

Abstract
RE-1 silencing transcription factor (REST), a master negative regulator of neuronal differentiation, controls neurogenesis by preventing the differentiation of neural stem cells. Here we focused on the role of REST in the early steps of differentiation and maturation of adult hippocampal progenitors (AHPs). REST knockdown promoted differentiation and affected the maturation of rat AHPs. Surprisingly, REST knockdown cells enhanced the differentiation of neighboring wild-type AHPs, suggesting that REST may play a non–cell-autonomous role. Gene expression analysis identified Secretogranin II (Scg2) as the major secreted REST target responsible for the non–cell-autonomous phenotype. Loss-of-function of Scg2 inhibited differentiation in vitro, and exogenous SCG2 partially rescued this phenotype. Knockdown of REST in neural progenitors in mice led to precocious maturation into neurons at the expense of mushroom spines in vivo. In summary, we found that, in addition to its cell-autonomous function, REST regulates differentiation and maturation of AHPs non–cell-autonomously via SCG2.
SIGNIFICANCE STATEMENT Our results reveal that REST regulates differentiation and maturation of neural progenitor cells in vitro by orchestrating both cell-intrinsic and non–cell-autonomous factors and that Scg2 is a major secretory target of REST with a differentiation-enhancing activity in a paracrine manner. In vivo, REST depletion causes accelerated differentiation of newborn neurons at the expense of spine defects, suggesting a potential role for REST in the timing of the maturation of granule neurons.
Keywords: adult neurogenesis, microfludics, neurosecretory, REST/NRSF, SCG2, stem cells
Introduction
The progression from a neural stem cell (NSC) to a mature, integrated functional granule neuron in the dentate gyrus (DG) during adult neurogenesis is regulated by cell-intrinsic factors (transcription factors) and extrinsic signals (WNT, SHH, and NOTCH as well as neurotransmitters and growth factors) within the niche (Gage, 2000, 2002; Lie et al., 2005; Suh et al., 2009; Ming and Song, 2011). Although we know of a number of genes and signaling pathways involved in adult DG neurogenesis, we are far from a comprehensive understanding of how these components orchestrate the birth and development of new neurons.
RE-1 silencing transcription factor (REST) is a transcriptional repressor that binds RE-1 sequences to negatively regulate neuronal gene expression during both embryogenesis and adult neurogenesis (Chong et al., 1995; Schoenherr and Anderson, 1995; Singh et al., 2008; Jørgensen et al., 2009; Gao et al., 2011; Satoh et al., 2013). REST represses >2000 targets, such as Mash1, Neurod1, Type II sodium channel, Na-K ATPase, Synapsin 1, neuronal Na, K ATPase subunit, and BDNF in non-neuronal cells (Chong et al., 1995; Schoenherr and Anderson, 1995). The expression of REST persists in the adult CNS (Palm et al., 1998), albeit at much lower levels compared with embryonic neuronal development (Ballas et al., 2005). The differences in REST protein levels, combined with the underlying local chromatin structure, may lead to cell type-dependent target selection and thus context-dependent roles for REST (Ballas et al., 2005). Moreover, Hsieh and colleagues recently showed that REST has a biphasic expression pattern, in quiescent adult NSCs and then in a subset of neurons, and is required to regulate neurogenesis (Gao et al., 2011).
In this study, we focused on the role of REST in the early steps of neuronal differentiation, and we provide evidence of a novel role for REST in non–cell-autonomous regulation of neuronal differentiation. The analysis of the repertoire of genes regulated by REST in adult hippocampal progenitors (AHPs) identified Secretogranin II (SCG2) as the main REST-regulated secreted factor that could potentially mediate the non–cell-autonomous, differentiation-enhancing activity. We established the cellular and fluidic compartmentalization of AHP culture in a microfluidic device and modeled the non–cell-autonomous processes that affect neuronal development of AHPs by REST-Scg2 regulation. In parallel, electrophysiological recordings from hippocampal neurons in response to REST knockdown and recombinant SCG2 provided a functional interpretation for its role in neuronal differentiation. Results from both knockdown of Scg2 in AHPs and immunodepletion of SCG2 from conditioned medium demonstrate that Scg2 is required for the differentiation of AHPs. In vivo, knockdown of REST in the DG not only resulted in accelerated differentiation but also caused significant defects in dendritic spine maturation. We propose that Scg2 plays a critical role in the proper differentiation and maturation of neural progenitors into functional neurons, both cell-autonomously and non–cell-autonomously.
Materials and Methods
Retrovirus-mediated knockdown in NSCs/progenitor cells in the mouse hippocampus.
Mouse Maloney retrovirus-based virus was prepared using human embryonic kidney cells (293T) as the packaging line, and virus was collected by high-speed ultracentrifugation. The concentrated viral solution (5 × 107 pfu/ml) was delivered to the DG of the mouse hippocampus via stereotaxic injection as described previously (van Praag et al., 2002). The mice were female C57BL/6, 6–8 weeks old at the time of injection. Animals were perfused at 8 d after injection of retrovirus (dpi), sliced tissue was stained with DAPI, and GFP was visualized by confocal microscopy. The Salk Institutional Animal Care and Use Committee approved all animal protocols.
Tissue preparation for immunohistochemical analyses.
Animals were anesthetized with a solution of ketamine/xylazine (100 mg/kg, 10 mg/kg) and were perfused transcardially with saline followed by cold 4% PFA. Brains were postfixed for 12 h with 4% PFA and equilibrated in 30% sucrose before slicing. A microtome was used to cut 40-μm coronal sections. Tissue sections were then stained with the following antibodies: anti-REST (Santa Cruz Biotechnology, C-15 and P-18), anti-NeuN (Millipore, MAB377), and anti-GFP (Aves Laboratories, 1020). Tissue stainings were performed by a BrdU method. Briefly, free-floating tissues were permeabilized with 2N HCl at 37°C for 30 min and neutralized in 0.1 m borate buffer, pH 8.5, at room temperature for 10 min. The tissues were blocked using a blocking solution containing Tris-buffered saline, Triton X-100 (0.1%), and horse serum (3%). Tissues were incubated at 4°C overnight in primary antibodies and then with secondary antibodies at room temperature for 4 h. DAPI was used to stain the nuclei.
Morphological analysis of GFP+ neurons and spine density analysis.
The morphology of the neurons from 8 dpi tissues was analyzed on a LSM 710 confocal microscope using z-series at 1 μm step intervals. Tissues for 28 dpi were analyzed on a Bio-Rad Blue Diode confocal microscope using z-series at 1 μm step intervals. The spine images for the 28 dpi tissue were captured using 0.5 μm step intervals for the 60× oil lens with a digital zoom of 4. Images were deconvolved using the Deconvolution Lab plug-in for ImageJ with a Tikhonov-Miller algorithm. Maximum intensity projections were formed using ImageJ. The length of each dendritic segment was calculated by tracing the shaft, and the number of spines was counted using the Cell Counter plug-in for ImageJ. Spine density was then calculated by dividing the total number of spines by the length of the dendritic shaft in microns. Microfluidic cultures for testing conditioned media and recombinant SCG2 used 2 or 3 devices per experimental condition. We imaged 12 regions of each device using the same parameters on an epifluorescence microscope; then these images were analyzed for relative TUJ1/Sox2 expression by counting the number of cells and measuring Map2(+) dendritic branches using NeuronJ. Immunodepletion assays were done in duplicate; we used 5 devices for each experimental condition and 6 regions per device were included in the statistics.
Statistics.
All statistics for dendritic spine density analysis and average dendrite length counts were performed using a two-tailed Student's t test. For REST overexpression studies, average dendrite lengths were measured using ImageJ for control (n = 33) versus REST overexpression tissue (n = 19). Dendritic length measurements and percentage of neurons with dendrite measurements at 8 dpi were performed for shSCR (n = 30) and shREST tissue (n = 22). Overall spine density and mushroom spine density measurements were performed in shREST (n = 24) versus control tissue (n = 26). p values for immunofluorescence analysis and qRT-PCR analysis were calculated using a one-tailed t test.
Immunofluorescence and microscopy.
AHPs were fixed for 15 min in 4% PFA and stained with the following antibodies: anti-REST (Santa Cruz Biotechnology, C-15), anti-SOX2 (Cell Signaling Technology, 2748S), anti-β-tubulin III (Covance, MMS-435P), and anti-GFP (Aves Laboratories, 1020). DAPI was used to stain nuclei. Images were taken using confocal microscopy (Zeiss, LSM 710).
Cell culture.
Rat AHPs were cultured on plates coated with poly-ornithine and laminin. Basic fibroblast growth factor 2 (FGF2) was added for proliferation, and retinoic acid (RA) or FBS was added for differentiation, as described previously (Ray and Gage, 2006). Cells under differentiation conditions were cultured for 3–6 d after addition of RA. Cells were transfected using Nucleofector (Amaxa). Recombinant SCG2 (MW 70.8 kDa, NP_003460) was purchased from Origene and used at 20 ng/ml, resuspended in 0.1% BSA in PBS. AHPs were cultured in FGF2 medium and supplemented with recombinant SCG2 every other day. Phenotypes were monitored starting from 3 to 4 d until 6 d. Quantitative analysis was done after 6 d of culture.
Preparation of microfluidics-based compartmentalized culture.
The microfluidic devices for compartmentalized culture of AHPs were fabricated using photolithography for the master mold and soft lithography for replicated devices. Two layers of negative photoresist, with 3 μm high microgrooves and 100 μm high chambers, were generated on a silicon wafer using standard photolithography techniques. To replicate the microfluidic devices, a mixture of curing agent and poly-dimethyl-siloxane (Sylgard 184, Dow Corning) was cured on the master mold. The poly-dimethyl-siloxane replica was cut out, and the reservoirs were punched out before assembling on the glass coverslips. To improve cell adhesion, glass coverslips (24 × 40 mm, Fisher Scientific) were coated with 0.5 mg/ml poly-l-lysine (MW 70,000–150,000, Sigma), and then the devices were assembled. Before plating cells, laminin diluted in basal culture medium was added to the assembled devices. The density of cells was 1 × 106 cells/ml; 20 μl of dissociated cells was plated in the cell chamber. To test the non–cell-autonomous effect by a secretion process, either the conditioned medium or recombinant SCG2 was added to the treatment chamber opposite the cell chamber containing AHPs.
qRT-PCR.
Total RNA was extracted using Trizol LS (Invitrogen), and ∼1 μg of RNA was used as a template to make cDNA. Super Script III (Invitrogen, 18080051) was then used for first-strand synthesis using random hexamers. Real-time PCR was performed with cDNAs using the SYBR Green PCR Master Mix from Applied Biosystems (catalog #4309155). PCR was then performed in triplicate on an ABI qPCR machine. Analysis was performed by normalizing Ct values to GAPDH. The following primers were used: REST forward, TCATCTAACTCGACACATGCG; REST reverse, ACAGTGAGGGCAATTAAGAGG; GAPDH forward, GGGTGGGAACCACGAGAAAT; GAPDH reverse, ATGGCATGGACTGTGGTCAT; Neurod1 forward, AGGCTCCAGGGTTATGAGATCGT; Neurod1 reverse, CGCTCTCGCTGTATGATTTGGTCA; Scg2 forward, CCAATGGTCATGGTATTGACA; and Scg2 reverse, TTTGCTCCAGAACTCCACAA.
Western blotting.
Nondenaturing extracts prepared using Triton X-100 were resuspended in 5 mm DTT and 4× NuPage SDS loading buffer and denatured at 95°C for 5–7 min. Samples were run on a 3%–8% Tris-acetate gel with the Novex Pre-Stained Sharp Marker (LC5800) in a Tris-acetate buffer system. The antibody used against REST was from Millipore (07-579). The other antibodies used were mouse anti-α-tubulin (Cell Signaling Technology, 2144S) and mouse anti-GAPDH (Fitzgerald).
shRNA and vector construction.
shRNA sequences were as follows: SCR, CGAGGGCGACTTAACCTTAGG; rat and mouse REST, AATTAAGAGGTTTAGGCCC; mouse REST, TAGAACTCCTGCATTTGTCTTC; and mouse Scg2, TCATACATGAGAGGGAACCG. shRNAs were then cloned into a retrovirus vector for U6 promoter-driven shRNA expression. The vector also expressed GFP driven by CAG promoter to track cells expressing shRNA.
RNA preparation, microarray analysis, and semiquantitative RT-PCR.
Transfected AHP cells were sorted for GFP+ cells on a Becton-Dickinson FACS Vantage SE DiVa. Trizol LS was used to extract total RNA from sorted cells. Samples were run on GeneChip Rat Gene 2.0 ST Arrays (Affymetrix). Partek Genomics Suite with RMA settings was used to analyze the microarray data. Gene ontology term analysis was done using the DAVID software (Huang da et al., 2009).
Chromatin immunoprecipitation.
Cells were treated with formaldehyde at a final concentration of 1%, scraped, pelleted, lysed in lysis buffer (1% SDS, 10 mm EDTA, and 50 mm Tris-HCl, pH 8.1) containing protease and phosphatase inhibitors (3 mm PMSF, 5 mg/ml aprotinine, 1 mg/ml pepstatin A, Wako Chemicals; and 2 mm sodium orthovanadate), and then incubated for 5 min on ice. Cell lysates were sonicated until the DNA fragments were 500–800 bp in length; 1% of the total volume was stored as input at −20°C until use. Immunoprecipitation was performed at 4°C overnight with 1 μg antibody/4–5 million cells after samples were precleared with beads (GE Healthcare). Immune complexes were collected by protein A Sepharose beads and washed with the following buffers: low salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 150 mm NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 500 mm NaCl), LiCl buffer (0.25 m LiCl, 1% NP-40, 1% deoxycholate, 1 mm EDTA, and 10 mm Tris-HCl, pH 8.1), and TE buffer (10 mm Tris-HCl and 1 mm EDTA, pH 8.0). DNA was further incubated with proteinase K (Roche) and RNase A (Invitrogen), purified by phenol extraction, and precipitated with ethanol. DNA pellets were dissolved in sterile H2O and used as a template for PCR with primers that amplified potential binding sites. Primers were designed using the Primer 3 software.
ELISA.
SCG2 protein levels in media collected from shSCR and shREST transfected cell culture were measured using a quantitative sandwich enzyme immunoassay kit (CUSABIO Biotech). Immobilized anti-SCG2 antibody onto a microplate detected SCG2 protein present in the medium. A biotin-conjugated antibody specific for SCG2 followed by avidin-conjugated HRP was added to the wells. Any unbound avidin-enzyme reagent was washed; a substrate solution was next added to the wells to develop a color in proportion to the amount of SCG2 bound. After stopping the color development, the intensity of the color was measured.
Electrophysiology.
Hippocampal neurons were cultured from E18 Wistar rats (Charles River) of both sexes. Neurons were isolated by mechanical dissociation following incubation in 0.5% trypsin (Thermo Scientific) in HBSS + 10 mm HEPES for 25 min at 37°C. Cells were then centrifuged for 4 min at 1.4 k rpm and then resuspended in rat Nucleofector Solution (Lonza). Cells were transfected with either shREST or shSCR constructs using an Amaxa Nucleofector Device (Lonza) and then plated onto poly-d-lysine-coated glass coverslips (Millipore). Cultures were maintained for 2–3 weeks in Neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen) and 1% glutamax. For the conditioned media experiments, cells were fed half fresh medium and half conditioned medium from either shREST- or shSCR-transfected neural cultures. Miniature excitatory post-synaptic current (mEPSC) activity was recorded using an Axopatch 200B amplifier. Cells were perfused in ACSF solution containing the following (in mm): 119 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, 11 glucose, 0.1 picrotoxin, and 0.001 tetrodotoxin. Three to 5 mΩ glass pipettes were filled with a Cs-Gluc internal solution containing the following (in mm): 130 CsOH, 130 d-gluconic acid, 5 NaCl, 12 phosphocreatine, 10 HEPES, 10 EGTA, 3 MgATP, 0.2 NaGTP, pH 7.3. mEPSCs were recorded at −70 mV and analyzed using Clampfit 10 (Molecular Devices).
Immunodepletion.
Rabbit anti-SCG2 antibody was purchased from Protein Tech. Conditioned media were obtained from AHPs transfected with shREST. Anti-SCG2 and control rabbit antibody were used for immunodepletions. Supernatants from immunoprecipitations were transferred to the treatment chambers of microfluidic devices for branching assays.
Results
REST knockdown promotes differentiation of AHPs in vitro
AHPs isolated from the hippocampus of adult rats have been instrumental in investigations of adult neurogenesis processes (Gage, 2000; Ray and Gage, 2006). We used these cells to confirm the presence and functionality of REST in neural progenitor populations in vitro. REST colocalized with Nestin and Sox2 in cultured AHPs under proliferation condition (Fig. 1A). Chromatin IP was used to confirm that known and predicted targets of REST, both coding and noncoding, were bound by REST (Fig. 1B). REST bound to a predicted target, miR-153, which in turn regulated SNAP25, a major REST target (Wei et al., 2013).
Figure 1.

REST knockdown accelerates differentiation of AHPs. A, REST is present in AHPs in vitro, as shown by immunofluorescent staining in the presence of FGF2. Cells express REST (P-18, red), Nestin (green), and Sox2 (cyan). B, REST binds to target RE-1 sites (BDNF, Scg10 and miR-153) in AHPs. Chromatin IP using rabbit anti-REST, anti-Histone3 antibody as a positive control and with nonspecific rabbit IgG as a negative control. C, REST knockdown and overexpression, including antibody confirmed by Western blot; anti-REST antibody (from Millipore, 09–019) detects REST band at 180 kDa. D, REST knockdown results in increased differentiation of AHPs in vitro. Immunofluorescent staining of AHPs transfected with shSCR or shREST and cultured with FGF2. Green represents cells expressing shRNA. Red represents TUJ1. E, Immunofluorescent staining of AHPs transfected with shSCR or shREST cultured in RA for 4 d. Color representations are as in D. F, REST knockdown results in increased neurite outgrowth (white represents shSCR; black represents shREST). *p < 0.05. G, H, REST knockdown results in increased neuronal differentiation, as quantified by both neurite outgrowth and percentage of GFP+/β-tubulin III+ cells (white represents shSCR; black represents shREST); these were strong trends that did not reach statistical significance. I, REST knockdown leads to a decrease in REST transcripts and a significant increase in Neurod1 transcripts as measured by qRT-PCR. *p < 0.05. J–L, Knockdown of REST leads to an increase in mEPSC signaling in rat primary hippocampal neurons. Primary neurons were transfected with either shREST or shSCR constructs at embryonic day 18 (E18), then recorded from 2 to 3 weeks later. J, shREST neurons (n = 16) exhibited enhanced mEPSC amplitudes (mean 17.9 ± 3.1 pA) compared with shSCR-transfected neurons (n = 18, mean 10.7 ± 1.2 pA). *p < 0.05 (Student's t test). K, shREST neurons also showed an increase in mEPSC frequency (mean 6.5 ± 1.0 Hz) compared with shSCR neurons (mean 3.2 ± 0.5 Hz). **p < 0.005 (Student's t test). L, Example of mEPSC events recorded from an shSCR- and a shREST-transfected hippocampal neuron.
To manipulate the expression level of REST in AHPs, both shRNA-mediated knockdown and overexpression constructs were prepared. We also used these constructs to confirm the identity of REST as the 180 kDa protein in these cells (Fig. 1C). The identity of the 180 kDa protein as REST was further verified by immunoprecipitation and subsequent mass spectrometry (data not shown). REST knockdown resulted in increased neuronal differentiation even under proliferating conditions (FGF2) within 60 h, as measured by an increase in β-tubulin III (TUJ1) staining (Fig. 1D). This effect was also observed upon differentiation in the presence of RA (Fig. 1E). REST knockdown caused a more than fourfold increase in the length of neurites in the presence of FGF2 and almost a twofold increase in the presence of RA (Fig. 1F,G). These results are consistent with the finding in a Down syndrome transgenic mouse model cell line that REST negatively regulated a number of genes involved in neurite formation (Lepagnol-Bestel et al., 2007). There was more than a 30% increase in β-tubulin III+ cells upon knockdown in different differentiation media, as quantified by GFP+/β-tubulin III+ double-labeled cells (Fig. 1H). In addition, the REST target gene Neurod1, an essential regulator of adult neurogenesis (Gao et al., 2009), was upregulated in AHPs upon knockdown of REST in proliferating conditions (Fig. 1I).
To functionally investigate the effect of REST knockdown on early neuronal differentiation, we used dissociated embryonic (E18) rat primary hippocampal neurons in vitro to record mEPSCs. Consistent with prior findings of an increase in dendritic development, loss of REST expression led to a marked upregulation in both mEPSC amplitude (p < 0.05) and frequency (p < 0.005) after 2–3 weeks in vitro (Fig. 1J–L). Together, these data suggest that reduction in REST expression promotes enhanced morphological and functional maturation in young neurons.
Secreted proteins downstream of REST promote differentiation and maturation of AHPs non–cell-autonomously
Which genes does REST regulate in AHPs? While RE-1 sequence-bearing neuronal genes are preferentially downregulated by REST, the actual target genes are context-dependent (Johnson et al., 2008). Thus, to gain a molecular signature associated with the changes upon REST knockdown, we performed gene expression analysis in AHPs. Gene ontology term analysis from our microarray data identified secretory genes as a class that is enriched upon REST knockdown (Table 1). Therefore, we hypothesized that secretory processes might contribute to adult neuronal progenitor differentiation non–cell-autonomously.
Table 1.
GO term analysis of microarray profile after REST knockdown
| Biological process | p | Benjamini | Cellular component/molecular process | p | Benjamini |
|---|---|---|---|---|---|
| Cellular component | |||||
| Transmission of nerve impulse | 1.20E-06 | 3.70E-04 | Synapse | 2.10E-06 | 3.90E-04 |
| Secretion by cell | 1.30E-06 | 3.30E-04 | Cell junction | 6.30E-04 | 1.70E-02 |
| Cell–cell signaling | 7.00E-06 | 1.50E-03 | Cytoplasmic vesicle | 1.60E-03 | 2.60E-02 |
| Neurotransmitter transport | 8.20E-06 | 1.50E-03 | Neuron projection | 5.30E-03 | 6.90E-02 |
| Exocytosis | 1.90E-04 | 2.30E-02 | Synaptosome | 8.30E-03 | 9.30E-02 |
| Cell projection organization | 3.30E-04 | 3.70E-02 | |||
| Neuron differentiation | 8.10E-04 | 7.70E-02 | |||
| Regulation of neuronal synaptic plasticity | 1.60E-03 | 1.20E-01 | |||
| Intracellular signaling cascade | 2.70E-03 | 1.80E-01 | Molecular process | ||
| MAPKKK cascade | 3.50E-03 | 2.10E-01 | SNARE binding | 3.80E-05 | 1.10E-02 |
| Cell surface receptor linked signal transduction | 5.80E-03 | 3.00E-01 | Syntaxin binding | 2.90E-04 | 2.80E-02 |
| Synaptic vesicle transport | 8.40E-03 | 3.90E-01 | Nucleotide receptor activity | 2.40E-03 | 1.60E-01 |
| Stress-activated protein kinase signaling pathway | 9.60E-03 | 4.00E-01 | Cytoskeletal protein binding | 7.80E-03 | 3.70E-01 |
| Behavior | 1.20E-02 | 4.50E-01 | Calcium ion binding | 1.00E-02 | 4.00E-01 |
| Chemical homeostasis | 1.20E-02 | 4.50E-01 | Magnesium ion binding | 1.20E-02 | 4.00E-01 |
| Axonogenesis | 1.20E-02 | 4.40E-01 | Nucleotide receptor activity, G-protein coupled | 2.20E-02 | 5.10E-01 |
| Vesicle-mediated transport | 1.50E-02 | 4.90E-01 | Gated channel activity | 2.30E-02 | 4.90E-01 |
| Cell morphogenesis involved in neuron differentiation | 2.00E-02 | 5.70E-01 | Ion channel activity | 2.50E-02 | 4.50E-01 |
| Protein kinase cascade | 2.00E-02 | 5.60E-01 | Substrate specific channel activity | 2.90E-02 | 4.50E-01 |
| Regulation of membrane potential | 2.10E-02 | 5.60E-01 | |||
| Cellular component | |||||
| Response to wounding | 4.10E-05 | 5.20E-02 | Extracellular space | 3.60E-05 | 6.20E-03 |
| Taxis | 5.50E-05 | 3.50E-02 | Extracellular region part | 4.10E-05 | 3.50E-03 |
| Locomotory behavior | 1.80E-04 | 7.50E-02 | Proteinaceous extracellular matrix | 5.70E-02 | 9.20E-01 |
| Regeneration | 4.70E-04 | 1.40E-01 | Extracellular matrix | 9.40E-02 | 9.70E-01 |
| Behavior | 4.90E-04 | 1.20E-01 | Endoplasmic reticulum | 9.90E-02 | 9.50E-01 |
| Response to glucocorticoid stimulus | 6.60E-04 | 1.30E-01 | |||
| Response to organic cyclic substance | 9.00E-04 | 1.50E-01 | |||
| Response to corticosteroid stimulus | 9.20E-04 | 1.40E-01 | Molecular process | ||
| Cell motion | 1.30E-03 | 1.70E-01 | Cytokine activity | 1.50E-03 | 1.40E-01 |
| Response to mechanical stimulus | 2.20E-03 | 2.30E-01 | Olfactory receptor activity | 9.50E-03 | 5.10E-01 |
| Neuron differentiation | 3.00E-03 | 2.80E-01 | Calcium ion binding | 1.90E-02 | 7.00E-01 |
| Bone development | 3.50E-03 | 2.80E-01 | Glutathione transferase activity | 3.40E-02 | 7.80E-01 |
| Immune response | 3.70E-03 | 2.80E-01 | O-acyltransferase activity | 5.00E-02 | 8.30E-01 |
| Inflammatory response | 4.20E-03 | 2.90E-01 | Enzyme inhibitor activity | 5.30E-02 | 8.10E-01 |
| Positive regulation of anti-apoptosis | 4.30E-03 | 2.80E-01 | Growth factor activity | 6.60E-02 | 8.20E-01 |
| Regulation of cell proliferation | 5.30E-03 | 3.10E-01 | Metalloenzyme Regulator activity | 7.60E-02 | 8.40E-01 |
| Detection of stimulus | 5.60E-03 | 3.00E-01 | Endopeptidase inhibitor activity | 8.60E-02 | 8.60E-01 |
| Detection of chemical stimulus | 6.00E-03 | 3.10E-01 | Carbohydrate binding | 8.70E-02 | 8.40E-01 |
| Cognition | 7.20E-03 | 3.50E-01 | |||
| Biological adhesion | 8.80E-03 | 3.70E-01 |
To tease out the non–cell-autonomous effect of REST knockdown, we introduced a shREST vector-expressing mCherry into AHPs that were stably expressing GFP (Fig. 2A). Interestingly, not only shREST cells (red or yellow) but also neighboring cells (green-only) that did not uptake the shRNA-mCherry plasmid had increased neurite outgrowth. This effect was specific to REST knockdown, as the increased neurite outgrowth was not observed in the neighbors of shSCR-transfected cells (Fig. 2A, white arrow), suggesting that REST knockdown potentially mediates non–cell-autonomous signaling.
Figure 2.
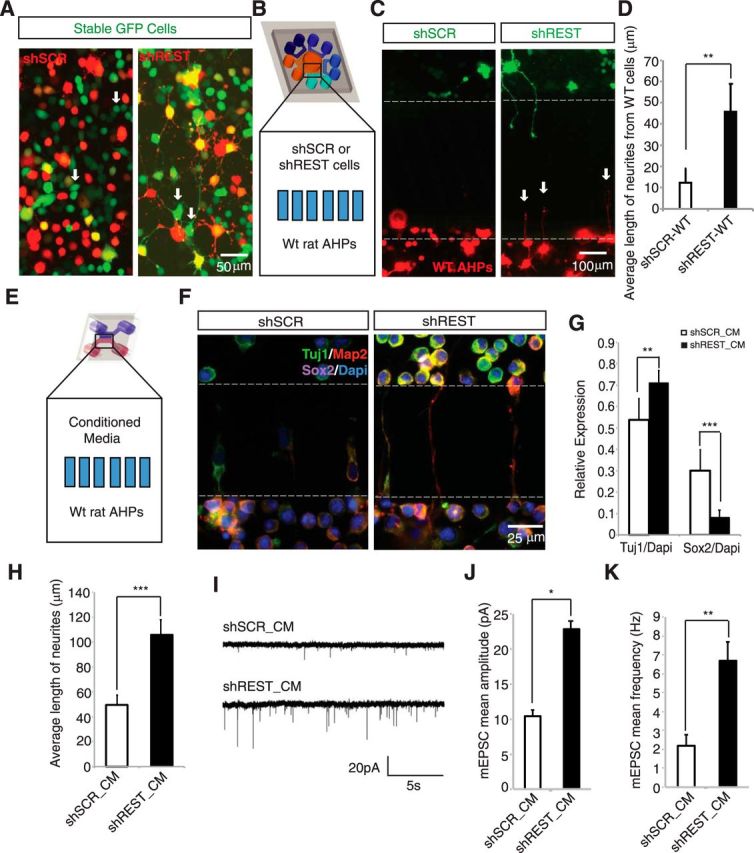
Conditioned medium from REST knockdown cells promote differentiation and maturation of AHPs non–cell-autonomously. A, A non–cell-autonomous effect was observed for REST knockdown cells. AHPs stably expressing GFP were transfected with shREST mCherry or shSCR mCherry plasmid. Electroporation efficiency was not 100%; thus, cells that did not take up the shRNA plasmids are green, whereas transfected cells are yellow. Green cells with neurites are seen in shREST but not in shSCR cultures (indicated by white arrows). B, Microfluidic compartmentalized coculture of either shSCR- or shREST-GFP transfected cells with wild-type (WT) cells-mCherry. Multicompartment design was introduced to see the non–cell-autonomous effect from shSCR and shREST toward the same population of WT cells simultaneously. C, Treatment chamber was plated with either shSCR- or shREST-GFP-transfected AHPs, and the response of WT AHPs (red cells) was monitored. WT AHPs showed neuronal morphology when the treatment chamber contained shREST-transfected cells (white arrows). D, Average length of processes from WT AHPs upon coculture with either shSCR or shREST cells (n = 12). **p < 0.005. E, Scheme showing two-chambered microfluidic device. In the top chamber (treatment chamber), we added conditioned medium from culture of shSCR- or shREST-transfected cells; WT AHPs were plated in the bottom chamber (cell chamber). F, shREST conditioned medium promotes differentiation of WT AHPs. Conditioned media were collected after 4 d of culture of shSCR- and shREST-transfected cells and then added to the treatment chamber for culturing WT AHPs (cell chamber). Map2-positive neurites (red) can be seen in the microgrooves in the shREST but not in the shSCR cultures. Green represents TUJ1. Magenta represents Sox2. Cells in the upper chamber (treatment chamber) result from migration through the microgrooves in both shSCR and shREST conditioned media. G, shREST conditioned medium (shREST_CM) caused an increase in TUJ1 expression (**p < 0.005, n = 24), whereas a reverse effect was observed for Sox2 expression (***p < 0.0001, n = 24). H, Average neurite length of AHPs cultured in conditioned medium taken from either shSCR- or shREST-transfected cells. A significant increase (***p < 0.0001, n = 50 for shREST_CM and n = 72 for shSCR_CM) in the average neurite length was observed in the treatment with shREST_CM in Map2(+) cells. I, Example of traces of mEPSCs recorded from primary neurons treated with conditioned medium from shSCR- or shREST-transfected cells. J, Treatment with shREST_CM led to a significant increase in mEPSC amplitude (mean 22.8 ± 4.3 pA, n = 10) compared with cells treated with shSCR_CM (mean 10.4 ± 0.8 pA, n = 8). *p < 0.05 (Student's t test). K, Cells treated with shREST_CM also exhibited an increase in mEPSC frequency (mean 6.7 ± 1.0 Hz) compared with shSCR_CM-treated cells (mean 2.2 ± 0.6 Hz). **p < 0.005 (Student's t test).
To confirm this non–cell-autonomous phenotype, we designed microfluidic coculture platforms to maintain controlled diffusion of secreted molecules. The platforms consisted of either 2 or multiple chambers (100 μm high) that were connected via a series of microgrooves (3 μm high). These microgrooves control the diffusion of soluble molecules via fluidic resistance. This setup allowed us to monitor how shREST cells influenced the differentiation of neighboring cells non–cell-autonomously. As shown in Figure 2B, wild-type AHPs were plated in the cell chamber (center chamber), which was connected via microgrooves to surrounding treatment chambers containing either shREST or shSCR cells. Wild-type AHPs in the cell chamber exhibited enhanced neurite outgrowth when shREST cells were in the treatment chamber, compared with those cultured with shSCR cells in the treatment chamber (Fig. 2C,D). Therefore, our results suggest that non–cell-autonomous activity of REST may play a role in neuronal differentiation.
To eliminate possible cell–cell contacts by either migrating cells or outgrowing neurites, we treated wild-type AHPs (the bottom chamber) with the conditioned medium (the top chamber) from cultures of either shSCR- or shREST-transfected cells (Fig. 2E). We found that adding conditioned medium taken from shREST-transfected AHPs to the treatment chamber was sufficient to enhance differentiation and maturation in the wild-type cells (Fig. 2F–K). In Figure 2G, H, these effects were quantified by measuring the average length of neurites and the relative expression of TUJ1 and Sox2. We observed a significant increase in TUJ1 expression (p < 0.005; Fig. 2G) and neurite length (p < 0.0001; Fig. 2H), whereas Sox2 expression decreased significantly (p < 0.0001; Fig. 2G) in AHPs cultured in conditioned medium from shREST-transfected cells compared with shSCR-transfected cells. These results indicate that the secreted components from the shREST-transfected AHPs are involved in the differentiation of neighboring wild-type AHPs.
To assess whether REST-regulated secreted factors influenced functional maturation, we performed electrophysiological recordings after treatment of isolated hippocampal neurons with conditioned medium from either shSCR- or shREST-transfected cells (Fig. 2I–K). Compared with the control cultures (CM_shSCR), neurons exposed to shREST-conditioned medium displayed significantly upregulated mEPSC amplitude (p < 0.05; Fig. 2J) and frequency (p < 0.005; Fig. 2K). Thus, the addition of conditioned medium from shREST-transfected cells is sufficient to enhance neuronal differentiation and spontaneous synaptic activity in hippocampal neurons non–cell-autonomously.
REST represses Scg2 in AHPs
The dramatic morphological changes observed upon REST knockdown were associated with a large number of changes at the mRNA level in our gene expression analysis (Fig. 3A). We focused our attention on the top upregulated genes expressed upon REST knockdown. Although a number of genes involved in neuronal processes were upregulated upon REST knockdown, a secreted gene, Scg2, stood out as the most significantly changed gene with an RE-1 element in its promoter region (Table 2). We confirmed the upregulation of Scg2 upon REST knockdown by qRT-PCR (data not shown). Using chromatin IP, we further confirmed that REST directly bound to the RE-1 sequence in the Scg2 promoter (Fig. 3B) and regulated its expression. Furthermore, we observed increased levels of SCG2 protein secreted in the medium upon REST knockdown. The relative amount of SCG2 present in the conditioned medium from REST knockdown AHPs was >4 times higher than that from control shSCR cells (Fig. 3C). These results show that REST directly represses Scg2 expression.
Figure 3.

REST represses Scg2 directly by binding to RE-1 sequence in the Scg2 promoter. A, The top 25 upregulated and downregulated genes upon REST knockdown in AHPs. * Denotes genes with RE-1 sequences in their promoter regions. B, Chromatin IP analysis shows REST binding to the Scg2 promoter. Primers designed to amplify the RE-1 site in the Scg2 promoter as well as two negative control sites (5 and 2 kb away) were used to assay REST binding. BDNF and Nav1.2 promoter primers were used as positive controls for REST binding. C, Relative amounts of SCG2 protein secreted in the conditioned medium measured by ELISA. **p = 0.0005.
Table 2.
List of upregulated and downregulated genes upon REST knockdown that code for secreted proteins
| Secreted | Fold increase or decrease |
|---|---|
| Upregulated | Increase |
| Scg2 | 21.71 |
| Vgf | 5.19 |
| Chgb | 4.51 |
| Hapln2 | 4.37 |
| Mmp24 | 3.89 |
| Resp18 | 3.76 |
| Scg3 | 3.75 |
| Calca | 2.85 |
| Ptgds | 2.81 |
| Nxph1 | 2.6 |
| Egflam | 2.59 |
| Cdnf | 2.31 |
| Nell2 | 2.1 |
| Enpp2 | 2.07 |
| Downregulated | Decrease |
| Ecm1 | 4.36 |
| Plaur | 4.31 |
| Kitlg | 4.02 |
| Ccl7 | 3.57 |
| Cxcl2 | 3.4 |
| Defb15 | 3.14 |
| Vcan | 3.05 |
| Prl6a1 | 2.86 |
| C1qtnf1 | 2.79 |
| Lif | 2.76 |
| Ccl20 | 2.66 |
| Cxcl10 | 2.63 |
| Spon2 | 2.62 |
| Masp1 | 2.37 |
| Lgi1 | 2.31 |
| Ccl2 | 2.2 |
| Ngf | 2.14 |
| Igf1 | 2.12 |
| Mmp9 | 2.11 |
| Cyss | 2.1 |
| Stc1 | 2.09 |
| Mmp11 | 2.08 |
| Chrdl1 | 2.06 |
| Timp1 | 2.02 |
Is Scg2 necessary for neuronal differentiation downstream of REST? To test whether Scg2 knockdown would rescue the cell-autonomous differentiation phenotype upon REST knockdown, we generated an Scg2 shRNA (shScg2) construct that led to efficient knockdown (data not shown). We transfected AHPs with different combinations of shSCR, shREST, and shScg2 and cultured them in the presence of FGF2 (Fig. 4). Cells transfected with control shRNAs showed minimal differentiation (Fig. 4A). Transfection of shREST into AHPs led to increased TUJ1 expression. Introduction of a shREST/shScg2 double knockdown caused a reduction in the percentage of GFP+TUJ1+ cells (Fig. 4B), suggesting that Scg2 is required for neuronal differentiation acting directly downstream of REST knockdown to induce the differentiation phenotype in these AHPs.
Figure 4.
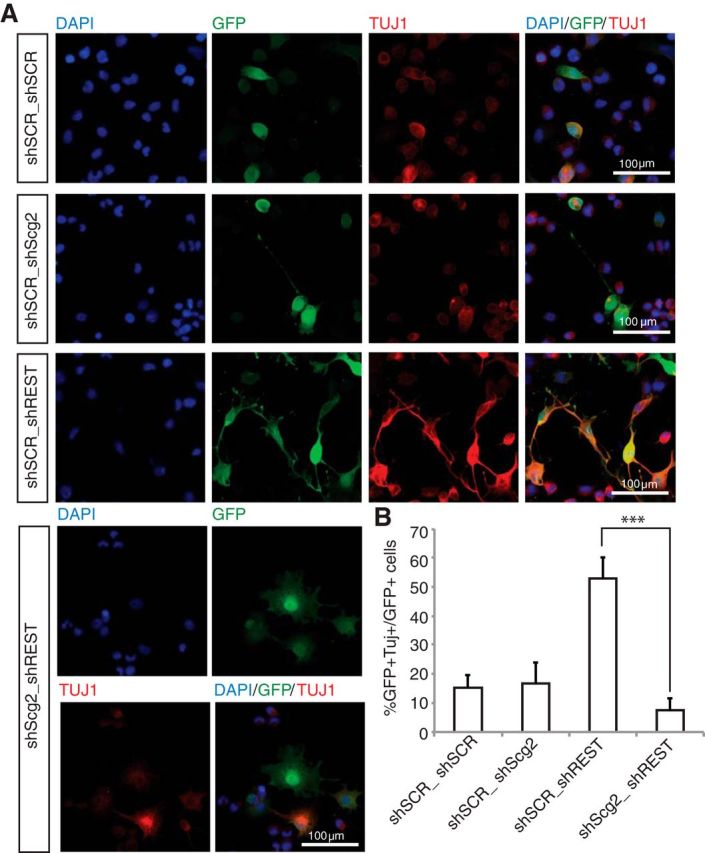
Scg2 upregulation is required downstream of REST for differentiation of AHPs. A, REST-Scg2 double knockdown rescues REST knockdown phenotype. Immunofluorescent staining of AHPs transfected with shSCR/shSCR, shSCR/shREST, shSCR/shScg2, or shREST/shScg2 cultured in the presence of FGF2 for 4 d. Green represents cells expressing shRNA. Blue represents DAPI. Red represents TUJ1. B, REST/Scg2 double knockdown results in decreased neuronal differentiation compared with REST knockdown alone, as quantified by percentage of GFP+/β-tubulin III+ cells. ***p < 5.68 × 10−7.
SCG2 is the major non–cell-autonomous factor affecting differentiation and maturation of AHPs downstream of REST
Following the identification of Scg2 as a prime candidate, we investigated whether this secretory molecule was sufficient to induce the non–cell-autonomous differentiation and maturation of AHPs. When AHPs were treated with SCG2 recombinant protein (top treatment chamber), we observed significantly increased neurite length (p < 0.0005) along with enhanced branching (Fig. 5A–D). Interestingly, AHPs started to differentiate upon application of SCG2, even though they were maintained in proliferation medium (FGF2). In addition, secondary, tertiary, and quaternary dendritic processes were highly developed following SCG2 treatment, whereas this phenotype was not observed in the control experiment. There were also functional consequences of the interplay between REST and SCG2. shSCR-transfected hippocampal neurons treated with SCG2 generated enhanced mEPSC signaling. SCG2 treatment enhanced the amplitude of mEPSCs in primary neurons, from a mean ± SE of 10.7 ± 1.2 pA in control cells (n = 18) to 16.6 ± 1.7 SE in SCG2-treated cells (n = 13) (p < 0.01). Although SCG2-treated cells also showed a tendency toward increased mEPSC frequency (6.4 ± 2.0 Hz) compared with controls (3.2 ± 0.5 Hz), this trend fell short of being significant (p = 0.08) (data not shown). Furthermore, SCG2 treatment on shSCR cells generated a comparable response following REST knockdown; indeed, treating shREST-transfected neurons with SCG2 had no additional effect upon mEPSC signaling (Fig. 5E–G).
Figure 5.

Exogenous SCG2 induces differentiation and maturation of AHPs. A, Recombinant SCG2 was added in the top chamber (treatment chamber) of a microfluidic device; WT AHPs were plated in the bottom chamber (cell chamber). B, Effect of recombinant SCG2 treatment (20 ng/ml) on AHPs. Recombinant SCG2-treated or untreated AHPs were stained with anti-TUJ1 (green), anti-Map2 (red), and DAPI (blue). C, Treatment with SCG2 promoted significant neurite outgrowth. ***p < 0.0005. n = 80. D, SCG2 promoted neurite branching of wild-type AHPs. The number of neurite sub-branches increased upon treatment with recombinant SCG2. E, Example of mEPSC recordings taken from shSCR_SCG2- and shREST_SCG2-treated neurons. F, Treating shSCR primary neurons with SCG2 (10 ng/ml) was sufficient to increase mEPSC amplitude to a level comparable with that of shREST-transfected neurons (shSCR_SCG2: n = 13, mean 17.0 ± 2.1 pA; shREST_SCG2: n = 8, mean 19.0 ± 1.7 pA). G, SCG2 treatment also abolished the difference in mEPSC frequency between shSCR- and shREST-transfected neurons (shSCR_SCG2: mean 6.4 ± 2.0 Hz; shREST_SCG2: mean 5.8 ± 1.7 Hz). H, Schematic representation of the microfluidic device. I–K, Fold changes in neurite outgrowth (length and branches) upon different combinations of double knockdown. AHPs were transfected with two different constructs encoded by fluorescent colors (mCherry and GFP) for each of the combinatory double knockdown setups (shSCR/scSCR, shSCR/shScg2, shREST/shSCR, and shREST/shScg2) and then cultured in the presence of FGF2 for 5 d. *p < 0.05. **p < 0.005. ns, Not significant.
To further investigate the non–cell-autonomous function of SCG2, we analyzed the neuronal differentiation of AHPs in a more controlled environment using REST/Scg2 double knockdown and recombinant SCG2 in microfluidic devices. We measured the average length and the number of branches per cell, presented as fold changes compared with the control set (n = 22–30; Fig. 5H–K). Loss of Scg2 caused a significant decrease in neurite length and number of branches per cell. Recombinant SCG2 treatment rescued this reduction, reverting back to levels comparable with those in the control cells (Fig. 5I). Indeed, AHPs with loss of Scg2 were unable to initiate differentiation even upon REST knockdown, exhibiting a significant decrease in both neurite length and number of branches per cell (Fig. 5J). Interestingly, recombinant SCG2 treatment could rescue the reduced differentiation phenotype and led to a significant increase in both neurite length and branches per cell, demonstrating an enhancement of differentiation phenotype (Fig. 5J). The REST knockdown phenotype of increased outgrowth and branching of neurites was also enhanced by exogenous recombinant SCG2 treatment (Fig. 5K). Moreover, we observed a greater effect on neurite branching compared with neurite length upon exogenous SCG2 treatment of REST knockdown cells (Fig. 5J,K; also compare with Fig. 5D). Together, these results suggest that exogenous SCG2 rescued the loss of differentiation phenotype of Scg2 knockdown, indicating an essential role for REST-Scg2 regulation controlling the dendritic development of AHPs.
SCG2 is required, but is it essential for the enhanced differentiation of neighboring cells in a non–cell-autonomous manner? To answer this question, we treated the wild-type AHPs with SCG2-immunodepleted conditioned medium from shREST-transfected cells (Fig. 6A, blue top side). Immunodepletion using anti-SCG2, but not a control antibody, removed the differentiation-enhancing activity from the conditioned medium (Fig. 6B). Compared with control cultures, TUJ1 expression was significantly decreased (p < 0.0001), whereas Sox2 expression was increased in SCG2-immunodepleted cultures (Fig. 6B,C). The average length of neurites also decreased significantly upon SCG2 immunodepletion (Fig. 6D), and SCG2-immunodepleted cultures failed to develop sub-branches, such as secondary, tertiary, and quaternary dendritic processes (Fig. 6E).
Figure 6.
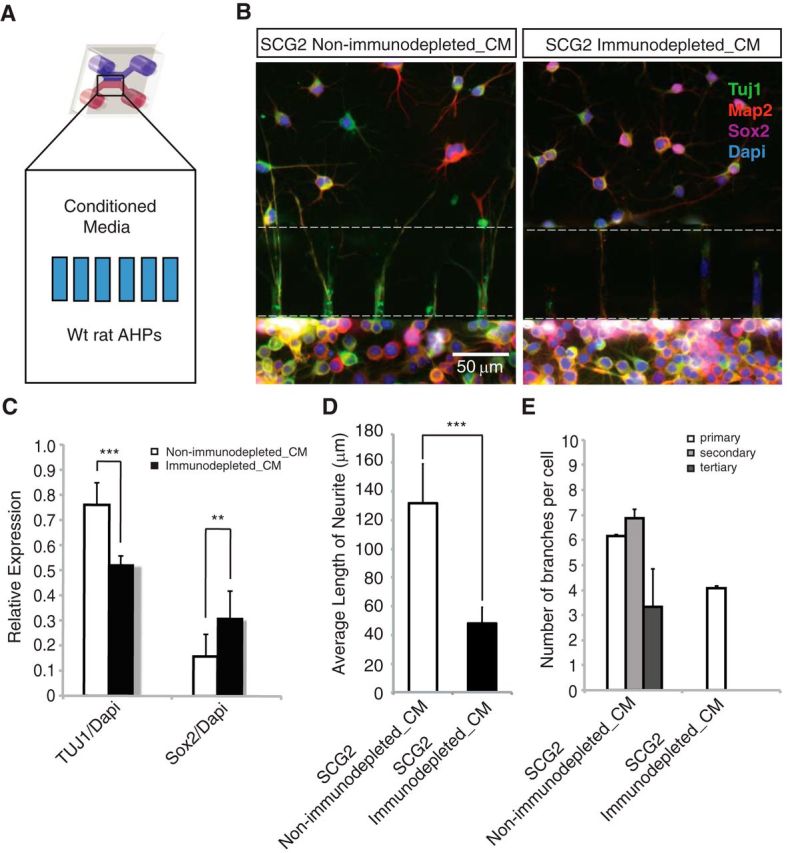
SCG2 is necessary and mediates the non–cell-autonomous effects of REST knockdown. A, In the top chamber of the microfluidic device (treatment chamber), conditioned medium was added; WT AHPs were plated in the bottom chamber (cell chamber). B, Anti-SCG2, but not control antibodies, can deplete the differentiation-inducing activity in shREST-conditioned medium. Conditioned medium from control antibody enhanced differentiation. C, An increase in Sox2 (**p < 0.001) and a decrease in TUJ1 (***p < 0.0001) expression were observed upon immunodepletion (n = 24). D, Average length analysis of neurites in immunodepletion experiments. A significant decrease in neurite length (***p < 0.0001) was observed upon SCG2 immunodepletion. E, Sub-branches of neurites are observed only in SCG2 non-immunodepleted samples.
Overall, our in vitro results suggest that SCG2 is the main factor responsible for increased differentiation in a non–cell-autonomous manner following REST knockdown, and it may play an important role in the maturation of adult hippocampal progenitors.
In vivo function of REST and Scg2 in neuronal differentiation and maturation
Hsieh and colleagues (Gao et al., 2011) previously demonstrated the presence of REST protein in the DG and its role as a regulator of neurogenesis driven by quiescent adult NSCs in vivo. This elegant study showed a requirement for REST function in these cells. Our in vitro results suggested a novel role of REST in promoting differentiation and maturation of neural progenitor cells. To test this hypothesis in vivo, we manipulated REST expression and analyzed its effect on neural progenitors in the DG. We first confirmed the expression of REST in the adult mouse hippocampus by immunohistochemical analysis. REST-positive cells were enriched in the subgranular zone of the DG (Fig. 7A). REST expressing-NeuN(+) mature granule neurons were also observed within the DG. Additionally, we manipulated REST expression in proliferative neural progenitors to examine the role of REST on the early differentiation of newborn granule cells in vivo. Neural progenitors in the DG were specifically targeted using a GFP-expressing retrovirus as described in Materials and Methods. Overexpression of REST resulted in decreased differentiation of cells in vivo. As shown in Figure 7B–D, a significant decrease in both the numbers and the lengths of the dendrites was observed in GFP-positive cells at 14 dpi. In contrast, REST knockdown at 8 dpi showed a significant (p < 0.004) increase in dendrite length compared with controls (Fig. 7E–G). The percentage of total neurons with dendrites upon infection with shSCR (sh-scrambled) (n = 30) or shREST (n = 22) was quantified. The number of neurons exhibiting dendrites was threefold higher in shREST-infected cells compared with the shSCR-infected controls (Fig. 7F), and more than a fourfold increase in the length of dendrites was observed upon REST knockdown (Fig. 7G).
Figure 7.
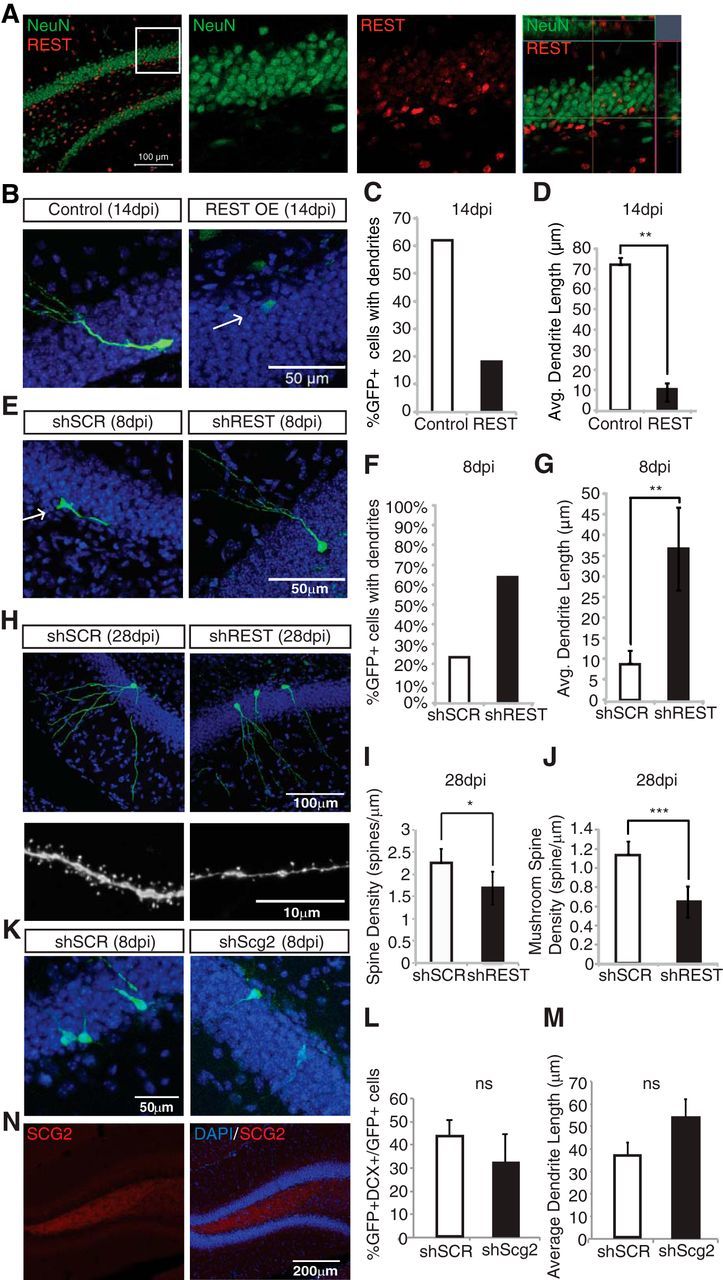
REST regulates neural differentiation in vivo. A, REST is enriched in the subgranular zone of the DG. Individual stains along with merged images are shown. The merged images are shown at lower magnification, and a portion of a boxed region is magnified. The magnified area is shown both as individual stains and as an orthogonal view of the merged stains. Immunohistochemical analyses of adult mouse DG using anti-REST (red) and NeuN (green). B, REST overexpression (REST-IRES-GFP) prevents differentiation of newborn neurons in vivo (14 dpi). Arrow indicates cells that failed to mature. C, D, REST overexpression results in a significant decrease in both the number of cells with dendrites and dendrite length in vivo. Average dendrite lengths measured using ImageJ are shown for control (n = 33) versus REST overexpression tissue (n = 19). **p < 0.001. E, REST knockdown results in accelerated differentiation in vivo. Immunohistochemical staining of tissue isolated from animals injected with shSCR and shREST with DAPI (blue) and GFP (green) 8 dpi. F, Percentage of neurons with dendrites for shSCR (n = 30) and shREST tissue (n = 22) 8 dpi. G, Average dendritic length in shSCR (n = 30) and shREST samples (n = 22). **p < 0.005. H, Gross morphologies of control and shREST neurons are similar at 28 dpi. Representative images of control and shREST spines at 28 dpi indicate decreases in spine density. I, REST knockdown results in a statistically significant decrease in spine density in shREST (n = 24) versus control tissue (n = 26). *p < 0.02. J, REST knockdown results in a statistically significant decrease in mushroom spine density in shREST (n = 24) versus control tissue (n = 26). ***p < 0.000005. K, Scg2 knockdown does not change the differentiation of newborn neurons in vivo (8 dpi). Immunohistochemical staining of tissue isolated from animals injected with shSCR and shScg2 with DAPI (blue) and GFP (green). L, The percentage of GFP+DCX+/GFP+ cells. M, Scg2 knockdown does not show a considerable difference in dendritic length in vivo. The average dendrite length of shSCR (n = 47) and shScg2 (n = 40). N, Immunohistochemistry of SCG2 in wild-type tissue shows expression of SCG2 in vivo (blue represents DAPI; red represents SCG2). ns, Not significant.
Interestingly, at the 28 dpi time-point, neurons with REST knockdown appeared morphologically similar to the shSCR-infected neurons (Fig. 7H, top). Average dendrite lengths calculated from scrambled and REST knockdown cells were also comparable (data not shown). Both sets of neurons appeared fully mature, with dendrites extending into the molecular layer. However, high-magnification analysis of the dendritic spines (Fig. 7H, bottom) revealed a statistically significant (p < 0.02) decrease in overall spine density in REST knockdown neurons (Fig. 7I). Furthermore, mushroom spines were significantly (p < 0.000005) decreased in these neurons (Fig. 7J). REST knockdown accelerates initial neuronal differentiation at the expense of proper dendritic spine formation in later development. These results suggest that temporal regulation of REST may be essential for the development and/or maintenance of dendritic spines during the early stages of neural differentiation.
Subsequently, we manipulated the expression of Scg2 in the proliferative neural progenitors in the DG using an shScg2-expressing retrovirus as described in Materials and Methods. We observed that the loss of Scg2 in vivo indeed did not have a significant effect on the neuronal differentiation in mouse brain at 8 dpi (Fig. 7K). There was also no significant difference in the percentage of GFP+DCX+ cells (Fig. 7L). The morphology, in particular the dendrite lengths, was not significantly affected as a result of Scg2 knockdown (Fig. 7M), suggesting that endogenous Scg2 knockdown did not have an effect on early neuronal differentiation in vivo. We observed expression of SCG2 protein in the hilus (Fig. 7N), which might be a potential source of exogenous SCG2 acting on neural progenitors. Combined with the observation that recombinant SCG2 rescues shScg2-induced phenotype in vitro, hilar SCG2 protein might compensate for the Scg2 knockdown in neural progenitors in vivo.
Discussion
Adult hippocampal neurogenesis is a process with potential links to learning and memory as well as neurodegenerative and neuropsychiatric disorders (Mu and Gage, 2011). Adult neurogenesis is regulated by cellular factors as well as signals from the niche. Although a number of molecules have been implicated in this process, the interplay between cell-intrinsic and cell-extrinsic factors remains largely undefined.
REST has been intensively studied as a key regulator of neurogenesis (Ballas and Mandel, 2005; Johnson et al., 2008; Singh et al., 2008; Jørgensen et al., 2009; Gao et al., 2011). In the adult hippocampus, Nestin-specific knock-out of REST results in depletion of NSCs and an eventual decrease in adult neurogenesis (Gao et al., 2011). Our results indicate that the untimely presence or loss of REST leads to impaired development of newborn neurons. How does REST knockdown lead to accelerated differentiation? The most parsimonious explanation is that REST-repressed genes are directly responsible for the phenotype. REST targets include an array of proneuronal genes that include transcription factors, ion channels, synaptic genes, and signaling molecules (Johnson et al., 2007). REST silencing was previously found to result in the deregulation of several other genes involved in neurite growth (L1cam, Elmo2, Ulip1, and Ulip2) (Lepagnol-Bestel et al., 2007).
Interestingly, accelerated differentiation of neurons in vivo was also followed by dendritic spine defects, suggesting that REST may not only serve as a negative regulator of neurogenesis but may also continue to temporally regulate proper maturation of granule cell neurons. How REST knockdown leads to spine defects at later time points is a more complex question and may well be tied to the maturation process of neurons. The rate of maturation of a newborn neuron may affect the response to the signals it receives during maturation and thus affect its final cellular connections and morphology. The information available on temporal regulation of adult neurogenesis is currently insufficient to fully support such a hypothesis. However, a trend seems to emerge from the few available studies. Ectopic expression of Tis21, which is also a REST target, leads to accelerated maturation of newborn neurons at the expense of defects in dendrite and spine morphology (Farioli-Vecchioli et al., 2008). Another example of accelerated differentiation (18 dpi) followed by later maturation defects (28 dpi) in adult neurogenesis comes from gain-of-function studies on amyloid-β (Sun et al., 2009). Amyloid-β-induced defects in adult neurogenesis were returned to normal by either early inhibition of GABAA receptors or late inhibition of calcineurin, both REST target genes, to enhance glutamatergic signaling and restore normal dendritic maturation. Another possible explanation is that REST plays a steady role in adult neurogenesis. Early on, a decrease in REST levels may be required to initiate the differentiation of neural progenitors. The fact that Neurod1, a REST target essential for hippocampal neurogenesis, is upregulated in early differentiation would be consistent with such a REST expression pattern. It is possible that, later on, REST gradually decreases in differentiating neurons in a way that temporally regulates neural development. Consistent with this hypothesis, REST targets are highly enriched in genes that code for voltage-dependent ion channels and synaptic proteins that could function later during maturation (Johnson et al., 2007).
In an attempt to identify REST targets in the context of adult neurogenesis, we performed a gene expression analysis upon REST knockdown in cultured AHPs that pointed to a surprising non–cell-autonomous role of REST target. Another key observation that attracted our interest, initially suggesting a non–cell-autonomous role, was the differentiating effect REST knockdown cells had on neighboring cells. Previously, REST had been shown to control neurosecretory and gliosecretory processes during differentiation in the phaeochromocytoma cell line PC12 and cortical astrocytes, respectively (Bruce et al., 2006; D'Alessandro et al., 2008; Prada et al., 2011; Tomasoni et al., 2011). Of the various secreted proteins upregulated upon REST knockdown, we identified and confirmed SCG2 as a major secreted factor that promoted differentiation and maturation of AHPs in vitro. Moreover, because secreted proteins, such as Wnt (Lie et al., 2005) and bone morphogenic protein signaling (Mira et al., 2010) have been implicated in adult neurogenesis, we set out to determine whether secretory components downstream of REST, in particular cargo proteins such as SCG2, played a role in non–cell-autonomous differentiation.
To our surprise, we observed no apparent changes in differentiation upon knockdown of endogenous Scg2 in vivo. In contrast, we detected impaired differentiation of AHPs upon Scg2 knockdown in vitro. What caused this difference? The combination of different cell types, signaling molecules, and the extracellular environment in vivo was absent from the cell culture setting. Currently, we do not know all the underlying factors leading to the differences of observed Scg2 knockdown phenotypes. However, we did observe that exogenous SCG2 could compensate for a decrease in Scg2 (shRNA-driven) in cultured neural progenitor cells. SCG2 immunoreactivity in the hippocampus was concentrated in the hilar region. It is possible that, analogous to the Wnt3 signaling originating from the hilus (Lie et al., 2005), SCG2 present in the hilus (Schwarzer et al., 1997) may be a potential exogenous factor acting on neural progenitors.
The present data do not provide a complete understanding of the function of SCG2 in vivo. We propose that SCG2 promotes differentiation of transit-amplifying NSCs, but it is also possible that the presence of extracellular SCG2 may be a way to synchronize the neurons in terms of their maturation and activity. For example, it is known that REST levels increase dramatically in the DG within 4 h after seizures in animals (Palm et al., 1998), perhaps as a way to reduce the overall activity by repressing target genes. It is worth investigating whether extracellular SCG2 is activity-dependent and responds to the upregulation of REST as it occurs in reaction to seizures.
In conclusion, we provide evidence that the knockdown of REST causes accelerated differentiation of newborn neurons at the expense of spine defects in vivo, suggesting a potential role for REST in the timing of the maturation of granule neurons. Our results reveal that REST regulates adult neurogenesis by orchestrating both cell-intrinsic and non–cell-autonomous factors and that Scg2 is a major secretory target of REST with a differentiation-enhancing activity in a paracrine manner.
Footnotes
This work was supported by National Institutes of Health RO1 Grant NS050217 and R01 MH090258, the Leona M. and Harry B. Helmsley Charitable Trust grant #2012-PG-MED00, a California Institute for Regenerative Medicine Training Grant, the G. Harold and Leila Y. Mathers Charitable Foundation, the JPB Foundation, and the McDonnell Foundation. We thank Eunice Mejia, Nada B. Abdallah, Star W. Lee, Anthony Vu, Stefan Aigner, Iryna Gallina, and Galit Benoni for technical assistance and sharing materials; and Mary Lynn Gage for editorial comments.
The authors declare no competing financial interests.
References
- Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol. 2005;15:500–506. doi: 10.1016/j.conb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Bruce AW, Krejcí A, Ooi L, Deuchars J, Wood IC, Dolezal V, Buckley NJ. The transcriptional repressor REST is a critical regulator of the neurosecretory phenotype. J Neurochem. 2006;98:1828–1840. doi: 10.1111/j.1471-4159.2006.04010.x. [DOI] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramírez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- D'Alessandro R, Klajn A, Stucchi L, Podini P, Malosio ML, Meldolesi J. Expression of the neurosecretory process in PC12 cells is governed by REST. J Neurochem. 2008;105:1369–1383. doi: 10.1111/j.1471-4159.2008.05259.x. [DOI] [PubMed] [Google Scholar]
- Farioli-Vecchioli S, Saraulli D, Costanzi M, Pacioni S, Cinà I, Aceti M, Micheli L, Bacci A, Cestari V, Tirone F. The timing of differentiation of adult hippocampal neurons is crucial for spatial memory. PLoS Biol. 2008;6:e246. doi: 10.1371/journal.pbio.0060246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ables JL, Lagace DC, Nave KA, Goebbels S, Eisch AJ, Hsieh J. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat Neurosci. 2009;12:1090–1092. doi: 10.1038/nn.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ding P, Nashaat M, Yuan L, Ma J, Hammer RE, Hsieh J. The master-negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J Neurosci. 2011;31:9772–9786. doi: 10.1523/JNEUROSCI.1604-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Johnson R, Teh CH, Kunarso G, Wong KY, Srinivasan G, Cooper ML, Volta M, Chan SS, Lipovich L, Pollard SM, Karuturi RK, Wei CL, Buckley NJ, Stanton LW. REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol. 2008;6:e256. doi: 10.1371/journal.pbio.0060256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen HF, Terry A, Beretta C, Pereira CF, Leleu M, Chen ZF, Kelly C, Merkenschlager M, Fisher AG. REST selectively represses a subset of RE1-containing neuronal genes in mouse embryonic stem cells. Development. 2009;136:715–721. doi: 10.1242/dev.028548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepagnol-Bestel AM, Maussion G, Ramoz N, Moalic JM, Gorwood P, Simonneau M. Nrsf silencing induces molecular and subcellular changes linked to neuronal plasticity. Neuroreport. 2007;18:441–446. doi: 10.1097/WNR.0b013e328011dc81. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira H, Andreu Z, Suh H, Lie DC, Jessberger S, Consiglio A, San Emeterio J, Hortigüela R, Marqués-Torrejón MA, Nakashima K, Colak D, Götz M, Fariñas I, Gage FH. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell. 2010;7:78–89. doi: 10.1016/j.stem.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer's disease. Mol Neurodegener. 2011;6:85. doi: 10.1186/1750-1326-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada I, Marchaland J, Podini P, Magrassi L, D'Alessandro R, Bezzi P, Meldolesi J. REST/NRSF governs the expression of dense-core vesicle gliosecretion in astrocytes. J Cell Biol. 2011;193:537–549. doi: 10.1083/jcb.201010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray J, Gage FH. Differential properties of adult rat and mouse brain-derived neural stem/progenitor cells. Mol Cell Neurosci. 2006;31:560–573. doi: 10.1016/j.mcn.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Satoh J, Kawana N, Yamamoto Y. ChIP-Seq data mining: remarkable differences in NRSF/REST target genes between human ESC and ESC-derived neurons. Bioinform Biol Insights. 2013;7:357–368. doi: 10.4137/BBI.S13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- Schwarzer C, Marksteiner J, Kroesen S, Kohl C, Sperk G, Winkler H. Secretoneurin: a market in rat hippocampal pathways. J Comp Neurol. 1997;377:29–40. doi: 10.1002/(SICI)1096-9861(19970106)377:1<29::AID-CNE4>3.0.CO%3B2-L. [DOI] [PubMed] [Google Scholar]
- Singh SK, Kagalwala MN, Parker-Thornburg J, Adams H, Majumder S. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–227. doi: 10.1038/nature06863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh H, Deng W, Gage FH. Signaling in adult neurogenesis. Annu Rev Cell Dev Biol. 2009;25:253–275. doi: 10.1146/annurev.cellbio.042308.113256. [DOI] [PubMed] [Google Scholar]
- Sun B, Halabisky B, Zhou Y, Palop JJ, Yu G, Mucke L, Gan L. Imbalance between GABAergic and glutamatergic transmission impairs adult neurogenesis in an animal model of Alzheimer's disease. Cell Stem Cell. 2009;5:624–633. doi: 10.1016/j.stem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasoni R, Negrini S, Fiordaliso S, Klajn A, Tkatch T, Mondino A, Meldolesi J, D'Alessandro R. A signaling loop of REST, TSC2 and beta-catenin governs proliferation and function of PC12 neural cells. J Cell Sci. 2011;124:3174–3186. doi: 10.1242/jcs.087551. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Thatcher EJ, Olena AF, Cha DJ, Perdigoto AL, Marshall AF, Carter BD, Broadie K, Patton JG. miR-153 regulates SNAP-25, synaptic transmission, and neuronal development. PLoS One. 2013;8:e57080. doi: 10.1371/journal.pone.0057080. [DOI] [PMC free article] [PubMed] [Google Scholar]