

Abstract
Because of the intimate association of obesity with type 2 diabetes mellitus (T2DM), during the last two decades, extensive research work is being conducted to find out whether the coexistence of the two is a simple association or there is a positive correlating link between the two. In this article, an attempt has been made to collect and analyse the recent developments in this field and to arrive at a conclusion on the subject. The possible role of several important factors (obtained from adipocytes/not of adipocyte origin) in linking the two has been discussed in detail. Some of the agents, specifically adiponectin, are beneficial (i.e., reduce the incidence of both), while others are harmful (i.e., increase their incidence). From the analysis, it appears that obesity and T2DM are intimately linked.
Keywords: Obesity, Insulin, Insulin resistance, Type 2 diabetes mellitus, Adipocyte
Core tip: The objective of this article is to establish the connection of obesity with that of insulin resistance (IR) and type 2 diabetes mellitus (T2DM) by analyzing the recent developments in this field. The factors linking the three have been found to be some adipocytokines as well as certain other factors not of adipocyte origin. Of these, adiponectin appears to play the most beneficial role (so also leptin, peroxisome proliferator-activated receptors, apelin, etc.), while others (tumour necrosis factor-alpha, interleukin-6, resistin, retinol binding protein-4, dipeptidyl peptidase-4, plasminogen activator inhibitor-1, visfatin, free fatty acid, angiotensin II and toll-like receptors) are harmful. Agonists and antagonists of these factors may be designed to fight against obesity, thereby achieving protection for IR and T2DM.
INTRODUCTION
It is practically established that type 1 diabetes mellitus is an autoimmune disorder where the tissue-specific antibodies target and cause complete or near complete destruction of islet-β-cells, leading to absolute insulin deficiency. In contrast, type 2 diabetes mellitus (T2DM) is usually a hereditary disorder, commonly (80%) associated with obesity, where deficient insulin action may be due to a real deficiency of insulin or a relative one associated with normal or even elevated plasma concentrations of insulin, i.e., insulin resistance (IR). Such simultaneous occurrence of the two (T2DM and obesity) suggests the possibility of a strong link between them, and during the past two decades several positive correlations between them have been established by many workers[1-4]. Besides obesity which is directly linked to T2DM via adipocytokines, some nonadipocytokines have been found to be related with T2DM indirectly by interfering with the growth, development and functions of adipocytes (mentioned later). In this article, an attempt has been made to collect and analyse some such authentic work-results together that will help the reader to comprehend and assess the developments in this field.
The intimate association of T2DM and obesity is a world-wide phenomenon. Though much knowledge about the pathophysiology, course and consequences of T2DM has been gathered, it is not so with obesity, which was almost practically considered as a cosmetic problem. But recently, because of its frequent association with T2DM as well as with hypertension, extensive work is being continued on the adipocyte anatomy, distribution pattern, physiological function, pathological role and its possible link with T2DM and hypertension.
PHYSIOLOGICAL ROLE OF ADIPOCYTES AND ADIPOSE TISSUE
Primary physiological role of adipose tissue is to insulate and cushion the body, to store fat when it is in excess and to supply it when needed[5]. The exogenous and endogenous pathways of lipid metabolism, during which free fatty acids (FFAs) are released from the lipoprotein (chylomicron, very low density lipoprotein, etc.) - triglyceride (TG) content upon hydrolysis by the enzyme lipoprotein lipase (LPL), their (FFAs) subsequent storage in fat depots as TG again, and their remobilisation into the periphery by hydrolysis of these stored TGs by the hormone sensitive lipase (HSL), is well established[5,6]. Insulin plays a major role for maintenance of adipocyte-fat content as it is a potent activator and inhibitor of LPL and HSL, respectively[5].
SECRETIONS OF ADIPOCYTES (ADIPOCYTOKINES)
Recently, adipocytes are considered as endocrine structures because of their wide variety of chemical secretions (adipocytokines), which affect many diverse physiological functions and related pathological processes of the body, like metabolism of carbohydrates and lipids, coagulation of blood, maintenance of blood pressure, feeding behaviour and inflammation, affecting almost all the organs of the body. Increased adipocyte number and adipose-tissue mass have been found to result in increased plasma adipocytokine level except adiponectin, whose plasma concentration is actually low in obesity[5]. Diseases like obesity, T2DM and metabolic syndrome are associated with altered plasma adipokine levels.
A brief discussion of the adipocytokines known till-date along with their possible roles in genesis or amelioration of IR and T2DM is made below. Besides the adipokines, possible involvement of certain other factors (not of adipocyte origin) has also been taken into account (Figure 1).
Figure 1.
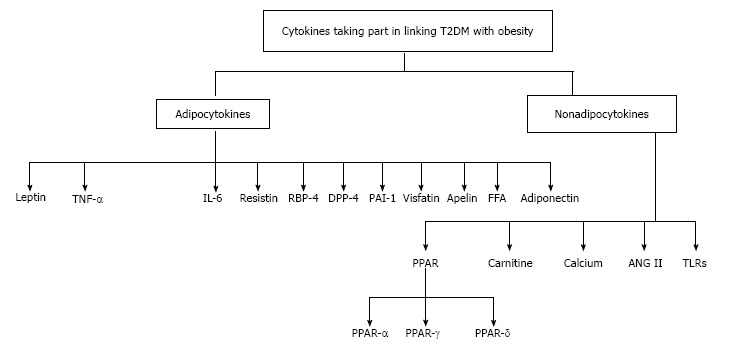
Cytokines linking type 2 diabetes mellitus with obesity. TNF-α: Tumour necrosis factor-alpha; IL-6: Interleukin-6; RBP-4: Retinol binding protein-4; DPP-4: Dipeptidyl peptidase-4; FFA: Free fatty acid; PPAR: Peroxisome proliferator-activated receptor; Ang II: Angiotensin II; TLRs: Toll-like receptors; T2DM: Type 2 diabetes mellitus; PAI-1: Plasminogen activator inhibitor-1.
Leptin
Several physiological functions of leptin along with its source and metabolism have been extensively discussed. This adipokine, which is a product of “ob” gene but mediates its function through the receptor coded by “db” gene, is involved in energy homeostasis of the body by interfering with the food-behaviour of the animal centrally (hypothalamus) via several hormones[7].
Many studies on mice and human beings have shown a beneficial and balancing complementary relationship between leptin and insulin where leptin has been found to reduce appetite, obesity and IR along with improvement of metabolic disturbances associated with T2DM. Moreover, mice with db/db gene (deficient leptin action) have been found to be obese and diabetic[7].
Though the receptors for insulin and leptin are different, both of them mediate their action through some common second messengers. Therefore, it is possible that leptin may trigger some of the same downstream events triggered by insulin. Increase in tissue sensitivity of insulin by leptin may be due to later’s action on oxidation of FFAs which is increased in skeletal muscles leading to its (FFAs) decreased blood concentrations[7].
Because of such functional cooperation, it may be assumed that obesity due to inadequate leptin action may predispose or get associated with IR and T2DM.
Tumour necrosis factor-α
The role of tumour necrosis factor-α (TNF-α) as a pro-inflammatory cytokine is well established[8]. It is produced by macrophages (mainly) as well as by some other cell types including visceral adipocytes[8-10]. Recently, it has been shown that besides its pro-inflammatory property, increased TNF-α inhibits insulin transduction mechanism, resulting in inadequate glucose metabolism, IR and obesity. Because visceral fat is a source of TNF-α, increase in such fat (obesity) leads to increased production of this cytokine, which aggravates obesity and a vicious cycle is established leading to predisposition, onset and progression of T2DM along with IR. Hence, reduction of obesity, which in turn may lead to decreased formation of TNF-α, may help to prevent genesis, progression and complications of T2DM[8]. Besides inhibiting insulin signalling mechanism, TNF-α also has been found to inhibit glucose-induced insulin secretion from β-cells, cause damage to insulin strand and enhance β-cell apoptosis. However, such functions of TNF-α have been demonstrated in vitro with concentrations of the cytokine, which was much higher than in vivo plasma concentrations[5]. Moreover, besides visceral adipocytes, macrophages and other cells also produce TNF-α, which may contribute towards the elevated level of this cytokine in obesity[10]. Therefore, obesity and increased TNF-α levels cannot be directly and definitely implicated with T2DM, although they seem to have a role which needs further investigations[5,8,9].
Interleukin-6
It is another pro-inflammatory cytokine produced by many cell types (fibroblast, endothelial cells, monocytes) in the body including adipocytes, the production (by adipocytes) being increased in obesity. In vitro studies as well as investigations on mice have shown interleukin (IL)-6 to upregulate the production of vascular endothelial growth factor, which is thought to support angiogenesis during adipose tissue growth, leading to increase in the production of IL-6 further (similar to TNF-α)[5,10].
IL-6 action is mediated through a cytokine class one receptor subtype involving Janus kinase/signal transducers and activators of transcription (JAK/STAT) signal transduction pathway, whereas insulin action is mediated through a receptor family having intrinsic tyrosine kinase activity, signal transduction being carried out through insulin receptor substrate (IRS) proteins. It has been clearly demonstrated that inspite of entirely different receptor involvement, a strong interaction occurs between the receptor signalling pathway of IL-6 and insulin, leading to impaired biological effect of the later. Though not fully clear, the interaction may involve activation of tyrosine phosphatase, leading to dephosphorylation and inactivation of tyrosine kinase activity or an interaction between suppressor of cytokine signalling proteins and insulin receptors, resulting in deficient insulin action[10]. Therefore, it appears that elevated plasma levels of IL-6 due to any cause (not necessarily of body fat) may get associated with IR and hence, increased risk of diabetes[5].
Resistin
This pro-inflammatory cytokine, besides monocytes and macrophages, is also produced by adipocytes. It is so named, because of its capacity to resist insulin action[1,10,11]. It has a molecular weight of 12.5 kDa and possesses 108 amino acid residues in humans. Unlike adiponectin, this polypeptide has a low circulatory level, which is increased in subjects with IR, T2DM and metabolic syndrome[3].
Several workers have demonstrated a definite role of resistin in linking obesity to T2DM, during which the cytokine has been found to modulate the insulin signalling pathway, leading to development of IR[2]. Increased production of resistin has been found to be a result of adipocyte differentiation as well as increase in their number. Locally (from adipocytes) released resistin may play a paracrine role, resulting in inhibition of insulin-induced glucose uptake by adipocytes, which prevents their (adipocytes) further differentiation, thereby reducing its own synthesis and release. This observation may suggest a reciprocal relationship between the two hormones which may further be supported by the fact that rosiglitazone (an oral antidiabetic drug) decreases the circulating concentration of resistin, whereas diet-induced and genetic forms of obesity increases it[11]. Moreover, neutralization of resistin has been found to increase the insulin-induced uptake of glucose by adipocytes, whereas resistin itself decreased it.
Recently, it has been observed that resistin-knockout mice show lower fasting blood sugar with increased glucose tolerance and insulin sensitivity associated with reduced hepatic output of glucose. The possible mechanism of this observation may be an overactivity of AMP-activated protein kinase (AMPK) resulting from lack of resistin, leading to reduced expression of genes responsible for hepatic neoglucogenesis. This possible mechanism suggests an opposite role of resistin to that of adiponectin. Again, it was observed that when these resistin-knockout mice were fed with high fat diet, they became obese and IR like their wild counterparts[10]. All these observations suggest a potential positive link between obesity and T2DM[1].
Retinol-binding protein-4
This adipocytokine, which is primarily a vitamin A -transport protein, has been recently shown to be linked with IR. Down-regulation of adipocyte GLUT-4 (glucose transporter) has been found to increase the secretion of retinol-binding protein-4 (RBP-4) from adipocytes. In mice, increased serum levels of RBP-4 has been found to be associated with decreased uptake of glucose by skeletal muscles and increased hepatic neoglucogenesis. On the other hand, insulin sensitivity was found to be increased when serum RBP-4 levels were low[12]. Similar positive correlations between raised plasma RBP-4 level and IR, plasma glucose, BMI and homeostatic model assessment-IR have also been shown in nondiabetics with a high genetic predisposition for T2DM. Interestingly, in this experiment, it was observed that serum RBP-4 levels were raised before significant appearance of diabetic markers[13]. Such an observation indicates the “elevated plasma RBP-4 level” to be a signal for development of insulin resistance and subsequent T2DM in future[12,13]. In another experiment, it has been shown that excess of RBP-4 relative to retinol (RBP to retinol ratio) is more accurate in predicting the development of T2DM than raised RBP-4 levels alone[14].
Dipeptidyl peptidase-4
The incretins (glucagon-like peptide-1 and glucose-dependent insulinotropic hormone) are known to possess favourable effect on carbohydrate and lipid metabolism as they increase postprandial insulin release along with a decrease in release of glucagon. The two incretins, like several other glycoprotein and peptide substrates, are metabolically degraded by the enzyme dipeptidyl peptidase-4 (DPP-4), which reduces their favourable metabolic effects in relation to diabetes and therefore may be considered as diabetogenic. Hence, DPP-4 inhibitors (sitagliptin, vildagliptin, etc.) are now used extensively for management of T2DM along with other antidiabetic agents[15].
Recently, it has been shown that like other cells, adipocytes also express DPP-4 and substantial overexpression is found in visceral fat of obese persons.Experiments have demonstrated that DPP-4 expression and circulating DPP-4 concentration are well-correlated with adipocyte size and adipose tissue inflammation. This may suggest a stimulatory role of pro-inflammatory adipokines on expression of DPP-4 from adipocytes and other tissues. Thus, increased release of DPP-4 from visceral adipocytes of obese persons may enhance the metabolic degradation of incretins in an autocrine or paracrine manner, thereby reducing their favourable effect on carbohydrate and lipid metabolism which in turn may predispose the concerned obese person for developmen of T2DM and metabolic syndrome. In another study, it has been shown that explants from subjects release more DPP-4 and the release is reduced after weight reduction[15]. Moreover, in insulin- sensitive obese patients, plasma concentration of DPP-4 has been found to be lower than those of insulin-resistant obese diabetics[16]. All these physiological and experimental observations suggest a strong link between T2DM and obesity, where the linking factor appears to be DPP-4.
Plasminogen activator inhibitor-1
This prothrombotic cytokine, besides being produced by vascular endothelial cells, is also produced by adipocytes, production being more from omental adipose tissue than that of subcutaneous adipocytes[17]. Some recent studies have found a direct contribution of this cytokine towards the complications of obesity like T2DM and coronary thrombosis, as well as increased accumulation of visceral fat[18]. Nowadays, plasminogen activator inhibitor-1 (PAI-1) is being considered as a strong predictor of T2DM, and has been found to stimulate adipocyte differentiation, which may be mediated through reducing peroxisome proliferator-activated receptor (PPAR)-γ activity, resulting in more production of resistin. It has been demonstrated that adipocyte-PAI-1 increases the production of TNF-α (an autocrine action) in adipocytes that reduces insulin action and predisposes to T2DM. Moreover, PPAR-γ receptor has been found to be downregulated both by PAI-1 and TNF-α. Hence, inhibition of PAI-1 action on adipocytes may prevent obesity and IR, and retard adipocyte differentiation and fat accumulation by removing not only its (of PAI-1) own antiinsulin action but also that of resistin and TNF-α[7,17].
Visfatin
This adipocytokine, a pro-inflammatory marker of adipose tissue, is mainly produced by visceral adipocytes of humans and mice, whose plasma concentration increases along with the progression of obesity[19-21]. Its production is upregulated by hypoxia, inflammation and hyperglycemia, and downregulated by insulin, somatostatin and cholesterol reducing statins. Besides visceral fat, intracellular presence of visfatin has also been demonstrated in many other tissues and organs, the location being both cytoplasmic and nuclear[21].
Functions of visfatin are difficult to explain as they appear to be contradictory. The cytokine has been found to possess insulinomimetic effect in cultured cells[19,20] and lowers plasma glucose concentration in mice[19]. It has also been shown to cause hypoglycaemia by reducing hepatic output of glucose and increasing utilisation of glucose in adipocytes and monocytes[21]. Inspite of such favourable insulinomimetic action[19,20], this cytokine has been found to be associated with IR and possesses a direct relationship between its plasma concentration and T2DM[21,22]. This anomaly may be explained by the fact that it also produces hyperlipidemia, which may be responsible for IR and hence T2DM (As T2DM may either be due to deficiency of insulin or IR)[22]. The resultant effect seems to be favouring the development of T2DM, which in turn suggests the pernicious role of visceral adipose tissue (VAT) in human obesity-related T2DM and accompanying metabolic disorders[20].
Besides these T2DM-related pathological functions, visfatin, by its endocrine, autocrine as well as paracrine function, has been found to cause increase in cell proliferation and biosynthesis of nicotinamide mono- and dinucleotides[16], significance of which is yet to be ascertained.
Apelin
Apelin, a small peptide adipokine, has also been found to be present in a number of tissues. It is the ligand of the G-protein-coupled receptor (GPCR) APJ, and has several active forms, which include apelin 13, apelin 17 and apelin 36. It is considered as a beneficial adipokine as it has been found to possess antiobesity and antidiabetic properties, because of its potent positive role[23,24] in energy metabolism and insulin sensitivity improvement[24]. Such actions appear to be due to promotion of complete lipid combustion[23] in muscle of IR mice through mitochondrial biogenesis and tighter matching between fatty acid oxidation and TCA cycle. Such apelin- stimulated improvement of FA oxidation led to decreased levels of acyl-carnitines and enhanced insulin-stimulated glucose uptake in soleus muscle[25]. For such beneficial actions, apelin may be considered as a promising useful therapeutic agent for T2DM and other metabolic disorders[23].
FFA
FFAs, which are produced during the metabolism of exogenous and endogenous lipids, play an important role in the development of IR and hence, genesis of T2DM, when their plasma concentration is abnormally raised[26].
Mechanisms of FFA-induced IR include inhibition of insulin-induced release of NO from endothelial cells, resulting in decreased blood flow, inhibition of insulin- stimulated glucose transport across the cell membrane and/or inhibition of intracellular phosphorylation of glucose by interfering with insulin signal transduction pathway. Acute elevation of FFA in plasma has been found to be associated with IR, which may account for 50% of IR in obese individuals with T2DM[27].
Intracellular mechanism of FFA-induced IR has been demonstrated both in vivo and in vitro, where there was an activation of pro-inflammatory nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. It has been shown in vivo that acute increase in FFA level resulted in activation of NF-κB pathway in human skeletal muscle and rat liver, leading to increased production of pro-inflammatory cytokines, i.e., TNF-α, IL-1β and IL-6, in both the test organs along with an increase in the concentration of macrophage chemoattractant protein-1 (MCP-1) in circulation. In health as well as in T2DM, insulin tends to reduce FFA-induced-IR by lowering the plasma concentration of FFAs through its lipogenic as well as antilipolytic action along with increased intracellular oxidation of FFA. However, in obesity, which is considered as an inflammatory state, there is not only an increase in FFA, but also an increase in the plasma concentration of pro-inflammatory cytokines, which together are liable to cause IR and T2DM[27].
Thus, obesity alone or along with increased FFA, can create and maintain a low grade inflammatory state by production of pro-inflammatory cytokines (TNF-α, IL-6, etc.), which may induce IR and T2DM. The condition may be further aggravated by antiinsulin action of FFA on glucose metabolism[27].
Adiponectin
This adipocytokine is being extensively studied world-wide since the past decade because of its remarkable insulin sensitizing property (IR is the major problem in T2DM) as well as antiatherogenic action (dyslipidemia, commonly associated with T2DM, is responsible for atherosclerotic complications of T2DM), thereby playing an important role in delaying and suppressing the metabolic derangements, which result in IR, T2DM, metabolic syndrome and complications of diabetes including vascular and cardiac. These two important functions of adiponectin involves myriads of interrelated molecular mechanisms, which interconnect it with other diabetogenic/antidiabetic adipokines as well as with many physiological and biochemical processes associated with maintenance of energy balance from metabolism of carbohydrates and lipids[3]. Because of such widespread metabolic involvement, an attempt has been made to discuss the pathophysiological role of this key adipocytokine in detail, which in concert with its siblings appears to play an important role in linking T2DM with obesity.
Source and location: Adiponectin, secreted by both white and brown adipose tissue, has several other names like gelatin binding protein-28, AdipoQ, Adipocyte complement-related protein-30 and OP-MI. Adiponectin mRNA presence is lower in VAT than that of subcutaneous adipose tissue[4,10]. Normal plasma concentration of this cytokine varies from 5-30 μg/mL and is inversely proportional to abdominal obesity, IR and T2DM. In some animal models, a decrease in plasma adiponectin concentration was found to precede the onset of T2DM and was parallel with decreased insulin sensitivity. The cytokine circulates in blood in multimeric forms, like trimeric, hexameric and high molecular mass species, each of which plays a specific role in maintenance of energy homeostasis[4,7].
Control of secretion: Control of adiponectin secretion is effected by: (1) some hormones; (2) many adipokines; (3) certain receptor families including its own; (4) endoplasmic reticulum (ER) and oxidative stresses; and (5) several other factors.
Hormonal control: Sex hormone: Adiponectin plasma concentration has been found to be higher in women than men, which may be due to the difference in concentration of oestrogen and androgen, suggesting a presumably stimulating role of oestrogen on synthesis and secretion of this adipokine[5,7]; Insulin: The relationship between plasma insulin concentration and adiponectin secretion appears to be peculiar, confusing and contradictory, as the experimental observations do not correlate as expected.
Though insulin favours adiponectin biosynthesis through PPAR-γ via inhibition of FOX01 (an inhibitor of PPAR-γ), type I diabetic patients who practically have no circulating insulin, contrary to the expectations, show elevated levels of plasma adiponectin. Moreover, patients, with defective insulin receptors due to abnormal genes coding for them, also show raised circulating adiponectin levels. Furthermore, adiponectin concentration, unlike other insulin-resistance-inducing adipokines, has been found to be decreased in obesity and insulin-resistant models. From such observations it seems that IR decreases plasma adiponectin concentration. This may be explained by taking into account the role of oxidative stress which is known to increase IR and to decrease adiponectin production. In obesity, adipocytes may develop oxidative stress, leading to decreased expression of adiponectin by them. That IR decreases adiponectin expression may further be supported by the observation that hyperinsulinemia associated with euglycemia (an IR state) significantly decreases the plasma adiponectin concentration and selectively downregulates its high molecular weight (HMW) form. The disparity between the above mentioned experimental observations in relation to role of insulin on adiponectin formation is not known and appears to be complicated[4].
Control by adipokines: TNF-α and IL-6 are considered to be established inhibitors of adiponectin synthesis[28]. As their synthesis and secretion increase in obesity, adiponectin plasma concentration decreases accordingly[4,29].
Control by certain receptors: PPAR-γ: This PPAR subfamily transcription factor, which is mainly found in adipocytes, has been shown to possess a positive regulatory role on adiponectin gene expression leading to increased production of proteins like Erol-La and DsbA-L, which take part in synthesis and secretion of adiponectin[4]; Own receptors: Circulating adiponectin concentration has been found to be inversely related to muscle AdipoR1/2 (receptor subtypes), but directly related to subcutaneous AdipoR2[4,30].
ER and oxidative stresses: ER is known to be an intracellular fine network of microtubules. It is continuous with the nuclear membrane, and is called sarcoplasmic reticulum in muscles. It controls intracellular calcium ion uptake and release besides its other functions, thereby effecting muscular contraction and relaxation. ER stress, which is produced in obesity, has been shown to be negatively related to adiponectin production by adipocytes. The molecular mechanism involved has been studied in 3T3LI-cells, where oxidative stress in ER lead to increased production of H2O2, which, via protein kinase B (Akt) and JAK/STAT pathway, appreciably suppressed the expression of adiponectin mRNA and consequent reduction in synthesis of proteins required for adiponectin formation. Moreover, in this model H2O2 has been found to increase the production of PAI-1 and IL-6, which are known to inhibit adiponectin synthesis[31].
Other factors: Obesity: Unlike other adipokines, adiponectin secretion has been found to be decreased in obesity. Though the exact cause of such reduction is not known, the suggested causes include increased production of TNF-α and IL-6[28], generation of a hypoxicmicroenvironment in the adipocyes of increased fat mass, and obesity-induced increased production of insulin like growth factor binding protein-3, which inhibits adiponectin transcription via hypoxia inducible factor-1α dependent pathway[4,32]; Drugs: PPAR-γ agonists (thiazolidinediones-TZDs), which increase insulin sensitivity, have been found to increase the plasma concentration of adiponectin, whereas anti-HIV drugs like protease inhibitors decrease it[29].
Physiological functions of adiponectin: Adiponectin, along with other adipokines, interferes in several metabolic functions, like lipid synthesis and storage, neoglucogenesis and peripheral utilisation of glucose, which have been demonstrated in skeletal and cardiac muscles, adipocytes and hepatocytes[31]. But, it differs from other adipokines in several aspects. Unlike others, its circulating concentration has been found to be decreased in obesity (particularly abdominal obesity) and T2DM, and instead of increasing insulin resistance, it decreases it in addition to possessing antiatherosclerotic effect. In animal models and in patients with obesity and T2DM, the cytokine has been shown to stimulate fatty acid (FA) oxidation, reduce lipid accumulation in muscles, decrease plasma FA concentration and increase insulin sensitivity. Because of such beneficial involvement in metabolic functions (lipids and carbohydrates), IR and atherosclerosis, this adipokine is expected to impart protection against coronary heart diseases, steatohepatitis, non-alcoholic fatty liver diseases and a wide variety of cancers[33].
Cellular basis of mechanism of action: Functions of adiponectin have been found to be mediated by three receptor subtypes namely, AdipoR1, AdipoR2 and T-cadherin. AdipoR1 and AdipoR2 are 7 transmembrane proteins but dissimilar to GPCRs. Its receptor distribution pattern varies from cell type to cell type - AdipoR1 being found abundantly in muscles, while AdipoR2 is mainly expressed in hepatocytes. Both the receptors are present in almost every tissue, but in a particular tissue, usually one type predominates. Moreover, degree of affinity of these receptors for different forms of adiponectin also varies[4,7]. AdipoR1 has high affinity for globular adiponectin (a cleaved part of full-length adiponectin) but low affinity for full-length adiponectin, whereas AdipoR2 has intermediate affinity for both forms. Hypoadiponectinemia, associated with IR, upregulates both the receptor types. Such upregulation also occurs in physical activity, suggesting an association between adiponectin hormone system and exercise-induced improvement in IR[7]. Adiponectin, binding to its cell surface receptors, activates several intracellular signalling molecules like p38MAPK, PPAR, the RAS-associated protein Rab5, PI3K, Akt and AMP-activated protein kinase (AMPK), of which AMPK system and PPARs play an important and dominant role leading to modification of lipid and carbohydrate metabolism[4,7,29].
As has already been mentioned, in this article emphasis would be given on two important protective physiological functions of adiponectin, i.e., protection against IR and artherosclerosis. Obesity, T2DM, dyslipidemia and IR are intimately related, where one leads to the other and once developed, aggravate each other thereby establishing a vicious cycle, leading to development of practically all the dangerous complications of T2DM[34]. Increased fatmass, as found in obesity, not only increases the production of bad adipokines who enhance this cycle further but also decreases the production of the good one-adiponectin, deficiency of which contributes significantly towards the development, continuation and aggravation of this cycle. Adiponectin has been shown to prevent the development as well as to break this dangerous cycle, thereby posing itself as a potential therapeutic agent in such condition.
Mechanisms of antiatherosclerotic and IR preventing actions of adiponectin: As these two actions are interrelated, it is convenient to discuss them together. It has already been mentioned that adiponectin increases FA oxidation in mitochondria that leads to a decrease in plasma concentration of FA. Reduced level of FA in circulation prevents the development and progression of atherosclerosis and IR. Multiple biochemical actions at cellular level are modified for this action of adiponectin that needs an extensive discussion and correlation between them to arrive at a conclusion.
Adiponectin-induced FA oxidation is primarily mediated by phosphorylation (activation) of AMPK - a multi-subunit protein kinase, which appears to be a sensor of intracellular energy status through activation of PPAR-α receptor. It has been demonstrated that when muscles were treated with adiponectin or when its receptors were expressed ectopically, there occurred an increase in AMPK phosphorylation and FA oxidation in the muscles that was abolished by dominant-negative AMPK use. Stressful conditions, like heat shock, hypoxia, starvation and exercise, etc., which need expenditure of more energy (denoted by high AMP - to - ATP ratio) have been found to cause AMPK activation. This important signalling molecule (AMPK) is also directly activated by other upstream kinases, where they cause phosphorylation of its threonine residue in the kinase domain. In skeletal muscle, activated AMPK increases FA oxidation by stimulating the phosphorylation (leading to inactivation) of the key enzyme acetyl-CoA carboxylase (ACC). Reduced ACC activity, in turn, decreases intracellular malonyl-CoA concentration along with stimulation of carnitine palmitoyl transferase 1 (CPT1) activity, leading to increased entry of long-chain FAs into mitochondria and hence, more of their peripheral oxidation. The fact, that adiponectin increases insulin sensitivity by decreasing plasma FA concentration, has been demonstrated in obese and T2DM patients, where serum adiponectin concentration is low. In such patients, administration of adiponectin has been found to increase insulin sensitivity by decreasing their plasma FA and TG[33].
Metabolic stressful conditions like muscle contraction, hypoxia, ischemia and hyperosmolality, etc., not only increase AMPK activation (as mentioned before), but also stimulate the activity of p38MAPK (a signalling molecule activated by inflammatory cytokines). This indicates an association between the two signalling molecules during signal transduction, though the agonists (adiponectin, inflammatory cytokines) inducing the signals are different. In fact, adiponectin has been found to stimulate the activity of not only AMPK but also that of p38MAPK and PPAR-α in target tissue though the subsequent signal transduction pathway following these three activations is not fully known. Other evidences in muscles suggest a sequential activity of these three, leading to increased FA oxidation and increased glucose uptake by muscles. But it has been shown that when primary hepatocytes are treated with adiponectin, their FA oxidation is not increased, which suggests a differential effect of the cytokine on FA oxidation of muscles and liver[33].
ER stress decreases adiponectin secretion: Several workers have shown that ER stress in adipocytes decreases adiponectin secretion. It has been demonstrated that properly integrated mitochondrial function in adipocytes is necessary for adequate secretion of adiponectin. Like other cells, growth and development of adipocytes occur through differentiation and hypertrophy, which need increased mitochondrial function, because of greater energy requirement. Newly differentiated adipocytes are small in size, because of less accumulated TG due to increased FA oxidation in them, as the mitochondrial content and activity are more[29].
It has been shown that these small adipocytes synthesise and secrete more adiponectin because of their high mitochondrial functional level, whereas large hypertrophied fat cells, as found in obesity, produced the cytokine to lesser extent because of impaired mitochondrial function. Though till now, adiponectin synthesis has not been properly correlated with increased mitochondrial function, it may be due to much greater consumption of energy for the synthesis of this cytokine protein in comparison with other proteins. Therefore, it appears that synthesis of adiponectin in adipocytes needs high consumption of energy, which is produced by elevated (adequate) mitochondrial function. In support of this, it has been shown that rosiglitazone and others agents like Ad-NFR-1, which increase mitochondrial biogenesis, also cause an increase in adiponectin synthesis. This observation points the finger towards mitochondrial dysfunction as the cause of low adiponectin level in obesity[29].
Moreover, several evidences have been put forward where obesity-induced mitochondrial dysfunction has resulted in ER stress, which decreases adiponectin secretion and development of IR. Both ER stress and mitochondrial dysfunction have been demonstrated to activate a series of reactions involving sequential activation of JNK and activating transcription factor 3 (ATF3), which in turn decrease the transcription of adiponectin. When JNK and ATF3 are inhibited, adiponectin transcription is restored. It has also been suggested that ER stress and impaired mitochondrial function are separately responsible for genesis of IR in various tissues of obese persons[29].
Adiponectin-induced increase in FA oxidation via activation of AMPK and phosphorylation of ACC is of short duration, as ACC phosphorylation is short-lived. Hence, this pathway cannot be considered to be fully responsible for the long term effect of adiponectin in causing weight loss and FA oxidation, for which action through PPAR-α is thought to be involved, because PPAR-α action has been found to persist even after initial signalling is over. This is so, because adiponectin has been found to increase transcriptional activity of PPAR-α and subsequent expression of its target genes via activation of AMPK. Involvement of AMPK is supported by the fact that when PPAR-α agonists were administered to obese animals, there occurred an equivalent and sufficient lowering of lipids, as was found with adiponectin. This fact was further supported by in vivo administration of 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) to lean and obese Zucker rats where the compound was found to decrease plasma FA and TG levels significantly, because AICAR is known to increase the transcriptional activity of PPAR-α via activation of AMPK[33].
Anti-inflammatory action of adiponectin: Mention has been made about the decreased secretion of inflammatory cytokine TNF-α by adiponectin from macrophages that contribute towards its antiatherogenic effect. This anti-inflammatory property is also likely to be involved in its IR reducing action, because TNF-α and IL-6 are known to decrease adiponectin formation and to induce IR[10,28].
Recently, it has been shown that NF-κB activation in endothelial and monocytic cells, which is involved in causation of inflammation and metabolic alteration in obesity, is suppressed in these cells by adiponectin. Moreover, both forms of adiponectin-globular as well as full-length, have been found to decrease the production of pro-inflammatory cytokines IL-6 and MCP1 from inflammed adipocytes that may be due to inhibition of NF-κB activity as well as PPAR-α expression[35].
Insulin sensitizing actions of adiponectin: Adiponectin aids to insulin sensitivity by several novel mechanisms, which include - increased FA oxidation, decreased ER stress, improvement in insulin signalling pathway, increased (improved) mitochondrial number and function, increased insulin secretion, decreased hepatic output of glucose, increased uptake of glucose by liver and muscle, and increased glucose metabolism.
Increase FA oxidation: Adiponectin-induced increase in FA oxidation has been demonstrated by several workers[5,10,33,36]. This action of adiponectin contributes significantly towards its insulin sensitizing action and prevention of development of IR, as increased plasma FA concentration is the most important cause of IR. In some animal models, adiponectin has been shown to decrease FFA concentration in plasma by increasing its uptake and oxidation in skeletal muscles. On the other hand, acute reduction of plasma FFA has been found to be associated with low adiponectin concentration, though the exact role of FFA in such action is not known[36]. It is well documented that the key enzyme responsible for FA oxidation is AMPK, which is activated by adiponectin[4,10,31,33]. It has already been mentioned that once activated, AMPK inhibits the activity of ACC, which not only leads to reduced contents of intracellular malonyl-COA but also increases activity of CPTI. Such an action increases the entry of long-chain FAs into mitochondria and hence, an increase in their oxidation. Works on FA oxidation in skeletal muscles have shown a sequential activation of AMPK, p38MAPK and PPAR-α to be responsible for increased FA oxidation. But the signalling pathways and components involved in such sequential activation is not known. PPAR-α, a ligand-activated nuclear receptor, plays an important role in FA oxidation. This receptor is abundantly expressed in tissues like liver, heart, kidney and skeletal muscles, who meet their metabolic energy consumption from oxidation of FAs. It has been shown that HMW adiponectin fraction increases the PPAR-α target gene expression. Moreover, in IR rodent models, PPAR-α ligands have been found to reduce lipid levels and to improve insulin sensitivity. Several studies on humans and rodents have shown that both forms of adiponectin, HMW as well as low molecular weight (LMW), not only increase target gene expression of PPAR-α but also increase the phosphorylation of AMPK and p38MAPK. But such activity is more pronounced and better correlated with HMW fraction than that of LMW, suggesting a differential efficacy between the two fractions or involvement of multiple pathways in increasing FA oxidation in muscles[33].
Decreased ER stress: It has already been mentioned that mitochondrial dysfunction in adipocytes induces ER stress, which in turn reduces adiponectin transcription, leading to decreased production of this adipokine along with development of IR[29,31]. Moreover, as discussed earlier, adiponectin, via activated AMPK, also improves mitochondrial number and function in skeletal muscles[29]. From these two observations it may be inferred that adiponectin, by counteracting mitochondrial dysfunction (through improvement of mitochondrial function), decreases ER stress and improves its own secretion, which in turn may contribute towards reduction of IR. Mention has already been made about the IR-inducing and diabetogenic adipocytokine resistin[1,10,11], whose plasma concentration is high in IR, T2DM, metabolic syndrome and cardiovascular diseases[3]. In contrast, its sibiling adiponectin plasma concentration is low in such conditions[4], and it has favourable effects on them. Such contrasting effects of the two adipokines may be due to their comparable domain architecture, assembled in a multimeric form, which suggests a common regulatory mechanism (opposite to each other) on insulin-signalling pathway, as well as on mechanisms involved in glucose and lipid homeostasis. In IR and T2DM, hypoadiponectinemia along with hyperrestinemia have been found to antagonise insulin signalling by causing dephosphorylation and deactivation of the key enzyme AMPK in skeletal muscles and liver along with increased expression of genes coding for the synthesis of neoglucogenic enzymes as well as reduced expression of IRS-2 and glucose transporter, GLUT-2. The resultant effects of such action were decreased FFA oxidation in muscles, decreased hepatic uptake of glucose, increased neoglucogenesis and glycogenolysis leading to hyperglycemia and increased plasma FFA. Impaired FFA oxidation may be further aggravated by downregulated PPAR-α action[3].
Improvement in insulin signalling pathway: It is well established that insulin resistance is very often associated with inadequate functioning of post receptor signalling molecules including IRS. It has been demonstrated that adiponectin upregulates IRS-2 by activation of STAT-3 in liver. Such activation was also associated with increased production of IL-6 from macrophages - an adiponectin action mediated through activation of NF-κB, which does not require activation of classical AdipoR1 and AdipoR2 receptors. Upregulation of IRS-2 definitely improves insulin sensitivity, but exact mechanisms of such upregulation are not known. Probably, it is effected by an IL-6 dependent pathway, which is initiated by adiponectin, through its combination with yet another unidentified adiponectin receptor. Moreover, though adiponectin activates AMPK and PPAR-α through activation of its classical AdipoR1 and AdipoR2 receptors leading to increased FA oxidation and insulin sensitisation, it has not been possible to link AMPK and PPAR-α activation with the proper functioning of post-receptor insulin signalling molecules[37]. Experiments on skeletal muscles have demonstrated that AMPK activation by adiponectin occurs by two pathways, out of which one is a major one while the other plays a minor role. In the major pathway (the APPL1/LKB1-dependent pathway), AMPK activation needs the binding of adapter protein APPL1, which promotes the translocation of APPL1-dependent LKB1 into the cytosol where it is anchored. The same pathway has been found to be followed by the insulin sensitising drug metformin. Through the minor pathway (the phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathway), via activation of phospholipase C, Ca2+ is released from the intracellular calcium ion stores that plays a minor role in activation of AMPK[38].
Increased mitochondrial number and function: Works on skeletal muscles have shown that adiponectin, through AMPK activation, not only increases mitochondrial function, but also increases their number. As activated AMPK in skeletal muscles has been found to stimulate mitochondrial biogenesis under conditions of chronic energy deprivation or endurance training, it appears that adiponectin-induced-increase in mitochondrial number is due to stimulation of mitochondrial biogenesis. This action of the adipokine points towards its insulin sensitising action, because mitochondrial function in skeletal muscles is taken as an indicator of whole-body insulin sensitivity. Thus, it may be presumed that adipocyte-mitochondrial action, which regulates adiponectin synthesis in adipocytes (already discussed), also regulates skeletal muscle-mitochondrial (or metabolic) activity and insulin action in skeletal muscles through adiponectin[29].
Increased insulin secretion: Though adiponectin does not have any effect on normal insulin secretion, the adipokine has been found to increase it in insulin resistant mice fed with high fat diet. But in such mice, augmentation of secretion occurs only in response to high plasma glucose, but actually inhibited when plasma glucose was low. Adiponectin appears to possess a protective effect on islet-β-cells, as it has been found to reduce the pro-apoptotic effect of FFAs and other cytokines on β-cells[5].
Decreased hepatic output of glucose: Several workers have demonstrated the capacity of adiponectin to decrease hepatic output of glucose, thereby contributing towards reduction of plasma glucose concentration and hence, increased insulin sensitivity[5,10]. One of the important causes of increased hepatic output of glucose in diabetes mellitus is increased neoglucogenesis due to inadequate insulin action. As mentioned earlier, adiponectin inhibits hepatic neoglucogenesis by decreasing the formation of two important enzymes concerned, through interference with the mRNA expression that is necessary for the synthesis of these enzymes[10]. Moreover, adiponectin, by increasing the oxidation of FAs, decreases their availability for utilization in the process of neoglucogenesis.
Increased uptake of glucose by liver and muscle: Adiponectin has been found to increase the uptake of glucose by liver and muscles which appears to result from improvement in insulin signalling pathway, leading to better insulin action and hence, decreased blood sugar and increased insulin sensitivity[5].
Increased glucose metabolism: It has been observed that in obese individuals with IR and in patients having metabolic syndrome (who are IR), adiponectin receptors are downregulated, which suggests inadequate adiponectin action as the cause of IR. In vitro and in vivo experiments on skeletal muscles have shown adiponectin to increase glucose metabolism and insulin sensitivity via activation of AMPK[31].
AR (adiponectin: Resistin) and IRAR indices: Upregulated resistin, which is followed by PPAR-α downregulation, has been found to impair adipocyte differentiation, leading to dramatic decrease in adiponectin formation. Because of such inverse relationship with respect to both secretion and function, it seems to be more predictive to use their ratio (AR index-adiponectin: Resistin) in linking obesity with T2DM than using either of them alone[3].
Besides AR index another novel IRAR index has been coined that seems to be a strong indicator of degree of IR in T2DM. The index appears to relate IR with AR. As expected, AR index value gets smaller and smaller according to the degree of obesity (which determines the magnitude of hypoadiponectinemia with hyperresistinemia), resulting in a parallel rise of IR. Hence, greater the IRAR index value, more is the degree of IR in T2DM.
As IR in T2DM is the major determinant of progression into metabolic syndrome, which in turn, laids the foundation for other complications of diabetes, this index may also be used to predict the arrival of T2DM complications[3].
FACTORS NOT OF ADIPOCYTE ORIGIN
In addition to these adipokines, there are some other factors (not of adipocyte origin), whose role in linking obesity with T2DM cannot be ignored. These factors include PPARs, carnitine, calcium, angiotensin II and toll-like receptors (TLRs).
PPARs
This nuclear receptor family, consisting of PPAR-α, PPAR-γ and PPAR-δ, are primarily related with lipid metabolism having fatty acids and their derivatives as their endogenous ligands.
PPAR-α: Besides interference with several steps of lipid metabolism, the main results of this receptor activation is increased oxidation of FA that leads to decreased plasma level of TG by decreasing its synthesis and storage in adipocytes. Moreover, PPAR-α activation, along with activation of PPAR-γ, has been found not only to increase the formation and secretion of adiponectin but also to upregulate AdipoR1/AdipoR2[7].
PPAR-γ: These receptors, mainly expressed in liver and adipose tissue, on stimulation, cause gene expression necessary for differentiation of fibroblasts into adipocytes, and for lipid synthesis and storage in adipocytes. Because of their lipogenicity, they seem to decrease insulin sensitivity rather than increase it. But, their exogenous agonists-TZDs, have been found to decrease IR and increase insulin sensitivity. Such paradoxical actions of TZDs, have been shown to be due to reduced lipotoxicity in liver and skeletal muscles because of lipid storage in adipocytes, and increase in number of small adipocytes, which are not only more sensitive to insulin action, but also secrete large quantity of adiponectin (insulin-sensitising), while decreasing the release of resistin and TNF-α (both are IR-inducing)[7].
PPAR-δ: Main result of this receptor activation is increased FA oxidation, which contributes towards decreasing IR and increasing insulin sensitivity[7].
It may be noted that the results of activation of these three receptors, particularly activation of those of PPAR-α and PPAR-γ, are beneficial in IR and insulin sensitivity through their interference with adipocyte number (increased number of small adipocytes) and function (increased production of adiponectin and decreased production of resistin and TNF-α), FA oxidation (which decreases TG formation in adipocytes resulting in decreased obesity) and upregulation of AdipoR1 and AdipoR2 (decreased IR and increased insulin sensitivity). As all these functions finally lead to reduced obesity, this receptor family can be considered to play a role in linking obesity and T2DM.
Carnitine
This vitamin and amino acid, which is derived from yeast, milk, liver and muscles (in large quantities), increases FFA oxidation through carnitine shuttle reactions. In this reaction, carnitine has been found not only to favour entry of long-chain FFAs across the mitochondrial membrane, but also facilitate the transport of fatty acyl-CoA into mitochondrial matrix for β-oxidation. Therefore, carnitine deficiency, which is commonly found in several IR cases, leads to increased concentration of plasma FFA and hence, their increased conversion into TG in adipocytes, resulting in obesity and further aggravation of IR. Moreover, relative carnitine deficiency may occur in prolonged metabolic stress, which may add to mitochondrial dysfunction, leading to reduced glucose tolerance. These two factors may contribute towards obesity-associated IR in T2DM. Therefore, like PPAR-receptor family action, carnitine function in the body may contribute towards linking obesity with diabetes as its deficiency is reflected upon genes of obesity and IR[7].
Calcium
Role of calcium in vario us cellular secretory processes[39], including secretion of insulin from islet β-cells, is well established. Improper regulation of intracellular calcium has been found to affect insulin secretion and its tissue sensitivity adversely[40]. High calcium intake alone or with vitamin D has been shown to reduce not only body weight and fat mass, but also to decrease weight gain and adipocyte fat accumulation. The mechanisms suggested for such beneficial actions include adipocyte apoptosis and reduced adipogenesis along with deranged lipid metabolism[40,41]. Moreover, epidemiological studies have shown that low calcium intake and poor vitamin D status are associated with increased risk of obesity[36]. From such observations, it may be inferred that obesity, thus developed, may lead to increased production of IR-inducing and diabetogenic adipokines, thereby linking it (obesity) with IR and T2DM.
Angiotensin II
Renin-angiotensin-aldosterone system, whose primary function is to maintain water and electrolyte balance of the body and to regulate blood pressure, is known to mediate its function by formation of angiotensin II (AngII). AngII formation occurs through several steps where renin of renal origin converts angiotensinogen of hepatic origin to AngI, which is then converted to AngII by the enzyme angiotensin-converting enzyme (ACE) of endothelial cell origin[42]. But recently, a local RAAS has been demonstrated in several tissues of the body including adipose tissue, which is involved in several functions of the adipocytes including adipose tissue growth and cell differentiation. It has been shown that when AT2 receptors (one of the subtypes of angiotensin receptor) are deleted from adipocytes, the cell size is reduced, and there is protection from diet-induced obesity and IR[43]. Such observations suggest an additional beneficial role of ACE inhibitors and AT2 receptor blockers, when used as antihypertensives in patients having hypertension with obesity and T2DM[44]. Moreover, like low Ca2+ and poor vitamin D status, locally generated AngII, via its action on adipocytes, may link obesity with T2DM.
TLRs
TLRs are transmembrane glycoprotein receptors whose known function is antigen recognition[6,45]. Recently, substantial evidences have been put forward which suggest their pathological role in genesis of obesity. In this respect, both TLR-2 and TLR-4 have been found to be overexpressed on adipocytes in obese persons having T2DM. Such overexpressed TLR receptors along with similarly overexpressed adipokines in adipose tissue of obese individuals may play an important role in obesity-associated meta inflammation resulting in IR and T2DM. It has been demonstrated that inhibition of TLR-2 in skeletal muscles and white adipose tissue of mice fed with high fat diet, improves insulin sensitivity and signalling[43].
Moreover, overexpression of TLRs on adipocytes may also suggest an important role of adipose tissue in the regulation of inflammation and innate immunity in human beings by modulating TLR/NF-κB regulatory pathway. Such observations suggest a modulatory role of TLRs in the interaction between the pathways of inflammation and metabolism[43]. The above- discussed roles of TLRs in genesis of obesity, reduction of insulin signalling and sensitivity, and modulation of the interacting pathways of inflammation and metabolism appear to support the correlation between obesity and T2DM.
CONCLUSION
From the discussions made so far, it may be observed that results obtained from extensive research work on the factors supposed to link obesity with T2DM, very clearly show an intimate relationship between the two, for which both adipocytokines as well as some factors not derived from adipocytes have been implicated. Of them, few (Adiponectin, Leptin, PPAR, Carnitine, Apelin and Calcium) are beneficial, while others (TNF-α, IL-6, Resistin, RBP-4, DPP-4, PAI-1, Visfatin, FFA, AngII and TLR) are harmful, but all of them play a definite role in linking obesity with T2DM (mentioned earlier). Among these, adiponectin has been found to play a crucial and seemingly complicated but definite role. Such studies may be extended to all concerned factors giving emphasis on mitochondrial and ER stresses. Finally, using these agents, drugs may be designed which will be helpful to prevent the development of obesity, thereby producing a beneficial response in prevention, progression and treatment of T2DM.
Footnotes
P- Reviewer: Chang ST, Tarantino G S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Conflict-of-interest statement: The author has no conflict of interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 14, 2015
First decision: August 25, 2015
Article in press: October 27, 2015
References
- 1.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 2.Sentinelli F, Romeo S, Arca M, Filippi E, Leonetti F, Banchieri M, Di Mario U, Baroni MG. Human resistin gene, obesity, and type 2 diabetes: mutation analysis and population study. Diabetes. 2002;51:860–862. doi: 10.2337/diabetes.51.3.860. [DOI] [PubMed] [Google Scholar]
- 3.Lau CH, Muniandy S. Novel adiponectin-resistin (AR) and insulin resistance (IRAR) indexes are useful integrated diagnostic biomarkers for insulin resistance, type 2 diabetes and metabolic syndrome: a case control study. Cardiovasc Diabetol. 2011;10:8. doi: 10.1186/1475-2840-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shehzad A, Iqbal W, Shehzad O, Lee YS. Adiponectin: regulation of its production and its role in human diseases. Hormones (Athens) 2012;11:8–20. doi: 10.1007/BF03401534. [DOI] [PubMed] [Google Scholar]
- 5.Hajer GR, van Haeften TW, Visseren FL. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J. 2008;29:2959–2971. doi: 10.1093/eurheartj/ehn387. [DOI] [PubMed] [Google Scholar]
- 6.Rang HP, Dale MM, Ritter JM, Flower RJ, Handerson G. Rang and Dale’s Pharmacology. 7th ed. Edinburgh: Churchill Livingstone; 2012. pp. 77–88. [Google Scholar]
- 7.Chakraborti CK. Possible links between obesity and type 2 diabetes mellitus. Int J Pharm Sci Res. 2012;3:1935–1945. Available from: http://www.ijpsr.com. [Google Scholar]
- 8.Swaroop JJ, Rajarajeswari D, Naidu JN. Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J Med Res. 2012;135:127–130. doi: 10.4103/0971-5916.93435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borst SE. The role of TNF-alpha in insulin resistance. Endocrine. 2004;23:177–182. doi: 10.1385/ENDO:23:2-3:177. [DOI] [PubMed] [Google Scholar]
- 10.Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12. [PubMed] [Google Scholar]
- 11.Kusminski CM, McTernan PG, Kumar S. Role of resistin in obesity, insulin resistance and Type II diabetes. Clin Sci (Lond) 2005;109:243–256. doi: 10.1042/CS20050078. [DOI] [PubMed] [Google Scholar]
- 12.Wolf G. Serum retinol-binding protein: a link between obesity, insulin resistance, and type 2 diabetes. Nutr Rev. 2007;65:251–256. doi: 10.1111/j.1753-4887.2007.tb00302.x. [DOI] [PubMed] [Google Scholar]
- 13.Bose KS, Gupta SK, Singh S. Is serum retinol binding protein-4: A predictor for diabetes in genetically high risk population? J Res Med Sci. 2012;17:1015–1019. [PMC free article] [PubMed] [Google Scholar]
- 14.Erikstrup C, Mortensen OH, Nielsen AR, Fischer CP, Plomgaard P, Petersen AM, Krogh-Madsen R, Lindegaard B, Erhardt JG, Ullum H, et al. RBP-to-retinol ratio, but not total RBP, is elevated in patients with type 2 diabetes. Diabetes Obes Metab. 2009;11:204–212. doi: 10.1111/j.1463-1326.2008.00901.x. [DOI] [PubMed] [Google Scholar]
- 15.Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, Eckardt K, Kaufman JM, Ryden M, Müller S, et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60:1917–1925. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blüher M. Adipokines - removing road blocks to obesity and diabetes therapy. Mol Metab. 2014;3:230–240. doi: 10.1016/j.molmet.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Correia ML, Haynes WG. A role for plasminogen activator inhibitor-1 in obesity: from pie to PAI? Arterioscler Thromb Vasc Biol. 2006;26:2183–2185. doi: 10.1161/01.ATV.0000244018.24120.70. [DOI] [PubMed] [Google Scholar]
- 18.De Taeye B, Smith LH, Vaughan DE. Plasminogen activator inhibitor-1: a common denominator in obesity, diabetes and cardiovascular disease. Curr Opin Pharmacol. 2005;5:149–154. doi: 10.1016/j.coph.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 20.Sethi JK, Vidal-Puig A. Visfatin: the missing link between intra-abdominal obesity and diabetes? Trends Mol Med. 2005;11:344–347. doi: 10.1016/j.molmed.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adeghate E. Visfatin/PBEF- a new natural insulin-mimetic adipokine. Curr Med Chem. 2008;15:1851–1862. doi: 10.2174/092986708785133004. [DOI] [PubMed] [Google Scholar]
- 22.Chang YC, Chang TJ, Lee WJ, Chuang LM. The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism. 2010;59:93–99. doi: 10.1016/j.metabol.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Castan-Laurell I, Dray C, Attané C, Duparc T, Knauf C, Valet P. Apelin, diabetes, and obesity. Endocrine. 2011;40:1–9. doi: 10.1007/s12020-011-9507-9. [DOI] [PubMed] [Google Scholar]
- 24.Castan-Laurell I, Dray C, Knauf C, Kunduzova O, Valet P. Apelin, a promising target for type 2 diabetes treatment? Trends Endocrinol Metab. 2012;23:234–241. doi: 10.1016/j.tem.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Attané C, Foussal C, Le Gonidec S, Benani A, Daviaud D, Wanecq E, Guzmán-Ruiz R, Dray C, Bezaire V, Rancoule C, et al. Apelin treatment increases complete Fatty Acid oxidation, mitochondrial oxidative capacity, and biogenesis in muscle of insulin-resistant mice. Diabetes. 2012;61:310–320. doi: 10.2337/db11-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Li X, Wang M, Ning H, A L, Li Y, Sun C. Lipoprotein lipase links vitamin D, insulin resistance, and type 2 diabetes: a cross-sectional epidemiological study. Cardiovasc Diabetol. 2013;12:17. doi: 10.1186/1475-2840-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18:139–143. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lubos E, Handy DE, Loscalzo J. Role of oxidative stress and nitric oxide in atherothrombosis. Front Biosci. 2008;13:5323–5344. doi: 10.2741/3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koh EH, Park JY, Park HS, Jeon MJ, Ryu JW, Kim M, Kim SY, Kim MS, Kim SW, Park IS, et al. Essential role of mitochondrial function in adiponectin synthesis in adipocytes. Diabetes. 2007;56:2973–2981. doi: 10.2337/db07-0510. [DOI] [PubMed] [Google Scholar]
- 30.Blüher M, Williams CJ, Klöting N, Hsi A, Ruschke K, Oberbach A, Fasshauer M, Berndt J, Schön MR, Wolk A, et al. Gene expression of adiponectin receptors in human visceral and subcutaneous adipose tissue is related to insulin resistance and metabolic parameters and is altered in response to physical training. Diabetes Care. 2007;30:3110–3115. doi: 10.2337/dc07-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boddu NJ, Theus S, Luo S, Wei JY, Ranganathan G. Is the lack of adiponectin associated with increased ER/SR stress and inflammation in the heart? Adipocyte. 2014;3:10–18. doi: 10.4161/adip.26684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marcotorchino J, Gouranton E, Romier B, Tourniaire F, Astier J, Malezet C, Amiot MJ, Landrier JF. Vitamin D reduces the inflammatory response and restores glucose uptake in adipocytes. Mol Nutr Food Res. 2012;56:1771–1782. doi: 10.1002/mnfr.201200383. [DOI] [PubMed] [Google Scholar]
- 33.Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55:2562–2570. doi: 10.2337/db05-1322. [DOI] [PubMed] [Google Scholar]
- 34.Shu AD, Myer MG. Pharmacology of the endocrine pancreas. In: Golan DE, Tashjian AH, Armstrong EJ, Galanter JM, Armstrong AW, et al., editors. Principles of Pharmacology: The pathophysiologic basis of drug therapy. Philadelphia: Lippincott Williams and Wilkins; 2005. pp. 457–469. [Google Scholar]
- 35.Zoico E, Garbin U, Olioso D, Mazzali G, Fratta Pasini AM, Di Francesco V, Sepe A, Cominacini L, Zamboni M. The effects of adiponectin on interleukin-6 and MCP-1 secretion in lipopolysaccharide-treated 3T3-L1 adipocytes: role of the NF-kappaB pathway. Int J Mol Med. 2009;24:847–851. doi: 10.3892/ijmm_00000302. [DOI] [PubMed] [Google Scholar]
- 36.Bernstein EL, Koutkia P, Ljungquist K, Breu J, Canavan B, Grinspoon S. Acute regulation of adiponectin by free fatty acids. Metabolism. 2004;53:790–793. doi: 10.1016/j.metabol.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 37.Awazawa M, Ueki K, Inabe K, Yamauchi T, Kubota N, Kaneko K, Kobayashi M, Iwane A, Sasako T, Okazaki Y, et al. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab. 2011;13:401–412. doi: 10.1016/j.cmet.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 38.Zhou L, Deepa SS, Etzler JC, Ryu J, Mao X, Fang Q, Liu DD, Torres JM, Jia W, Lechleiter JD, et al. Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J Biol Chem. 2009;284:22426–22435. doi: 10.1074/jbc.M109.028357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penner R, Neher E. The role of calcium in stimulus-secretion coupling in excitable and non-excitable cells. J Exp Biol. 1988;139:329–345. doi: 10.1242/jeb.139.1.329. [DOI] [PubMed] [Google Scholar]
- 40.Pittas AG, Dawson-Hughes B, Li T, Van Dam RM, Willett WC, Manson JE, Hu FB. Vitamin D and calcium intake in relation to type 2 diabetes in women. Diabetes Care. 2006;29:650–656. doi: 10.2337/diacare.29.03.06.dc05-1961. [DOI] [PubMed] [Google Scholar]
- 41.Song Q, Sergeev IN. Calcium and vitamin D in obesity. Nutr Res Rev. 2012;25:130–141. doi: 10.1017/S0954422412000029. [DOI] [PubMed] [Google Scholar]
- 42.Jackson EK. Renin and angiotensin. In: Brunton LL, Lazo JS, Parker KL, et al., editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. New York: Mc Graw-Hill Medical Publishing Division; 2006. pp. 789–821. [Google Scholar]
- 43.vinh quốc Lu’o’ng K, Nguyễn LT. The beneficial role of vitamin D in obesity: possible genetic and cell signaling mechanisms. Nutr J. 2013;12:89. doi: 10.1186/1475-2891-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnston CI. Angiotensin receptor antagonists for the treatment of hypertension. Aust Prescr. 1998;21:95–97. [Google Scholar]
- 45.Stafford JL, Ellestad KK, Magor KE, Belosevic M, Magor BG. A toll-like receptor (TLR) gene that is up-regulated in activated goldfish macrophages. Dev Comp Immunol. 2003;27:685–698. doi: 10.1016/s0145-305x(03)00041-7. [DOI] [PubMed] [Google Scholar]