

Summary
S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH) link one-carbon metabolism to methylation status. However it is unknown whether regulation of SAM and SAH by nutrient availability can be directly sensed to alter the kinetics of key histone methylation marks. We provide evidence that the status of methionine metabolism is sufficient to determine levels of histone methylation by modulating SAM and SAH. This dynamic interaction led to rapid changes in H3K4me3, altered gene transcription, provided feedback regulation to one-carbon metabolism and could be fully recovered upon restoration of methionine. Modulation of methionine in diet led to changes in metabolism and histone methylation in liver. In humans, methionine variability in fasting serum was commensurate with concentrations needed for these dynamics and could be partly explained by diet. Together these findings demonstrate that flux through methionine metabolism and the sensing of methionine availability may allow for direct communication to the chromatin state in cells.
Keywords: metabolomics, one carbon metabolism, epigenetics, chromatin biology
Graphical Abstract
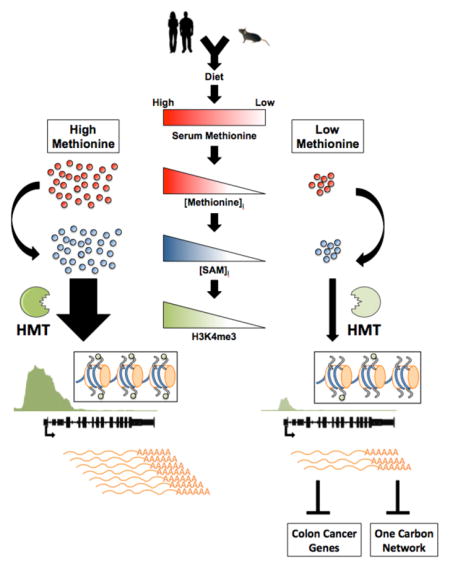
Introduction
Alterations in the methylation status of proteins, nucleic acids, and metabolites contribute to the pathogenesis of many of the major human pathophysiological conditions including cancer, obesity, and aging (Bergman and Cedar, 2013; Greer and Shi, 2012; Kraus et al., 2014). When these changes affect the methylation of histones and nucleic acids that determine the epigenetic status in cells, they can affect the expression of thousands of genes (Barth and Imhof, 2010). Changes in methylation status occur due to differences in the enzyme activity of methyltransferases and demethylases. Genes that encode these enzymes are frequently altered in pathological states leading to alterations in methylation (Chi et al., 2010; Dawson and Kouzarides, 2012). It has also been long established that S-adenosylmethionine (SAM) is the universal methyl donor for these enzymes that transfer its methyl group to yield S-adenosylhomocysteine (SAH) and a methylated substrate (Finkelstein, 1990). The methylation of this substrate provides a link between the metabolism that regulates SAM and SAH, which may act through product inhibition of a methyltransferase, and the epigenetic status of cells (Gut and Verdin, 2013; Katada et al., 2012; Teperino et al., 2010).
SAM and SAH are intermediate metabolites in a metabolic pathway that is a subset of a larger network collectively referred to as one carbon metabolism (Locasale, 2013). One carbon metabolism integrates nutrients from diverse sources such as glucose, serine, threonine, methionine, and choline and processes them into distinct outputs that achieve diverse biological functions. Whether the concentrations of SAM and SAH or their ratio ever reach values that could affect methyltransferase activity has been controversial. Some studies have concluded that their concentrations do not reach limiting values (Hoffman et al., 1979). Recent studies however have provided evidence that aberrant expression of NNMT, an enzyme that metabolizes SAM, has profound biological consequences resulting from changes in histone methylation (Kraus et al., 2014; Ulanovskaya et al., 2013). Others have found that only the levels of SAH correlated with methylation status including a recent finding that investigated threonine metabolism in mouse pluripotent stem cells that demonstrated threonine catabolism affected both pyruvate and glycine metabolism and altered histone methylation (Shyh-Chang et al., 2013). Although this study was the first to our knowledge documentation of an amino acid affecting histone methylation, this effect was shown to occur through indirect pathways that involve energy production and acetyl-coA metabolism. Further studies have demonstrated in human pluripotent stem cells that a depletion of methionine that is the precursor to SAM could lead to changes in histone methylation (Shiraki et al., 2014). However, these changes are also accompanied by widespread induction of stress response pathways and induction of cell death confounding the interpretation of whether the changes in histone methylation occurred through the sensing of SAM/SAH status.
Given these previous findings, we hypothesized that there exists a direct mechanism whereby the status of one carbon metabolism could alter the concentrations of SAM and SAH to confer, through their interaction with methyltransferases, the output of a defined methylation state. We focused on histones since key methylation modifications on their tails such as trimethylation at Lysine 4 are known to be required for the maintenance of defined cellular states (Benayoun et al., 2014; Ruthenburg et al., 2007) and have been shown to be modulated by metabolism (Shiraki et al., 2014; Shyh-Chang et al., 2013). We provide evidence in cells and mice that both SAM levels and the SAM/SAH ratio can be quantitatively tuned through changes in the metabolic flux of the methionine cycle to affect a critical component of chromatin status. This regulation occurred at physiologically relevant concentrations and appeared to control numerous physiological processes including the feedback regulation for the maintenance of homeostasis in one carbon metabolism and the activity of genes involved in cancer and cell fate. Together these findings are consistent with a model whereby the status of one carbon metabolism is in communication with the chromatin state of cells through its ability to modify the kinetics of enzymes that mediate histone methylation.
Results
Methionine metabolism quantitatively affects histone methylation
Since methionine is the closest substrate in the methionine cycle that affects SAM levels, we therefore tested whether cells depleted of methionine have alterations in the methionine cycle and whether this would confer effects on the methylation of histones. We generated a media formulation in which methionine was depleted from the culture media. HCT116 cells were placed in methionine restricted (3μM) media for 24 hours. We then utilized a liquid chromatography, high-resolution mass spectrometry (LC-HRMS) metabolomics technology we have recently developed (Liu et al., 2014b) to generate a quantitative profile of over 300 metabolites in response to methionine restriction that is visualized in a volcano plot (Figure 1A). It was found that metabolites in the methionine cycle exhibited dramatic changes in their concentrations with only moderate compensation from other pathways that fuel the one carbon cycle (Figure 1B). Notably, both SAM and SAH are depleted under these conditions. To test whether these alterations were sufficient to induce changes in histone methylation, we considered the relative levels of several histone methylation marks involving trimethylation at lysines 4,9, 27 that are each known to have substantial roles defining chromatin states, most notably at active and inactive genes, and mediating gene expression (Shilatifard, 2006) (Figure 1C). In addition, the kinetic properties, such as SAM binding affinity (Km), of these histone methyltransferases (An et al., 2011; Chin et al., 2005; Horiuchi et al., 2013; Obianyo et al., 2008; Patnaik et al., 2004; Xiao et al., 2003) suggests SAM concentration may play a role in their regulation and directly affect the rate of histone methylation reactions in the cell (Figure S1A). Each modification exhibited decreased methylation with trimethylation at lysine 4 on histone H3 (H3K4me3) exhibiting the largest change. To investigate whether these changes were specific to methionine restriction, we considered the removal of several other amino acids (W, Q, K, H, L) that include essential amino acids and glutamine that has been shown to be essential for cell proliferation (Figure 1D). We found that in each case no change in histone methylation was observed. To further test the generality of this observation, we considered the response of H3K4me3 to methionine restriction on a panel of six human cell lines subjected to methionine restriction for 24 hours. In each case, H3K4me3 was responsive to methionine restriction (Figure 1E). Additionally, we observed decreases in H3K9ac after methionine restriction (Figure S1B). Having shown that extreme withdrawal of methionine was sufficient to alter histone methylation, we investigated the concentration of methionine needed to achieve this effect. We considered a titration of differing concentrations of methionine in the culture media ranging from 3 to 500 μM. The relative levels of the methionine cycle and one carbon metabolism-related metabolites including methionine, SAM, SAH, homocysteine, betaine, and dimethylglycine (DMG) were measured (Figure 1F). It was found that the levels of these intermediates exhibited a graded response to changes in methionine concentration in the media. SAM and SAH were altered at concentrations below 25 μM - concentrations well below those present in typical culture media. We next investigated the dose response of changes in histone methylation to changes in methionine availability (Figure 1G). Histone methylation responded differently to differing concentrations of methionine. H3K4me3 was affected at concentrations between 10 and 25 μM consistent with concentrations needed to deplete SAM and SAH in the methionine cycle. One possibility is that these changes were due to non-specific effects on cell proliferation. We therefore considered the effects of methionine restriction on cell proliferation over a course of three days (Figure 1H, S1C). At extremely low concentrations of methionine, there were marked defects in cell morphology and cell proliferation but no activation of p53 when H3K4me3 is changing (Figure S1C, D) indicating that H3K4me3 dynamics occur before a cellular stress response. At 10 μM, cells remained viable with no gross alterations in morphology and only a modest decrease in cell proliferation that was fully recoverable at 25 μM confirming that the changes in histone methylation are not likely due to defects in cell proliferation. Together these findings demonstrate that methionine affects histone methylation and the dynamics of the methionine cycle.
Figure 1. Methionine metabolism alters histone methylation status.

a.) Metabolomics profile of methionine restricted HCT116 cells. Log2 fold change versus –log10(P value) (Student’s t-test two-tailed, n = 3). b.) Effects on methionine restriction on one carbon cycle metabolism. c.) Effects of methionine restriction on histone methylation measured by immunoblotting. Band intensities are normalized to total H3 levels and relative intensity was calculated compared to control cells. d.) Effects of deprivation of other amino acids on histone methylation. Data are normalized to control media. e.) Effects of methionine restriction on histone methylation across a panel of cell lines. f.) Relative concentration of methionine cycle metabolites in cells cultured in different concentrations of methionine. g.) Concentration dependent effects of methionine restriction on histone methylation. Data are normalized to the 100 μm condition. h.) Cell proliferation of HCT116 cells for differing levels of methionine. All error bars are computed from standard error of measurement (n=3) and all quantitation is normalized to the total H3 and the fold change between experimental and control groups are reported.
Methionine cycle and histone methylation dynamics in response to methionine restriction
Having observed alterations in the methionine cycle and concomitant changes in histone methylation, we sought to understand the dynamics of the process and its connection to cellular metabolism. We reasoned that the kinetics could provide further insights into how changes in metabolism alter histone methylation.
We considered the metabolic dynamics of methionine restriction across twelve time points (0, 5, 15, 30, 60, 90, 120, 240, 360, 540, 720, and 1440 minutes) from very early time up to times leading to cell death (Figure 2A). At each of these time points, we profiled over 300 metabolites spanning pathways across central carbon and secondary metabolism. A hierarchical clustering of the time course revealed coordinated dynamics across the metabolome in response to methionine restriction. Waves of coordinated changes that involve different metabolic pathways were observed. We utilized a recently developed algorithm for assessing the contribution to different pathways (i.e. pathway impact) defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Xia and Wishart, 2010) (Figure 2B). Pathway impact is computed by considering a weighted score of each metabolite with greater weights being assigned to metabolites that are centrally located in a given metabolic pathway. At early times, a module related to cysteine and methionine exhibited decreasing dynamics that were sustained across the time course. At intermediate times alterations in metabolites belonging to phenylalanine, tryptophan, and lysine metabolism were observed. At later times arginine and proline metabolism appeared altered together suggesting that complex compensatory amino acid metabolic response at later times are the primary response to methionine restriction. Interestingly, pathways such as glycolysis, TCA, and anabolic metabolism associated with nucleotide and lipid metabolism appeared not to exhibit coordinated dynamics suggesting that general affects on metabolism and cell proliferation do not appear altered throughout the kinetics. A further inspection of the module related to methionine cycle dynamics revealed that within minutes of methionine restriction, methionine is depleted within cells (Figure 2C). SAM levels exhibited a decay that occurred within an hour (Figure 2D). SAH levels exhibited more complex behavior involving a fast decrease, a recovery at two hours and a later decrease (Figure 2E). These dynamics are likely due to additional pathways such as remethylation from one carbon metabolism, methionine salvage, and flux into the transsulfuration pathway. These changes contributed to complicated dynamics of the SAM/SAH ratio that decreased at two hours (Figure 2F). In addition, 5-methyltetrahydrafolate did not contribute to the methionine cycle as measured by 13C serine, indicating that the folate cycle is uncoupled to the methionine in these cells (Figure S2). We next monitored the dynamics of histone methylation to assess its interaction with metabolic dynamics. We considered the dynamics of the levels of H3K4/9/27 in response to methionine restriction (Figure 2G,H). Surprisingly, it was found that within two to four hours, a depletion of histone methylation was observed. These kinetics exhibited only a one to two hour lag between depletion of the methionine cycle and the subsequent changes in histone methylation. These dynamics are generally too short for a transcriptional or translational regulatory response. Together these findings conclude that the dynamics of methionine metabolism result primarily in amino acid compensatory responses and not general features that result in disruption of cell proliferation. They also indicate that methionine dynamics can be coordinately sensed by histones to determine the levels of histone methylation.
Figure 2. Histone methylation is dynamically regulated by methionine metabolism.

a.) Global metabolic dynamics in response to methionine restriction. b.) Pathway analysis of dynamics shown in (a). c.) Dynamics of methionine. d.) Dynamics of SAM. e.) Dynamics of SAH. f.) Dynamics of the SAM/SAH ratio. g.) Dynamics of histone methylation. h.) Quantitation of results in (g). Integrated intensities are normalized to total H3 and the fold change represents differences compared to 24 hours. All error bars are computed from standard error of measurement (n=3).
Methionine metabolism and histone methylation dynamics are reversible
Thus far we have shown that methionine metabolism can induce alterations in histone methylation levels and that this effect is dynamic and occurs on time scales consistent with that of a sensing mechanism. Another necessary requirement of such a signal transduction mechanism is that the dynamics are reversible. We therefore questioned whether metabolism and histone methylation can be recovered after methionine restriction. Cells were subjected to methionine restriction for 24 hours (between day 1 and day 2) at which point we allowed the cells to be cultured for an additional 24 hours in media containing the full concentration of methionine (between day 2 and day 3) (Figure 3A). We carried out metabolomics at each time point as considered previously (Figure 3B). Hierarchical clustering of each profile revealed marked changes across the metabolic network at days 1, 2, and 3. The gross measured differences in metabolism from day 1 and day 3 revealed that overall metabolism is not reversible under these conditions. An assessment of the pathways involved indicated that several amino acid metabolism pathways were not recoverable at 3 days (Figure 3C). Of the pathways recovered after reincorporation of methionine into the culture media, cysteine and methionine metabolism was one of three pathways shown to be reversible. This effect is illustrated in Figure 3D where the methionine cycle is plotted and for each metabolite in the cycle, some recovery of its concentration and its salvage pathway (Figure 3E) was observed. To investigate whether these dynamics correlate with histone methylation we measured histone methylation levels (Figure 3F) and found that methylation levels exhibited a concomitant recovery back to levels observed in the original media. Together these findings indicate that the methionine cycle is reversible even when the remainder of global metabolism is altered. Furthermore, these results show that the recovery of the levels of the metabolites in the methionine cycle is sufficient to restore the levels of histone methylation.
Figure 3. Histone methylation and methionine cycle dynamics are reversible.

a.) Experimental setup of methionine restriction and recovery. b.) Effects of methionine restriction and recovery on global metabolism. c.) Pathway analysis of methionine recovery. d.) Effect of methionine restriction and recovery on methionine cycle metabolism. e.) Effect of methionine resctiction and recovery on methionine salvage and transsulfuration. f.) Response of histone methylation from methionine restriction and recovery. Integrated intensities are normalized to total H3 and fold change was calculated. All error bars are computed from the standard error of measurement (n=3).
Methionine restriction decreases H3K4me3 signature peaks and alters gene expression
Although results from immunoblotting identified global alterations in histone methylation after methionine restriction, it remained unclear whether methionine metabolism affected specific marks on the genome to mediate gene expression. We therefore sought to determine the precise location H3K4me3 on the genome using chromatin immunoprecipation with sequencing (ChIP-seq) and the consequences on gene expression using RNA-sequencing (RNA-seq). The specificity of the H3K4me3 antibody was first confirmed with modified histone peptide array and sensitivity tested using serial titrations of H3K4me3 peptides as suggested in the ENCODE guidelines (Bailey et al., 2013) (Figure S4A, B). Cells were subjected to methionine restriction (−MET) or normal methionine (+MET) conditions for 24 hours and cross-linked chromatin was collected for H3K4me3 ChIP-seq analysis with total RNA collected for RNA-seq in parallel. After peak calling (methods), replicate samples for the +MET and −MET conditions cluster together suggesting the genome-wide profile of H3K4me3 marks are similar between replicates but vastly different between the conditions (Figure S4C). Next, we investigated the change in H3K4me3 peaks at the transcription start site (TSS) of genes, since H3K4me3 at gene promoters has been associated with gene transcription. Overall, we found that the H3K4me3 peaks around the TSS are bimodal and decrease, on average, after methionine restriction (Figure 4A). In addition, H3K4me3 peaks decreased breadth after methionine restriction (Figure S4D) but no significant change in H3K4me3 binding site distribution around the TSS was observed (Figure S4E).
Figure 4. Methionine restrictions decreases H3K4me3 and alters gene expression.

a.) Change in H3K4me3 distribution around the transcription start site (TSS) after 24hr methionine restriction b.) Genome wide distribution of H3K4me3 peaks in +MET (red) and −MET (blue) conditions. c.) Enlargement of chromosome 17 d.) Changes in H3K4me3 ChIP-seq signal with corresponding changes in gene expression from RNA-seq for colon cancer genes. e.) Enzymes with decreased H3K4me3 and gene expression essential in one-carbon metabolism and related pathways are highlighted in red. f.) H3K4me3 ChIP-seq and RNA-seq signals for enzymes in (e) before and after methionine restriction.
Next, peak counts were plotted for each chromosome showing H3K4me3 peak distribution across the genome is relatively consistent (Figure 4B). Closer inspection of individual chromosomes revealed that particular genomic regions are differentially marked by H3K4me3 (Figure 4C) and further differential binding analysis indicated significantly depleted regions of H3K4me3 (Figure S4F).
Since promoter H3K4me3 is a signature for active transcription we further investigated how decreased H3K4me3 at promoters affects gene transcription. To this end we performed RNA-seq (Figure S4G) under methionine-restricted conditions in Hct116 and compared these results to our ChIP-seq analysis. Interestingly, we found that colorectal cancer (CRC)-associated genes were enriched for loss of H3K4me3 at promoters with resulting decreased expression (p = 0.02, Fisher’s Exact Test. Notably AKT1, MYC, and MAPK, among others are responsive to decreases in promoter H3K4me3 (Figure 4D). In addition, CRC genes that have significant decreases in gene expression also show significant decreases in H3K4me3 over the promoter region when compared to genome wide loss of H3K4me3 (Figure S4H). Overall, these results suggest that nutrient status, has a dramatic effect on the chromatin state and gene regulation in cells and affects pathophysiological outcomes.
To further evaluate specific physiological consequences of this mechanism, we identified a network of enzymes involved in one-carbon metabolism that show decreased H3K4me3 with concomitant decreases in expression (Figure 4E) suggesting a feedback mechanism that decreases consumption of SAM by alternate pathways and an attempt to maintain H3K4me3 by regulation of JARID1B (KDM5B), H3K4me3 demethylase that is deregulated in cancer (Yamane et al., 2007). This finding is indicative of a signal transduction mechanism by which alterations in H3K4me3 directly feedback to one carbon metabolism and methylation-related enzymes to maintain physiological homeostasis.
Methionine cycle alterations can be sustained by diet in vivo
We next questioned whether changes in methionine can occur through and are maintained in response to alterations in nutrient availability in a longer-term physiological setting. We considered an analysis of the effects of methionine restriction in vivo at a concentration that has been shown to have health-promoting effects (Ables et al., 2012). Seven-week-old C57Bl6 mice were randomized into two groups and one arm was fed a standard diet consisting of 0.84% (w/w) methionine and the other a methionine-restricted (MR) diet consisting of over seven times less methionine (0.12%, w/w) (Figure 5A). We first confirmed that the MR diet specifically decreased serum methionine compared to all other amino acids (Figure 5B). At twelve weeks, mice were sacrificed and no gross phenotypic alterations were observed as measured by liver histology and as previously described (Ables et al., 2012) (Figure 5A). An examination of the metabolite profile in the fasting serum and liver by carrying out unsupervised hierarchical clustering revealed global alterations in metabolism in liver and plasma (Figure S5A) with a pathway analysis revealing that Cysteine and Methionine metabolism were predominantly altered in plasma (Figure S5B). Other amino acid metabolism pathways to a lesser extent exhibited alterations. Inspection of metabolism in liver exhibited sustained alterations in cysteine and methionine along with alterations in taurine metabolism and other amino acid metabolism pathways (Figure S5C). Globally, an analysis of the quantile-quantile (Q-Q) plot showed skewness towards more changes being observed in liver but with more significant changes occurring in the serum (Figure 5SD). These differences were further exemplified by comparing correlations of metabolite levels between liver and plasma (Figure S5E) with metabolites related to beta oxidation such as acylcarnitines, amino acid metabolism, and one carbon metabolism found to correlate with the extent of methionine levels in the plasma (Figure S5F).
Figure 5. Alterations in methionine metabolism can be sustained by diet in vivo.

a.) Methionine restriction in mice with representative histology in methionine-restricted and control mice. Images are at 40X with Hematoxylin and Eosin staining. b.) Fold change of serum amino acids in −MET compared to +MET, measured my LC-MS (*denotes p<0.001). c.) Methionine cycle metabolites in liver of methionine-restricted mice. d.) Methionine cycle metabolites in plasma of methionine-restricted mice e.) Histone methylation in liver of methionine-restricted mice. Integrated intensities were normalized to total H3 f.) Quantitation of results histone methylation from n=8 mice in each cohort. P value is obtained from a student’s t-test equal variance. g–h) Correlation of histone methylation and methionine cycle metabolites in liver and plasma.
Together these findings indicate that the regions of the metabolic network that are altered in response to sustained methionine-restriction in vivo are comparable to those aspects of the metabolic network that are dynamic in response to acute methionine restriction in cells. Furthermore, the metabolic state of fasting liver is dramatically different in the animals that were maintained on different diets for the long term.
Methionine restriction in vivo sustains altered methionine cycle and histone methylation
Having demonstrated that cysteine and methionine metabolism exhibit sustainable alterations in mice, we then asked whether these changes led to differences in the methionine cycle. In liver (Figure 5C), methionine concentrations exhibited no significant changes suggesting that compensatory fluxes involving its consumption or intake are occurring. These changes in fluxes resulted in decreases in SAM, SAH, the SAM/SAH ratio, cystathionine, methylthioadenosine (MTA) and increases in the methyl donors dimethylglycine (DMG) and betaine. However, in plasma (Figure 5D), methionine levels were maintained at lower levels. In addition, cystathionine and 2-keto-4-methylthiobutyrate (KMTB) a component of the methionine salvage pathway were also significantly decreased. Conversely, betaine and DMG each exhibited increases suggestive of their possibility as plasma biomarkers. Methionine cycle metabolites SAM and SAH did not show changes in plasma. This observation was expected since cells are not thought to secrete these metabolites at appreciable concentrations (Agrimi et al., 2004). We next asked whether these sustained changes in the methionine cycle were sufficient to induce changes in histone methylation. We observed decreases in H3K4me3 in the methionine restricted liver (Figure 5E) that we quantified (Figure 5F) indicating that the alterations in SAM and SAH levels observed in the liver are sufficient to induce global changes in histone methylation. We found that SAM, the SAM/SAH ratio, cystathionine positively correlated with H3K4me3 levels and methionine, betaine, and DMG negatively correlated with H3K4me3 levels (Figure 5G, S5G). In plasma, the levels of methionine and cystathionine were positively correlated and betaine and DMG were negatively correlated (Figures 5H, S5H). Together these findings provide evidence that the levels of SAM and SAM/SAH ratio are predictive of histone methylation levels, that plasma metabolites in methionine cycle metabolism are predictive of histone methylation in liver and that these levels can be directly modulated by diet in mice.
Humans exhibit variability in methionine levels
We next questioned whether variability in methionine metabolism exists in humans and how it can be regulated. Standard clinical parameters and a record of dietary intake over four days was considered to reflect variations in habitual diet as is standard practice in clinical nutrition (Levine et al., 2014) across a cohort of healthy human subjects. Fasting serum was collected and subjected to a metabolomics analysis (Figure 6A, S6A). We performed an unsupervised hierarchical clustering of the nutrient intake for each subject and found sets of defined modules that were able to classify the subjects into discrete dietary behaviors such as groups that are high in fruits and vegetables or carbohydrates (Figure 6B). We next measured the concentration of methionine along with a panel of amino acids in these subjects (Figure 6C). Strikingly, the concentration of methionine exhibits substantial variation with values ranging from 3 to 30 μM with methionine exhibiting the largest variation (Figure 6D). This variation in concentration is on the same order as that needed to induce changes in histone methylation in cells and in mice. In addition, methionine in the serum correlates with N,N,N-trimethyllysine and N-methylglycine (sarcosine), both of which are methylated by the transfer of methyl groups from SAM, suggesting that methionine levels in the serum are indicative of cellular methylation status (Figure 6E) with clear patterns of food intake that corresponded to both high and low methionine (Figure 6F, S6B). Vegetable-based nutrients such as fiber correlated with low methionine levels and age, body weight and fat intake correlated with high methionine levels. Surprisingly, protein intake exhibited no correlation with methionine. An analysis of the metabolites and pathways that correlated with methionine levels (Figure 6G, S6C) revealed pathways related to ketogenesis and amino metabolism. Taken together, these findings demonstrate that the variability in methionine concentration in humans is on the same scale as that needed to induce alterations in histone methylation and that these differences correlate with changes in diet and health status.
Figure 6. Methionine and metabolic variation in human subjects.

a.) Measurement of serum methionine and clinical and dietary variables in human subjects. b.) (left) Hierarchical clustering of the distance matrix diet variables. (right) k-means clustering of subjects and diet variables (N=24). c.) Absolute concentrations of amino acids in fasting serum in 38 human subjects. d.) Coefficients of variation of amino acids in fasting serum. e. Correlation of methionine in the serum with methylated serum metabolites, N,N,N- trimethyllysine and sarcosine.f.) Correlation of methionine concentrations with dietary variables obtained from habitual diet records. g.) Correlation of methionine concentrations with fasting serum metabolite levels.
A computational model identifies factors that contribute to methionine variability
Finally, having identified associations of methionine with diet and other factors, we built a computational model to identify the direct influences on methionine concentration. We considered a mixed effects model that aims to identify causal features in high dimensional data and thus identify factors that give rise to the variation in methionine levels (Figure 7A). We considered dietary intake variables, clinical variables such as age, gender, and body composition measured by Duel X-ray Absorptiometry (DEXA). To reduce the dimensionality of the data matrix, we filtered the variables according to their correlations with methionine, carried out a principle components analysis on the DEXA variables (Figure S7A), and checked the resulting twelve variables for collinearity (Figure S7B). We then carried out a regression using least-squares minimization with maximum likelihood estimation (methods) and obtained a model with good fit (P < 10−4, F test) to the experimental methionine concentrations (Figure 7B).
Figure 7. A computational model identifies determinants of methionine variability.

a.) Overview of variable selection for the computational model. b.) Predicted versus measured methionine levels in human subjects. c.) Regression coefficients. Error bars are obtained from maximum likelihood estimates. d.) Schematic of dietary factors that contribute to each modeled variable. e.) Results from variance partitioning.
An analysis of the coefficients revealed (Figure 7C) several contributions to methionine variation including age, body composition and gender (with male-ness contributing to a positive influence) and diet including variables known to be associated with methionine including zinc and tocepherol. Using established guidelines (methods), it was found that the diet variables could be related to major sources of food intake (Figure 7D) with for example, fats, seafood and meat contributing to higher concentrations of methionine. Finally we performed a variance partitioning calculation (Figure 7E) and found that about thirty percent of the variation is explained by diet, about thirty percent explained by clinical variables including gender and age and the remaining unaccounted variance likely due to genetic factors. These results indicate that methionine concentrations observed in humans can be decomposed into several factors including diet.
Discussion
These findings together provide evidence for a dynamic regulatory mechanism whereby the status of the methionine component of one carbon metabolism is sensed by histones to determine the levels of methylation on critical residues that mediate cellular epigenetic status. We first demonstrated that methionine deprivation and subsequent depletion of SAM and SAH induce changes in histone methylation. Furthermore, we tested whether these dynamics occurred in a manner consistent with a signal transduction mechanism. Necessarily, we found that changes in histone methylation occurred at quantitatively relevant concentrations of methionine. The kinetics of turnover of histone methylation in response to SAM and SAH availability were found to occur within hours and thus are much shorter than times corresponding to changes in cell cycle progression or global stress responses that also affect histone methylation. Importantly these dynamics were found to be reversible as the depletion of histone methylation could be recovered upon restoration of methionine availability even when the remainder of cellular metabolism was not recovered. Each of these properties is consistent with a mechanism whereby the status of key histone marks such as H3K4me3 that mediate chromatin structure, gene expression, and cell identity are sensed by the metabolic status of one carbon metabolism.
In vivo, the dynamic response to methionine restriction further provided support of such a mechanism since alterations in SAM and SAH could be sustained in liver and these changes were sufficient to induce changes in histone methylation. Interestingly, although this dietary intervention maintained reduced methionine levels in the serum, the levels in the liver were not altered suggesting that compensatory adaptations in methionine fluxes occurred. These likely serve to maintain other essential processes involved in methionine metabolism possibly providing a basis for why methionine restriction can be sustained in healthy animals even when changes in histone methylation are observed. Notably, methylation status and metabolism were measured in fasting conditions where methionine restriction was shown to alter histone methylation. This fasted condition we expect sets a lower bound on the extent that the methionine cycle can be depleted with feeding contributing to increases in methionine levels. Given this observation, it is tempting to speculate that feeding or a diurnal rhythm might regulate this effect, perhaps raising methionine levels to recover depletions in histone methylation.
In humans, the variation of methionine in healthy individuals was found to be largest across a panel of serum amino acids. Although these measurements are by no means exhaustive of human population dynamics, concentrations in many individuals were found to be far lower than the concentration required to induce changes in methylation levels. Computational modeling of diet and clinical variables found that about thirty percent of the variation could be due to fundamental clinical variables such as age, body composition, and gender, and about thirty percent was modulated by diet. In the future the results from these models could further be integrated with genomics data to specifically define the genetic contributions as have been identified to associate with serum metabolite levels (Suhre et al., 2011). Nevertheless it is tempting to speculate that basic dietary factors such as vegetable and fat intake could mediate human epigenetics through modulation of methionine metabolism.
One carbon metabolism as a metabolic signal transduction mechanism
Cellular decision-making occurs through signal transduction that utilizes post-translation modifications. For example, protein kinases establish networks of protein phosphorylation with multiple layers of nonlinear regulation that control almost all aspects of physiology. Despite the ubiquitous importance of histone and other macromolecular methylation, relatively little is known about how methylation is regulated. In contrast to protein kinases where the substrate, ATP, is in vast excess, the concentrations of SAM or SAH are on the order of the kinetic parameters that determine enzyme activity. Thus much of histone and other methylation could be regulated in large part by the status of methionine metabolism and one carbon cycle flux consistent with relatively few numbers of methyltransferases compared to the over 500 protein kinases that exist. This type of regulation by virtue of its biochemistry would have fundamentally different control properties and information capacity than that of protein kinase-mediated signaling. This regulation is evident in the feedback control observed in genes in one carbon metabolism whose expression and H3K4me3 signature is modulated in response to a decrease in SAM and SAH levels, likely as a specific mechanism to maintain homeostasis in one carbon metabolism. In particular, enzymes important in regenerating methionine by utilizing SAM are some of the most down regulated genes with loss of H3K4me3 at their promoters.
Methionine metabolism in pathophysiology through its modulation of chromatin state
Numerous studies have documented a contribution of methionine metabolism and its restriction to beneficial metabolic health including the extension of mammalian life span, the acquisition of resistance to diet-induced obesity, and the therapeutic efficacy of ketogenic diets (Kraus et al., 2014; Malloy et al., 2006; Orentreich et al., 1993; Pissios et al., 2013). In each of these cases, the biological mechanisms contributing to these outcomes are largely unknown. Although alterations in methylation status are likely not accounting for all of these effects, it is possibly if not likely that the alterations in gene expression of key genes important for the relevant physiological effect in each of these cases. Indeed chronic diseases including obesity, diabetes, and cancer often stem from the inability of the organism to adapt to the demands of their nutritional load (Hotamisligil, 2010; Laplante and Sabatini, 2012). These adaptations require signal transduction mechanisms that integrate nutritional status to achieve the desired physiological demand. There are numerous signal transduction pathways that sense nutritional status and it is likely that the sensing of one carbon metabolism by histones is one of these mechanisms.
Interestingly, this mechanism affected gene regulation of several cancer-promoting genes. Many cancers are vulnerable to disruptions in one carbon metabolism (Locasale, 2013), and our results demonstrate the necessity of methionine metabolism for the maintenance of histone methylation and the consequences for expression of cancer gene programs. Thus it is tempting to speculate that some of the anti-tumor effects of targeting one carbon metabolism may occur through changes in cancer epigenetics.
Methods
Methionine Restriction in Cell Culture
Briefly, colorectal cancer cells were grown in RPMI supplemented with 10% FBS and 100U/mL penicillin and streptomycin. For methionine restriction experiments, media was changed to either conditional media lacking methionine (−MET) or media with methionine (+MET) for indicated times. Detailed protocol and media recipes are provided in the Supplemental Information.
Mouse Feeding and Tissue and Plasma Analysis
Mouse feeding and sample collection was performed as previously described (Ables et al., 2012). Briefly, C57BL/6J mice were randomly assigned to either an isocaloric, 0.85% (w/w) methionine (control; C) or 0.12% (w/w) methionine (methionine restricted; MR) diets and sacrificed after 12 weeks. Detailed methods are described in the Supplemental Information.
Metabolite Extraction and LC-HRMS Analysis
The procedures for cultured cells, serum, and tissues were described in previous studies (Liu et al., 2014a; Liu et al., 2014b). Additional methods for liquid chromatography mass spectrometry (LC-MS) analysis are provided in the Supplementary Information.
RNA Sequencing
Total RNA from +MET and −MET conditions was polyA selected and libraries were prepared according to a standard protocol provided by Illumina and sequenced on the Illumina HiSEQ 2500 Rapid Run sequencer.
Chromatin Immunoprecipation and Sequencing
Hct116 cells from +MET and −MET conditions were cross-linked and each IP was performed as previously described, (Ercan et al., 2007; Landt et al., 2012) using 1.5×106 cells. Libraries were made and pooled according to Illumina instructions, then sequenced on an Illumina HiSEQ 2500 Rapid Run sequencer.
Human Studies
Serum samples, 4-day diet records, and body composition results (via DXA scan) on 24 de-identified healthy older adults were provided by Marcas Bamman at the University of Alabama at Birmingham (UAB). As part of the UAB Institutional Review Board-approved parent project, all 24 subjects agreed to have their samples and data used for future research.
Statistical Analysis
Error bars represent standard error of the measurement (SEM). Significance reported are results from Student’s t-test unless otherwise noted.
Computational Modeling
In brief, a set of 12 predictor variables were obtained after a variable selection process that incorporates methionine correlations, a dimensional reduction of the Duel X-ray Absorptiometry (DEXA) data using principal components analysis, and a collinearity assessment using the variance inflation factor. We next generated a mixed effects linear model including the set of predictor variables, a random effect term and noise. Model selection was carried out using exhaustive sampling and optimization of the Akaike information criteria. Variance contributions for each variable and random effect were summed to define the total contribution of variance for each variable and unexplained factor. The full details of the modeling are contained in the Supplemental Information.
Supplementary Material
Acknowledgments
Work was supported by R00CA168997 and RO1AI110613 to JWL and T32GM007273 to SJM from the National Institutes of Health and a Future Leader award from the International Life Sciences Institute to JWL. We gratefully acknowledge members of the Locasale lab for their support and helpful discussions. We thank Heather Roman for technical assistance and Sylvia Lee and Erin Jones for help with ChIP-seq experiments and analysis.
Footnotes
Author Contributions
SJM, JWL designed the study. SJM, SN, DG, MM, LH, XL, PG, DM, GA, SN performed experiments and contributed to data analysis. ATM, MBB, DG carried out the diet record and serum collection. SN led the feeding studies and SN, DM, GA carried out the feeding studies and harvested tissues in mice. SJM, JWL wrote the paper.
Accession Numbers
The ChIP-seq and RNA-seq datasets collected for this study have been deposited under the Gene Expression Omnibus (GEO) accession number GSE72131.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ables GP, Perrone CE, Orentreich D, Orentreich N. Methionine-restricted C57BL/6J mice are resistant to diet-induced obesity and insulin resistance but have low bone density. PloS one. 2012;7:e51357. doi: 10.1371/journal.pone.0051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrimi G, Di Noia MA, Marobbio CM, Fiermonte G, Lasorsa FM, Palmieri F. Identification of the human mitochondrial S-adenosylmethionine transporter: bacterial expression, reconstitution, functional characterization and tissue distribution. The Biochemical journal. 2004;379:183–190. doi: 10.1042/BJ20031664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Yeo KJ, Jeon YH, Song JJ. Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. The Journal of biological chemistry. 2011;286:8369–8374. doi: 10.1074/jbc.M110.203380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, Madrigal P, Taslim C, Zhang J. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS computational biology. 2013;9:e1003326. doi: 10.1371/journal.pcbi.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends in biochemical sciences. 2010;35:618–626. doi: 10.1016/j.tibs.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, Hitz BC, Gupta R, Rando TA, Baker JC, Snyder MP, Cherry JM, Brunet A. H3K4me3 Breadth Is Linked to Cell Identity and Transcriptional Consistency. Cell. 2014;158:673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nature structural & molecular biology. 2013;20:274–281. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- Caudill MA, Wang JC, Melnyk S, Pogribny IP, Jernigan S, Collins MD, Santos-Guzman J, Swendseid ME, Cogger EA, James SJ. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. The Journal of nutrition. 2001;131:2811–2818. doi: 10.1093/jn/131.11.2811. [DOI] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nature reviews Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin HG, Pradhan M, Esteve PO, Patnaik D, Evans TC, Jr, Pradhan S. Sequence specificity and role of proximal amino acids of the histone H3 tail on catalysis of murine G9A lysine 9 histone H3 methyltransferase. Biochemistry. 2005;44:12998–13006. doi: 10.1021/bi0509907. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Ercan S, Giresi PG, Whittle CM, Zhang X, Green RD, Lieb JD. X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nature genetics. 2007;39:403–408. doi: 10.1038/ng1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein JD. Methionine metabolism in mammals. The Journal of nutritional biochemistry. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature reviews Genetics. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- Hoffman DR, Cornatzer WE, Duerre JA. Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can J Biochem. 1979;57:56–65. doi: 10.1139/o79-007. [DOI] [PubMed] [Google Scholar]
- Horiuchi KY, Eason MM, Ferry JJ, Planck JL, Walsh CP, Smith RF, Howitz KT, Ma H. Assay development for histone methyltransferases. Assay and drug development technologies. 2013;11:227–236. doi: 10.1089/adt.2012.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–28. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508:258–262. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome research. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Suarez JA, Brandhorst S, Balasubramanian P, Cheng CW, Madia F, Fontana L, Mirisola MG, Guevara-Aguirre J, Wan J, Passarino G, Kennedy BK, Wei M, Cohen P, Crimmins EM, Longo VD. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell metabolism. 2014;19:407–417. doi: 10.1016/j.cmet.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, Cluntun AA, Mentch SJ, Locasale JW. A strategy for sensitive, large scale quantitative metabolomics. Journal of visualized experiments : JoVE. 2014a doi: 10.3791/51358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Analytical chemistry. 2014b;86:2175–2184. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nature reviews Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malloy VL, Krajcik RA, Bailey SJ, Hristopoulos G, Plummer JD, Orentreich N. Methionine restriction decreases visceral fat mass and preserves insulin action in aging male Fischer 344 rats independent of energy restriction. Aging cell. 2006;5:305–314. doi: 10.1111/j.1474-9726.2006.00220.x. [DOI] [PubMed] [Google Scholar]
- Obianyo O, Osborne TC, Thompson PR. Kinetic mechanism of protein arginine methyltransferase 1. Biochemistry. 2008;47:10420–10427. doi: 10.1021/bi800904m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low methionine ingestion by rats extends life span. The Journal of nutrition. 1993;123:269–274. doi: 10.1093/jn/123.2.269. [DOI] [PubMed] [Google Scholar]
- Patnaik D, Chin HG, Esteve PO, Benner J, Jacobsen SE, Pradhan S. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. The Journal of biological chemistry. 2004;279:53248–53258. doi: 10.1074/jbc.M409604200. [DOI] [PubMed] [Google Scholar]
- Pissios P, Hong S, Kennedy AR, Prasad D, Liu FF, Maratos-Flier E. Methionine and choline regulate the metabolic phenotype of a ketogenic diet. Molecular metabolism. 2013;2:306–313. doi: 10.1016/j.molmet.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Molecular cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annual review of biochemistry. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell metabolism. 2014;19:780–794. doi: 10.1016/j.cmet.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wagele B, Altmaier E, Deloukas P, Erdmann J, Grundberg E, Hammond CJ, de Angelis MH, Kastenmuller G, Kottgen A, Kronenberg F, Mangino M, Meisinger C, Meitinger T, Mewes HW, Milburn MV, Prehn C, Raffler J, Ried JS, Romisch-Margl W, Samani NJ, Small KS, Wichmann HE, Zhai G, Illig T, Spector TD, Adamski J, Soranzo N, Gieger C. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teperino R, Schoonjans K, Auwerx J. Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell metabolism. 2010;12:321–327. doi: 10.1016/j.cmet.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nature chemical biology. 2013;9:300–306. doi: 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wishart DS. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics. 2010;26:2342–2344. doi: 10.1093/bioinformatics/btq418. [DOI] [PubMed] [Google Scholar]
- Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, Taylor-Papadimitriou J, Tempst P, Zhang Y. PLU-1 Is an H3K4 Demethylase Involved in Transcriptional Repression and Breast Cancer Cell Proliferation. Molecular cell. 2007;25:801–812. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.