.Liu D, Lon HK, Dubois DC, Almon RR, Jusko WJ. Population pharmacokinetic pharmacodynamic-disease progression model for effects of anakinra in Lewis rats with collagen-induced arthritis. J Pharmacokinet Pharmacodyn. 2011 Dec;38(6):769–86. doi: 10.1007/s10928-011-9219-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. The New England journal of medicine. 2000 May 4;342(18):1301–8. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 48.Luce JM, Montgomery AB, Marks JD, Turner J, Metz CA, Murray JF. Ineffectiveness of High-Dose Methylprednisolone in Preventing Parenchymal Lung Injury and Improving Mortality in Patients with Septic Shock. American Review of Respiratory Disease. 1988 Jul;138(1):62–8. doi: 10.1164/ajrccm/138.1.62. [DOI] [PubMed] [Google Scholar]
- 49.Steinberg KP, Hudson LD, Goodman RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. The New England journal of medicine. 2006 Apr 20;354(16):1671–84. doi: 10.1056/NEJMoa051693. [DOI] [PubMed] [Google Scholar]
- 50.Meduri GU, Golden E, Freire AX, et al. Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest. 2007 Apr;131(4):954–63. doi: 10.1378/chest.06-2100. [DOI] [PubMed] [Google Scholar]
- 51.Iwata K, Doi A, Ohji G, et al. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern Med. 2010;49(22):2423–32. doi: 10.2169/internalmedicine.49.4010. [DOI] [PubMed] [Google Scholar]
- 52.Takano Y, Mitsuhashi H, Ueno K. 1alpha,25-Dihydroxyvitamin D(3) inhibits neutrophil recruitment in hamster model of acute lung injury. Steroids. 2011 Nov;76(12):1305–9. doi: 10.1016/j.steroids.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 53.Parekh D, Dancer RC, Lax S, et al. Vitamin D to prevent acute lung injury following oesophagectomy (VINDALOO): study protocol for a randomised placebo controlled trial. Trials. 2013;14:100. doi: 10.1186/1745-6215-14-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher BJ, Kraskauskas D, Martin EJ, et al. Attenuation of Sepsis-Induced Organ Injury in Mice by Vitamin C. Jpen-Parenter Enter. 2014 Sep;38(7):825–39. doi: 10.1177/0148607113497760. [DOI] [PubMed] [Google Scholar]
- 55.Ware LB, Matthay MA. Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am J Physiol Lung Cell Mol Physiol. 2002 May;282(5):L924–40. doi: 10.1152/ajplung.00439.2001. [DOI] [PubMed] [Google Scholar]
- 56.Wu H, Suzuki T, Carey B, Trapnell BC, McCormack FX. Keratinocyte growth factor augments pulmonary innate immunity through epithelium-driven, GM-CSF-dependent paracrine activation of alveolar macrophages. The Journal of biological chemistry. 2011 Apr 29;286(17):14932–40. doi: 10.1074/jbc.M110.182170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paine R, 3rd, Standiford TJ, Dechert RE, et al. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med. 2012 Jan;40(1):90–7. doi: 10.1097/CCM.0b013e31822d7bf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW. A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction. Am J Respir Crit Care Med. 2002 Jul 15;166(2):138–43. doi: 10.1164/rccm.2009005. [DOI] [PubMed] [Google Scholar]
- 59.Rosenbloom AJ, Linden PK, Dorrance A, Penkosky N, Cohen-Melamed MH, Pinsky MR. Effect of granulocyte-monocyte colony-stimulating factor therapy on leukocyte function and clearance of serious infection in nonneutropenic patients. Chest. 2005 Jun;127(6):2139–50. doi: 10.1378/chest.127.6.2139. [DOI] [PubMed] [Google Scholar]
- 60.Orozco H, Arch J, Medina-Franco H, et al. Molgramostim (GM-CSF) associated with antibiotic treatment in nontraumatic abdominal sepsis: a randomized, double-blind, placebo-controlled clinical trial. Arch Surg. 2006 Feb;141(2):150–3. doi: 10.1001/archsurg.141.2.150. discussion 4. [DOI] [PubMed] [Google Scholar]
- 61.Meisel C, Schefold JC, Pschowski R, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. 2009 Oct 1;180(7):640–8. doi: 10.1164/rccm.200903-0363OC. [DOI] [PubMed] [Google Scholar]
- 62.Mason RJ, Leslie CC, McCormick-Shannon K, et al. Hepatocyte growth factor is a growth factor for rat alveolar type II cells. Am J Respir Cell Mol Biol. 1994 Nov;11(5):561–7. doi: 10.1165/ajrcmb.11.5.7524567. [DOI] [PubMed] [Google Scholar]
- 63.Mason RJ, McCormick-Shannon K, Rubin JS, Nakamura T, Leslie CC. Hepatocyte growth factor is a mitogen for alveolar type II cells in rat lavage fluid. Am J Physiol. 1996 Jul;271(1 Pt 1):L46–53. doi: 10.1152/ajplung.1996.271.1.L46. [DOI] [PubMed] [Google Scholar]
- 64.Panos RJ, Patel R, Bak PM. Intratracheal administration of hepatocyte growth factor/scatter factor stimulates rat alveolar type II cell proliferation in vivo. Am J Respir Cell Mol Biol. 1996 Nov;15(5):574–81. doi: 10.1165/ajrcmb.15.5.8918364. [DOI] [PubMed] [Google Scholar]
- 65.Singh-Kaw P, Zarnegar R, Siegfried JM. Stimulatory effects of hepatocyte growth factor on normal and neoplastic human bronchial epithelial cells. Am J Physiol. 1995 Jun;268(6 Pt 1):L1012–20. doi: 10.1152/ajplung.1995.268.6.L1012. [DOI] [PubMed] [Google Scholar]
- 66.Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med. 2003 Apr;31(4 Suppl):S213–20. doi: 10.1097/01.CCM.0000057846.21303.AB. [DOI] [PubMed] [Google Scholar]
- 67.Dixon B, Schultz MJ, Smith R, Fink JB, Santamaria JD, Campbell DJ. Nebulized heparin is associated with fewer days of mechanical ventilation in critically ill patients: a randomized controlled trial. Crit Care. 2010;14(5):R180. doi: 10.1186/cc9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Erlich JM, Talmor DS, Cartin-Ceba R, Gajic O, Kor DJ. Prehospitalization antiplatelet therapy is associated with a reduced incidence of acute lung injury: a population-based cohort study. Chest. 2011 Feb;139(2):289–95. doi: 10.1378/chest.10-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Igonin AA, Protsenko DN, Galstyan GM, et al. C1-esterase inhibitor infusion increases survival rates for patients with sepsis*. Critical care medicine. 2012 Mar;40(3):770–7. doi: 10.1097/CCM.0b013e318236edb8. [DOI] [PubMed] [Google Scholar]
- 70.Islam MN, Das SR, Emin MT, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012 May;18(5):759–65. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matthay MA, Goolaerts A, Howard JP, Lee JW. Mesenchymal stem cells for acute lung injury: preclinical evidence. Crit Care Med. 2010 Oct;38(10 Suppl):S569–73. doi: 10.1097/CCM.0b013e3181f1ff1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. Am J Respir Crit Care Med. 2013 Apr 1;187(7):751–60. doi: 10.1164/rccm.201206-0990OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007 Aug 1;179(3):1855–63. doi: 10.4049/jimmunol.179.3.1855. [DOI] [PubMed] [Google Scholar]
- 74.Zheng G, Huang L, Tong H, et al. Treatment of acute respiratory distress syndrome with allogeneic adipose-derived mesenchymal stem cells: a randomized, placebo-controlled pilot study. Respir Res. 2014;15:39. doi: 10.1186/1465-9921-15-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilson JG, Liu KD, Zhuo H, et al. Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. The Lancet Respiratory medicine. 2015 Jan;3(1):24–32. doi: 10.1016/S2213-2600(14)70291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu KD, Wilson JG, Zhuo H, et al. Design and implementation of the START (STem cells for ARDS Treatment) trial, a phase 1/2 trial of human mesenchymal stem/stromal cells for the treatment of moderate-severe acute respiratory distress syndrome. Annals of intensive care. 2014;4:22. doi: 10.1186/s13613-014-0022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eisner MD, Thompson T, Hudson LD, et al. Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001 Jul 15;164(2):231–6. doi: 10.1164/ajrccm.164.2.2011093. [DOI] [PubMed] [Google Scholar]