

Abstract
Almost all known members of the cytochrome P450 (CYP) superfamily conserve a key cysteine residue that coordinates the heme iron. Although mutation of this residue abolishes monooxygenase activity, recent work has shown that mutation to either serine or histidine unlocks non-natural carbene- and nitrene-transfer activities. Here we present the first crystal structure of a histidine-ligated P450. The T213A/C317H variant of the thermostable CYP119 from Sulfolobus acidocaldarius maintains heme iron coordination through the introduced ligand, an interaction that is accompanied by large changes in the overall protein structure. We also find that the axial cysteine C317 may be substituted with any other amino acid without abrogating folding and heme cofactor incorporation. Several of the axial mutants display unusual spectral features, suggesting that they have active sites with unique steric and electronic properties. These novel, highly stable enzyme active sites will be fruitful starting points for investigations of non-natural P450 catalysis and mechanisms.
Introduction
Axial ligation to the iron center is the principal mechanism through which hemoproteins tune the electronic properties of the heme cofactor, with a diverse set of amino acid residues (His, Tyr, Met, Lys, and Cys) called upon to act as ligands in naturally occurring hemoproteins.1 Among cytochrome P450s (CYPs), axial thiolate ligation plays a key role at several points during the catalytic cycle. Thiolate ligation enables redox-gating of oxygen activation,2 favors O–O bond heterolysis rather than homolysis,3 and markedly enhances the basicity of compound II,4 which favors hydrogen atom abstraction chemistry rather than 1e– oxidations typical of peroxidases. Although the Cys at the axial position is almost universally conserved among known naturally occurring P450s, a few engineered proteins with mutations at this position have been characterized as mechanistic probes of P450 monooxygenase chemistry.5 P450 axial mutants no longer catalyze monooxygenation and are also frequently impaired with respect to heme incorporation and soluble expression. Recently, this laboratory has shown that mutation of the axial ligand in several P450s strongly promotes non-natural carbene- and nitrene-transfer activities in the presence of carbene and nitrene precursors and suitable acceptor substrates.6 The conservative Cys-to-Ser substitution increases those activities, particularly in whole E. coli cells.6 An axial histidine mutant of P450BM3 was also shown to be highly active as a carbene-transfer catalyst.7
The similar sizes and side-chain functionalities of Ser and Cys would suggest that Ser coordinates in the same way as the native Cys ligand, and indeed, this has been confirmed in the X-ray crystal structures of two variants of P450BM3 containing the axial Cys-to-Ser mutation.6a,8 The hemes are superimposable in the Cys- and Ser-ligated structures, and there is no change in the conformation of the proximal loop that contains the axial ligand. In contrast, it is difficult to envision how the proximal pocket can accommodate heme coordination by the much bulkier side chain of histidine. In fact, previous attempts to prepare P450 axial variants with bulky ligands such as His or Tyr have resulted in low expression and poor heme incorporation,5a−5c,9 perhaps owing to unfavorable steric interactions imposed by these residues.
Despite longstanding interest in the structures of P450 axial mutants specifically and P450 structure and function more generally,10 no X-ray crystal structural data have been reported that reveal how the protein maintains heme binding and ligation when the axial Cys is substituted by an amino acid of such different size and chemical properties. Given the fundamental interest in exploring how enzyme active sites may be retuned for alternative catalytic functions, either in nature or the laboratory and the fact that accurate structural data are often vital to successful engineering efforts, we sought to determine the structure of an axial Cys-to-Ser mutant of a CYP. Because thermophilic enzymes have a greater tendency to yield high-quality protein crystals suitable for X-ray crystallography, we chose to focus our efforts on the highly thermostable and well-studied CYP119 from S. acidocaldarius.11 Here we describe the crystal structure of a histidine-ligated CYP119 and show that histidine heme iron ligation is accompanied by large local and global conformational changes in the protein. We further show that the universally conserved axial cysteine may be substituted in CYP119 with any other amino acid without abrogating heme incorporation or production of soluble enzyme. These results demonstrate an impressive level of structural adaptability in the P450 fold.
Results and Discussion
We set out to crystallize the axial Cys-to-His single mutant (C317H) of CYP119 as well as a variant containing an additional mutation to the conserved distal threonine residue (double mutant T213A/C317H). Mutations to both of these conserved residues have been shown to be highly activating of non-natural P450 catalysis.6,12 We were delighted when the double mutant yielded protein crystals that diffracted to 2.7 Å resolution, revealing an unusual conformation of the P450 fold and the first structure of a histidine-ligated P450.
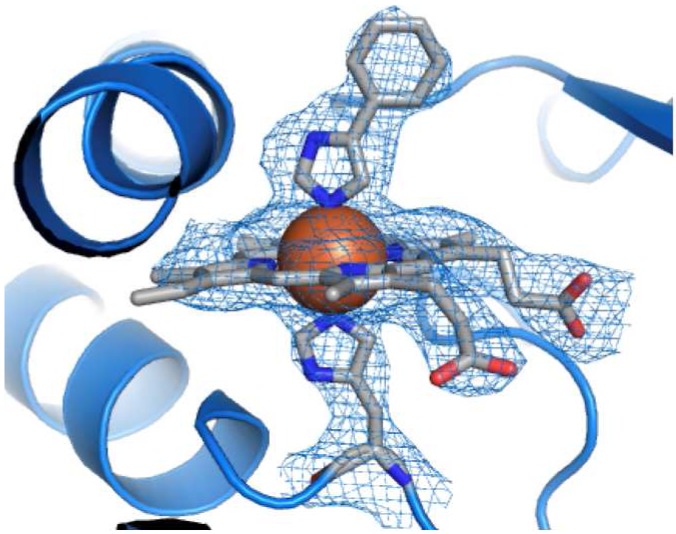
We cocrystallized the T213A/C317H variant with the P450 inhibitor phenylimidazole used in the determination of the crystal structure of wild-type CYP11911c so that we could compare the two structures. In each of the four protein molecules in the asymmetric unit of the T213A/C317H structure, the heme is present and is clearly histidine-ligated, with well-resolved electron density for H317 extending onto the heme iron (Figure 1; structure statistics presented in Table S1). Although there are some differences among the protomers, each shows a significant rearrangement of the proximal loop where H317 resides. This movement of the backbone shifts Cα of H317 by 1.2 Å away from the heme center (as measured to the wild-type CYP119 Fe atom). However, the rearrangement of the proximal loop alone is apparently insufficient to accommodate non-native ligation because histidine ligation also induces a striking 17° tilt in the plane of the porphyrin ring away from the axial residue (Figure 2A). The porphyrin tilt, in turn, requires a large compensating adjustment by the protein backbone in order to maintain coordination of the heme propionate side-chains. The interaction between the heme 7-propionate and R259 of the protein is easily accommodated by alternate rotamers of the heme propionates and enzyme amino acid side-chains (Figure 2B), which apparently obviate the need for any large-scale movement in the β sheet that contains R259. In contrast, there is a large adjustment of the C helix, in which sits a key histidine residue (H76) that forms a salt bridge to the heme 6-propionate at a distance of 2.8 Å in the wild-type structure (Figure 2A). This salt bridge is maintained in 2 out of 4 protomers in the mutant structure via a 7 Å displacement of the C helix (measured from Cα to Cα). This interaction is not observed in the other two protomers because of increased disorder in the C helix. Additionally, H315, which in the wild-type structure also forms a salt bridge to the heme 6-propionate at a distance of 2.7 Å, is shifted by 4 Å in the structure of the double mutant (Cα–Cα). As a result, this interaction is significantly longer in the mutant structure (4.5 Å).
Figure 1.
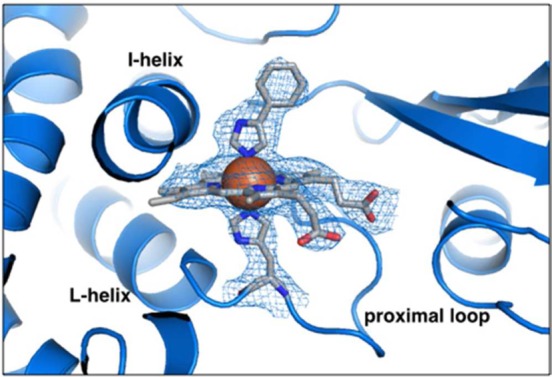
His coordination of heme center in CYP119 T213A/C317H.
Figure 2.
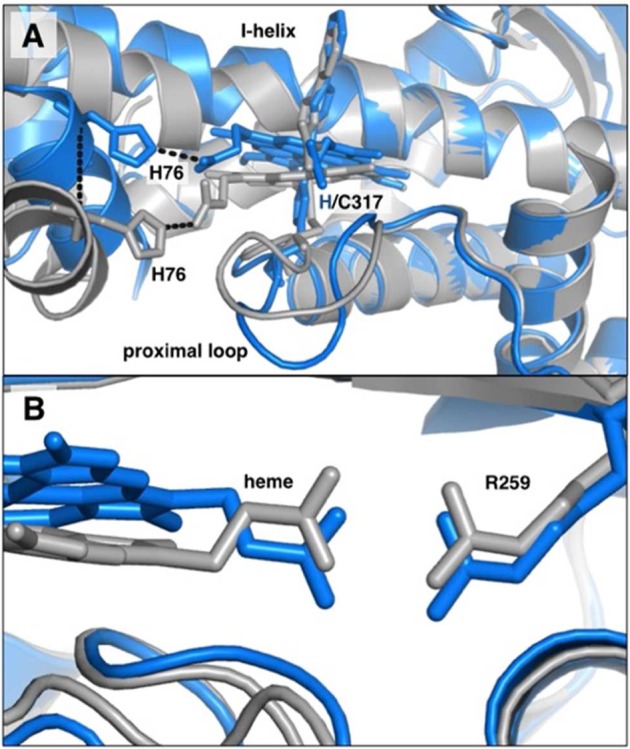
(A) His coordination of 6-propionate in wild-type CYP119 (gray) and T213A/C317H mutant (blue). (B) Arg coordination of 7-propionate in wild type (gray) and T213A/C317H mutant (blue).
These local perturbations are amplified at a global level, leading to significant distortions of the overall structure of the double mutant relative to that of wild type (Figure 3). Comparison of the gross conformational changes between wild-type CYP119 and its T213A/C317H variant is highly dependent upon the method used to superimpose the two structures. Nevertheless, least-squares (LSQ) alignment across all Cα atoms reveals that the wild-type and mutant structures differ by an astounding 4.7 Å RSMD.
Figure 3.

Overall structure of CYP119 T213A/C317H (left, blue) and wild-type CYP119 (right, gray) with selected helices labeled; only regions that are structured in the mutant are shown in the wild-type structure.
Several portions of the protein are unresolved in the T213A/C317H structure, including the F–G loop as well as the F and B′ helices, which are resolved in the wild-type structure 1F4T. Perhaps the most dramatic change is observed in the G helix, which is displaced by 14 Å in a hingelike movement away from the heme relative to the wild-type 4-phenylimidazole-bound structure (presumed to be a closed conformation). This shift is very different from the open–closed conformational motion of wild-type CYP119, which involves a significant movement (>10 Å) of the F–G loop but a relatively minor hingelike movement of the G helix (∼1 Å shift measured from K166 Cα–Cα using 4-phenylimidazole-bound (1F4T) and substrate-free (1IO7) structures).11c,13 In contrast, CYP119 T213A/C317H undergoes a much more significant hingelike movement of the G helix relative to that of the wild-type inhibitor-bound structure (14 Å as measured from K166 Cα–Cα), perhaps resulting from the significant adjustment forced by the porphyrin tilt.
Also unusual is the reorientation of the H helix and complete removal of the kink in the I helix, which results in a roughly 8 Å hinge movement at the beginning of the helix. Within the portion of the I helix that is closest to the heme also containing the T213A mutation, the helix geometry is very different from that of wild type. In particular, several water molecules are inserted within the I helix of the wild-type protein that disrupt direct intrahelix hydrogen bonds. In the His-ligated structure, the helix has expelled these waters and shifted register by a full residue to facilitate new direct backbone–backbone N–H·O=C hydrogen bonds (Figure 4). It is tempting to attribute the I-helix changes to the distal T213A mutation, but previous structures of P450s containing distal threonine-to-alanine mutations (T213A equivalent) are virtually superimposable with corresponding wild-type structures, both globally and locally. For example, P450BM3 and its distal threonine T268A mutant (2IJ2 and 1YQO, respectively) differ by a RMSD of 0.5 Å, whereas P450nor and its T243A mutant (1ROM and 1F24, respectively) differ by 0.4 Å. If we consider only the I helix, then the divergences in previously described distal threonine mutants appear even smaller: 0.2 Å for P450BM3 and its T268A mutant and 0.3 Å for P450nor and its T243A mutant. In contrast, the T213A/C317H structure shows a much greater distortion within the I helix (RMSD of 3.2 Å relative to wild type using only backbone atoms), which strongly suggests that the observed effect on the I helix is due to the axial mutation and the accompanying porphyrin tilt. Notably, the heme cofactor would clash with the I helix in its tilted state if there were not some compensatory change in the protein structure; the T213A mutation may enable this movement to some degree (see below).
Figure 4.
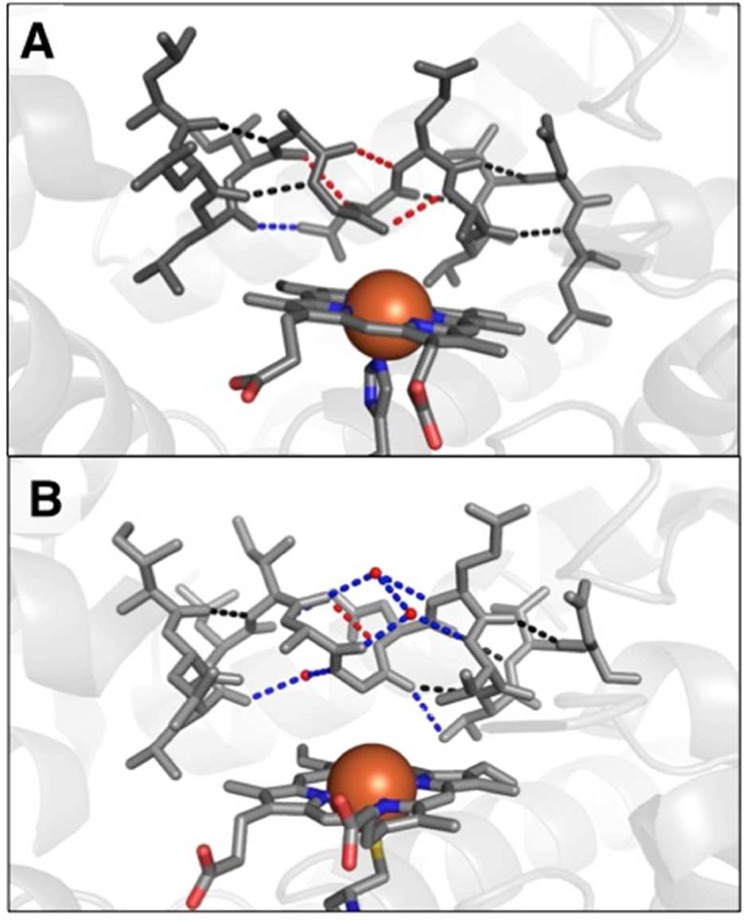
I-helix structure for (A) CYP119 T213A/C317H mutant (PDB: 5BV5) and (B) wild type (PDB: 1F4T) showing ordered waters in wild-type structure and the compression and register shift of the I helix in the T213A/C317H mutant. Iron atom shown as van der Waals sphere; phenylimidazole inhibitor not shown for clarity.
On the whole, large distortions in the overall fold observed in the T213A/C317H structure point to an impressive flexibility in the P450 fold that allows it to accommodate what would seem to be a highly disruptive mutation at the axial position. Reasoning that other substitutions should also be tolerated, we generated all possible single-axial mutants of wild-type CYP119 and expressed and purified the resulting proteins. To our surprise, all 19 possible substitutions at the universally conserved axial position resulted in expression of heme-containing holoprotein.
We first characterized the ferric, ferrous, and ferrous CO-bound spectra of the axial variants by UV–vis spectroscopy to determine whether we could discern different active site coordination environments. On the basis of these data, we grouped the axial mutants into two broad categories. The first group (C317A/G/S/T) displayed UV–vis spectral properties distinct from those of the His-ligated T213A/C317H enzyme as well as from one another. (Complete spectra are presented in Table S2 and Figure S1.) Particularly striking are the differences between the C317A and C317G variants because one might intuitively expect both to be water-ligated and thus to evince very similar spectra. However, the C317A and C317G enzymes are spectrally different from one another and from previously characterized Ala and Gly hemoprotein mutants.14 The C317A variant has a dark-brown color, a ferric Soret maximum at 405 nm, and a featureless α,β region. This variant also has an extremely blue-shifted ferrous CO-bound Soret maximum at 400 nm. C317G, by contrast, is bright red and has a ferrous CO Soret peak at 414 nm. In the ferric state, C317G has well-resolved α and β bands, suggesting either a mixed- or low-spin heme state.14a Axial serine and threonine variants also have distinct spectra, though both are fairly similar to those of previously described axial serine mutants.5a,5b,6a,9,15 In particular, C317S showed a roughly 5 nm blueshift in the ferric and ferrous CO-bound states relative to C317T.
The second group, which encompasses the majority of the axial variants, had ferric, ferrous, and ferrous CO-bound spectra that were quite similar to those of the structurally characterized histidine-ligated T213A/C317H double mutant and the C317H single mutant (Figure S1). These observations suggest that these C317X variants undergo a rearrangement within the proximal heme pocket that results in an alternative residue, presumably a histidine, assuming the coordinating role. Such rearrangements have been invoked to explain the observation of inactive “P420” preparations giving ferrous CO-bound Soret peaks at ∼420 nm.16 Indeed, like many other P450 enzymes, CYP119 has a histidine (H315) two residues upstream from the axial cysteine; H315 is a likely suspect for this alternative coordinating ligand.
Having observed the apparent resilience of CYP119 to mutation at C317, we wished to determine the effects of the C317X mutations on protein stability and cofactor incorporation. Relative levels of heme incorporation were measured by taking the ratio of the 276 nm (protein) and 395 nm (heme) absorbance for acid-denatured CYP119 variants. (See Supporting Information for assay description.) Surprisingly, we found that many of the C317X variants incorporated heme at levels even higher than did the wild-type enzyme, whereas a few showed lower levels (Table S3). Previous reports have noted that wild-type P450 and other hemoprotein preparations often contain a significant fraction of apoprotein,17 consistent with our observation of incomplete heme incorporation in wild type CYP119.
All 19 axial mutants of CYP119 were less thermostable than the wild-type enzyme, as measured by T50, the temperature at which half the protein is irreversibly denatured after a 10 min incubation (Table S4 and Figure S2). The maximum destabilization was 8 °C in T50 for C317A relative to that of wild type (T50 = 84.6 °C), with an average loss of ∼6 °C. The T213A/C317H double mutant experiences a relatively small loss of 1.2 °C in its T50, despite the very significant structural distortion. Although the T213A mutation has a slight stabilizing effect (+1.1 °C relative to that of wild type), it is significantly more stabilizing in the context of the C317H mutant (+4.9 °C). This suggests that the T213A mutation indeed serves to reduce a steric clash between the porphyrin and the I helix and to compensate for the C317H substitution in the T213A/C317H double mutant.
The exceptional thermostability of CYP119 undoubtedly aided our efforts to determine the effects of axial mutations on the P450 structure and spectral properties by providing a buffer for the significantly destabilizing effects of the axial mutations. Making corresponding axial mutations in mesophilic P450 enzymes, many of which are only marginally stable, may be challenging unless compensatory mutations (such as the distal threonine-to-alanine mutation) are also included. Indeed, P450cam axial histidine mutants are much more difficult to prepare than the corresponding wild-type enzyme, requiring either enzyme refolding5e or reconstitution.5d Even where bulky axial substitutions are reasonably well-tolerated, as in P450BM3,7 they may lead to increased conformational heterogeneity, making structural determination especially challenging. In fact, despite repeated efforts, we have been unable to obtain crystal structures of P450BM3 axial histidine mutants.
Conclusions
CYP119 accommodates heme iron coordination by the bulky histidyl side chain with a large tilt in the porphyrin ring and a significant rearrangement in the overall protein fold. Despite this, the His-ligated protein is still highly thermostable and efficiently incorporates heme. Given the impressive adaptability of the P450 fold revealed by this structure, we generated all 19 possible axial mutants of CYP119 and found that they could all be isolated as heme-containing holoproteins. This result further underscores the high degree of structural adaptability in the P450 fold.
It was believed until recently that the axial Cys ligand was universally conserved among CYPs. Several P450 sequences have been identified, however, that appear to lack an axial cysteine,18 which means that there is not a single universally conserved motif or residue in the vast and functionally diverse CYP superfamily.18a Although the structures and functions of these putative non-Cys-ligated CYPs are unknown, it is likely that new chemistry remains to be discovered among nature’s astonishing CYP diversity. Meanwhile, the beneficial effects of P450 axial mutations in conferring non-natural carbene- and nitrene-transfer activities6,7,10b clearly demonstrates the ability of this scaffold to take on new functions. Having all the axial mutants of CYP119 will allow exploration of a greater range of structural and heme electronic properties than has been possible in the past; we anticipate that this will facilitate the continued expansion of P450 catalysis.
Acknowledgments
We thank Jon Rittle, Hans Renata, Todd Hyster, Sheel Dodani, Jackson Cahn, Austin Rice, and Angelo Di Bilio for helpful discussions and comments on earlier versions of this manuscript. We would also like to thank Sabine Brinkmann-Chen, Pavle Nikolovski, and Jens Kaiser for assistance with crystallography screening and structure determination. J.A.M. and A.R.B. are supported by Ruth L. Kirschstein NRSA postdoctoral fellowships from the National Institutes of Health (F32GM101792 and F32GM110851, respectively). T.H. is supported by a FWF Schroedinger fellowship (J3327-B21). We also acknowledge support from the National Science Foundation, Office of Chemical, Bioengineering, Environmental and Transport Systems SusChEM Initiative (grant CBET-1403077), the Caltech Molecular Observatory, which is supported by the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis Bioengineering Research Program at Caltech. Acknowledgement for the cover art: Florian Kinigadner / http://www.labworkstudio.com.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b07107.
Methods, structure statistics, UV–vis spectral data, T50 curves, and heme incorporation data. (PDF)
Author Contributions
J.A.M. and T.H. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Li T.; Bonkovsky H. L.; Guo J.-T. BMC Struct. Biol. 2011, 11, 13. 10.1186/1472-6807-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogliaro F.; de Visser S. P.; Shaik S. J. Inorg. Biochem. 2002, 91, 554. 10.1016/S0162-0134(02)00437-3. [DOI] [PubMed] [Google Scholar]
- Dawson J. H. Science 1988, 240, 433. 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Science 2013, 342, 825. 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vatsis K. P.; Peng H. M.; Coon M. J. J. Inorg. Biochem. 2002, 91, 542. 10.1016/S0162-0134(02)00438-5. [DOI] [PubMed] [Google Scholar]; b Vatsis K. P.; Peng H. M.; Coon M. J. Arch. Biochem. Biophys. 2005, 434, 128. 10.1016/j.abb.2004.10.015. [DOI] [PubMed] [Google Scholar]; c Murugan R.; Mazumdar S. ChemBioChem 2005, 6, 1204. 10.1002/cbic.200400399. [DOI] [PubMed] [Google Scholar]; d Auclair K.; Moenne-Loccoz P.; Ortiz de Montellano P. R. J. Am. Chem. Soc. 2001, 123, 4877. 10.1021/ja0040262. [DOI] [PubMed] [Google Scholar]; e Yoshioka S.; Takahashi S.; Hori H.; Ishimori K.; Morishima I. Eur. J. Biochem. 2001, 268, 252. 10.1046/j.1432-1033.2001.01872.x. [DOI] [PubMed] [Google Scholar]
- a Coelho P. S.; Wang Z. J.; Ener M. E.; Baril S. A.; Kannan A.; Arnold F. H.; Brustad E. M. Nat. Chem. Biol. 2013, 9, 485. 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McIntosh J. A.; Coelho P. S.; Farwell C. C.; Wang Z. J.; Lewis J. C.; Brown T. R.; Arnold F. H. Angew. Chem., Int. Ed. 2013, 52, 9309. 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. J.; Renata H.; Peck N. E.; Farwell C. C.; Coelho P. S.; Arnold F. H. Angew. Chem. 2014, 126, 6928. 10.1002/ange.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyster T. K.; Farwell C. C.; Buller A. R.; McIntosh J. A.; Arnold F. H. J. Am. Chem. Soc. 2014, 136, 15505. 10.1021/ja509308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera R.; Sono M.; Voegtle H. L.; Dawson J. H. Arch. Biochem. Biophys. 2011, 507, 119. 10.1016/j.abb.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Whitehouse C. J.; Bell S. G.; Wong L. L. Chem. Soc. Rev. 2012, 41, 1218. 10.1039/C1CS15192D. [DOI] [PubMed] [Google Scholar]; b Hyster T. K.; Arnold F. H. Isr. J. Chem. 2015, 55, 14. 10.1002/ijch.201400080. [DOI] [Google Scholar]; c Munro A. W.; Leys D. G.; McLean M. A.; Marshall K. R.; Ost T. W.; Daff S.; Miles C. S.; Chapman S. K.; Lysek D. A.; Moser C. C.; Page C. C.; Dutton P. L. Trends Biochem. Sci. 2002, 27, 250. 10.1016/S0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]; d Pochapsky T. C.; Kazanis S.; Dang M. Antioxid. Redox Signaling 2010, 13, 1273. 10.1089/ars.2010.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a McLean M. A.; Maves S. A.; Weiss K. E.; Krepich S.; Sligar S. G. Biochem. Biophys. Res. Commun. 1998, 252, 166. 10.1006/bbrc.1998.9584. [DOI] [PubMed] [Google Scholar]; b Koo L. S.; Tschirret-Guth R. A.; Straub W. E.; Moenne-Loccoz P.; Loehr T. M.; Ortiz de Montellano P. R. J. Biol. Chem. 2000, 275, 14112. 10.1074/jbc.275.19.14112. [DOI] [PubMed] [Google Scholar]; c Yano J. K.; Koo L. S.; Schuller D. J.; Li H.; Ortiz de Montellano P. R.; Poulos T. L. J. Biol. Chem. 2000, 275, 31086. 10.1074/jbc.M004281200. [DOI] [PubMed] [Google Scholar]; d Rabe K. S.; Kiko K.; Niemeyer C. M. ChemBioChem 2008, 9, 420. 10.1002/cbic.200700450. [DOI] [PubMed] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Science 2013, 339, 307. 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- a Park S.-Y.; Yamane K.; Adachi S.-i.; Shiro Y.; Weiss K. E.; Maves S. A.; Sligar S. G. J. Inorg. Biochem. 2002, 91, 491. 10.1016/S0162-0134(02)00446-4. [DOI] [PubMed] [Google Scholar]; b Nishida C. R.; Ortiz de Montellano P. R. Biochem. Biophys. Res. Commun. 2005, 338, 437. 10.1016/j.bbrc.2005.08.093. [DOI] [PubMed] [Google Scholar]; c Basudhar D.; Madrona Y.; Kandel S.; Lampe J. N.; Nishida C. R.; Ortiz de Montellano P. R. J. Biol. Chem. 2015, 290, 10000. 10.1074/jbc.M114.627935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a McRee D. E.; Jensen G. M.; Fitzgerald M. M.; Siegel H. A.; Goodin D. B. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 12847. 10.1073/pnas.91.26.12847. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sun J.; Loehr T. M.; Wilks A.; Ortiz de Montellano P. R. Biochemistry 1994, 33, 13734. 10.1021/bi00250a026. [DOI] [PubMed] [Google Scholar]
- Heel T.; McIntosh J. A.; Dodani S. C.; Meyerowitz J. T.; Arnold F. H. ChemBioChem 2014, 15, 2556. 10.1002/cbic.201402286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sun Y.; Zeng W.; Benabbas A.; Ye X.; Denisov I.; Sligar S. G.; Du J.; Dawson J. H.; Champion P. M. Biochemistry 2013, 52, 5941. 10.1021/bi400541v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wells A. V.; Li P.; Champion P. M.; Martinis S. A.; Sligar S. G. Biochemistry 1992, 31, 4384. 10.1021/bi00133a002. [DOI] [PubMed] [Google Scholar]
- a Sudhamsu J.; Kabir M.; Airola M. V.; Patel B. A.; Yeh S.-R.; Rousseau D. L.; Crane B. R. Protein Expression Purif. 2010, 73, 78. 10.1016/j.pep.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Healy F. G.; Krasnoff S. B.; Wach M.; Gibson D. M.; Loria R. J. Bacteriol. 2002, 184, 2019. 10.1128/JB.184.7.2019-2029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Porter T. D. Biochemistry 1994, 33, 5942. 10.1021/bi00185a035. [DOI] [PubMed] [Google Scholar]
- a Sezutsu H.; Le Goff G.; Feyereisen R. Philos. Trans. R. Soc., B 2013, 368, 20120428. 10.1098/rstb.2012.0428. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Guo Y.; Zhang J.; Yu R.; Zhu K. Y.; Guo Y.; Ma E. Chemosphere 2012, 87, 709. 10.1016/j.chemosphere.2011.12.061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.