

Abstract
Neuronal positioning is a fundamental process during brain development. Abnormalities in this process cause several types of brain malformations and are linked to neurodevelopmental disorders such as autism, intellectual disability, epilepsy, and schizophrenia. Little is known about the pathogenesis of developmental brain malformations associated with abnormal neuron positioning, which has hindered research into potential treatments. However, recent advances in neurogenetics provide clues to the pathogenesis of aberrant neuronal positioning by identifying causative genes. This may help us form a foundation upon which therapeutic tools can be developed. In this review, we first provide a brief overview of neural development and migration, as they relate to defects in neuronal positioning. We then discuss recent progress in identifying genes and brain malformations associated with aberrant neuronal positioning during human brain development.
Keywords: Neuron positioning, Brain malformation, Neuron migration, Lissencephaly, Heterotopia, Polymicrogyria, Microcephaly, Cortical dysplasia, LIS1, DCX, Reelin, TUBA1A
Background
Neuronal positioning is an integral part of the coordinated steps comprising neural circuit formation in embryonic and neonatal development [1]. This process takes place throughout the nervous system at different time points depending on the type of neuron. Although neuronal positioning and migration occurs throughout the central nervous system, we will focus on neuronal positioning in the neocortex of the developing brain. We will present basic information on the process of neuronal positioning and describe the abnormalities that may occur in the human brain. Additionally, genes associated with neuronal positioning abnormalities will be discussed.
Correct positioning of neurons by normal migration plays a critical role in establishing cognitive functions and emotion. Human cognitive activity depends on appropriate brain circuit formation. Disrupted brain wiring due to abnormal neuronal development such as improper neuronal positioning can result in brain malformations, cognitive dysfunction, and seizures [2–4]. The causes of brain malformations associated with positioning and migration defects are varied and include genetic mutations and environmental toxins [1, 5, 6]. Studies of neuronal migration disorders have progressed due to advances in molecular genetics and brain magnetic resonance imaging. The commonly identified disorders of neuronal positioning include lissencephaly and heterotopia [7].
Neural progenitors as a source of migrating neurons in the human cerebral cortex
Neural progenitors can undergo self-renewal or give rise to neurons at the ventricular/subventricular zone in the developing cerebral cortex [8–10]. Reduced numbers of neural progenitors caused by depletion of progenitor pools or slow proliferation result in microcephaly with otherwise normal brain structure [11, 12]. However, microcephaly can also occur in combination with a migration defect, i.e., microcephaly with pachygyria (Norman-Roberts syndrome) [13]. Thus, the disruptive functions of neural progenitor renewal and neurogenesis may interfere with later developmental aspects such as neuronal migration and positioning in the developing brain.
Neuronal migration modes
After neurons are born, they migrate from their birthplaces to their final destinations (Fig. 1). There are two types of embryonic neuronal migration: radial and tangential. The migration of excitatory pyramidal neurons from the cortical ventricular zone (where they are born) is an example of radial migration (Fig. 1a). These neurons migrate into the cortical plate alongside radial glial processes [14–17]. The layers of the cortex form in an “inside-out” manner with later-born pyramidal neurons migrating past earlier-born predecessors in the cortical plate so that they are more superficial in their final position than earlier born neurons [5, 18–20]. In humans, neuronal migration takes place predominantly between 12 and 20 weeks in gestation. The migration of inhibitory interneurons (GABAergic neurons) from the medial ganglionic eminence of the ventral telencephalon (where they are born) is an example of tangential migration (Fig. 1b). Interneurons migrate tangentially to the dorsal telencephalon and then change direction to enter the cortical plate radially [20–23]. Subsets of these cells display ventricle-directed migration followed by radial movement to the cortical plate. Thus, neuronal migration determines the positioning of developing neurons into cortical layers and thereby is important in generating lamina-specific neural circuits. Normal development and function of the neocortex critically depends on the coordinated production and positioning of excitatory and inhibitory neurons [24–27]. Abnormal neuronal migration can arrest different types of neurons at the wrong positions along the migratory path resulting in brain malformations and neurological disorders.
Fig. 1.
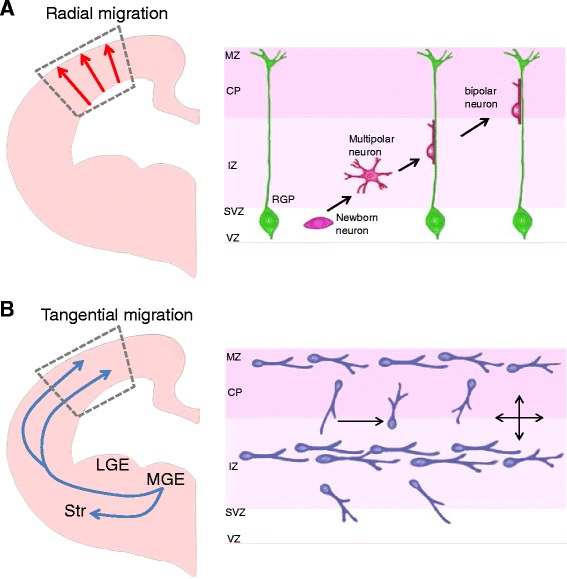
Two modes of neuronal migration in the developing brain. a Radial migration. Excitatory pyramidal projection neurons migrate from the ventricular zone to the cortical plate in the developing brain. The right panel shows what happens in the rectangular box in the left panel. Newly-born neurons from radial glial progenitors (RGP) at the ventricular zone (VZ) migrate along the radial processes of RGPs. MZ: marginal zone. CP: cortical plate. IZ: intermediate zone. SVZ: subventricular zone. b Tangential migration: Interneurons originate from distinct proliferative zones in the developing brain. Inhibitory interneurons are born in the medial ganglionic eminence (MGE) of ventral brain and migrate in multiple streams into the cerebral wall. Once interneurons reach appropriate spots in the cerebral cortex, they establish their final positions by local adjustment of radial and tangential movement. Unlike pyramidal neurons, these neurons extend multiple leading branches during migration. LGE: lateral ganglionic eminence. LGE: lateral ganglionic eminence. Str: striatum
In addition to these well-defined modes of embryonic neuronal migration, a limited number of neurons and neuronal precursors have been shown to migrate and differentiate in the early postnatal rodent and human cerebellum and hypothalamus [28, 29]. Another, more extensive mode of neuronal migration has been observed in adult rodents and non-human primates, in which neuronal precursors migrate along glial projections from the subventricular zone into the olfactory bulbs. This particular passage is referred to as the rostral migratory stream (RMS) [28–30], which continues well into adulthood, but has not been observed in humans [28, 31]. In the RMS, neuronal precursors migrate via a “tunnel” made up of astrocytes into the olfactory bulb, where they then radially migrate in a glial-independent manner toward the glomeruli and differentiate. The majority of these cells eventually become inhibitory neurons, mainly GABAergic granule neurons [28, 32]. Because the application of research tools is currently limited in humans, there is still ongoing debate about whether the RMS exists in humans [28, 31, 32].
In this review we will focus solely on brain malformations thought to be due to abnormal embryonic neuronal migration, although many of the genes and proteins discussed are no doubt involved in both embryonic and postnatal neuronal migration. It is important, however, that further research be done to understand the mechanisms of neuronal migration and the maintenance of neuronal precursor pools in adults, because of the potential to promote regeneration and repair in individuals with neuronal positioning disorders, neurodegenerative disorders, and severe brain injuries.Brain malformations and genes associated with abnormal neuron positioning are listed in Table 1.
Table 1.
Brain malformations and genes associated with abnormal neuron positioning
| Type | Gene | Location | Description |
|---|---|---|---|
| Lissencephaly type I | |||
| Lissencephaly (Autosomal dominant) | LIS1 | 17p13.3 | Microtubule-associated protein |
| Isolated lissencephaly sequence (ILS) or subcortical band heterotopia (SBH) | TUBA1A | 12q13.12 | Constituent of microtubules |
| Miller-Dieker syndrome | LIS1 + YWHAE | 17p13.3 | Microtubule-associated protein |
| Lissencephaly (X-linked) | |||
| ILS or SBH | DCX | Xq22.3-q23 | Microtubule-associated protein |
| X-linked lissencephaly with abnormal genitalia | ARX | Xp21.3 | Transcription factor |
| Lissencephaly (Autosomal recessive) | |||
| Lissencephaly with cerebellar hypoplasia (LCH) group b | RELN | 7q22 | Extracellular matrix serine protease |
| VLDLR | 9q24 | Binds VLDL and transports it into cells by endocytosis | |
| Lissencephaly type II: Cobblestone complex (Autosomal recessive) | |||
| Fukuyama congenital muscular dystrophy or Walker–Warburg syndrome (WWS) | FKTN | 9q31.2 | Involved in glycosylation |
| Muscle–eye–brain disease (MEB) or WWS | POMT1 | 9q34.13 | Protein-O-mannosyltransferase 1 |
| POMT2 | 14q24.3 | Protein-O-mannosyltransferase 2 | |
| POMGNT2 | 3p22.1 | O-linked mannose acetylglucosaminyltransferase | |
| FKRP | 19q13.32 | Involved in glycosylation | |
| MEB | LARGE | 22q12.3 | Glycosyltransferase |
| POMGnT1 | 1p34.1 | Participates in O-mannosyl glycosylation | |
| Bilateral frontoparietal polymicrogyria | GPR56 | 16q21 | G protein-coupled receptor 56 |
| CEDNIK syndrome | SNAP29 | 22q11.21 | Synaptosomal-associated protein |
| Muscular dystrophy | ISPD | 7q21.2 | Required for protein O-linked mannosylation |
| GTDC2 | 3p22.1 | O-linked mannose acetylglucosaminyltransferase | |
| TMEM5 | 12q14.2 | Glycosyltransferase function | |
| B3GALNT2 | 1q42.3 | Beta-1,3-N-acetylgalactosaminyltransferase | |
| SGK196 | 8q11.21 | Protein O-mannose kinase | |
| B3GNT1 | 11q13.2 | Synthesis of the linear poly-N-acetyllactosaminoglycans | |
| GMPPB | 3p21.31 | GDP-mannose pyrophosphorylase | |
| Polymicrogyria | |||
| TUBB2 | 6p25 | Major constituent of microtubules | |
| GPR56 | 16q21 | G protein-coupled receptor 56 | |
| SRPX2 | Xq22.1 | Plays a role in angiogenesis | |
| TBR2 | 3p24.1 | Transcriptional activator | |
| PAX6 | 11p13 | Transcription factor | |
| KIAA1279 | 10q22.1 | Organization of axonal microtubules | |
| RAB3GAP1 | 2q21.3 | RAB3 GTPase Activating Protein Subunit | |
| Adams-Oliver syndrome (AOS) | ARHGAP31 | 3q13.33 | Required for cell spreading |
| AOS | RBPJ | 4p15.2 | Plays a central role in Notch signaling |
| AOS | DOCK6 | 19p13.2 | Atypical guanine nucleotide exchange factors |
| AOS | EOGT | 3p14.1 | EGF domain-specific GlcNAc transferase |
| AOS | NOTCH1 | 9q34.3 | Play multiple roles during development |
| Heterotopia | |||
| Heterotopia (X-linked Autosomal dominant) | |||
| Classical bilateral periventricular heterotopia (PH) | FLNA | Xq28 | Actin-binding protein |
| PH with fragile-X syndrome | FMR1 | Xq27.3 | Translation repressor |
| PH and Williams syndrome | WBSCR16 | 7q11.23 | Guanine nucleotide exchange factor |
| PH | PVNH3 | 5p15.1 | Periventricular Nodular Heterotopia 3 |
| PH | PVNH5 | 5q14.3-q15 | Periventricular Nodular Heterotopia 5 |
| Heterotopia (Autosomal recessive) | |||
| PH with microcephaly | ARFGEF2 | 20q13.13 | Intracellular vesicular trafficking |
| PH with Donnai–Barrow syndrome | LRP2 | 2q31.1 | Low density lipoprotein-related protein 2 |
| Microcephaly | |||
| WDR62 | 19q13.12 | Required for cerebral cortical development | |
| KIAA1279 | 10q22.1 | Organization of axonal microtubules | |
| RAB3GAP1 | 2q21.3 | RAB3 GTPase Activating Protein Subunit | |
| ARFGEF2 | 20q13.13 | Intracellular vesicular trafficking | |
| Focal cortical dysplasia | |||
| TSC1 | 9q34.13 | Negatively regulating mTORC signaling | |
| TSC2 | 16p13.3 | Negatively regulating mTORC signaling | |
| Hemimegalencephaly | |||
| PIK3CA | 3q26.32 | Serine/threonine kinase - component of PI3K/AKT signaling | |
| AKT3 | 1q44 | Serine/threonine kinase - component of PI3K/AKT signaling | |
| MTOR | 1p36.22 | Serine/threonine kinase – component of PI3K/AKT signaling |
Genes and brain malformations associated with defective neuron positioning
Type I lissencephaly
Perhaps the best known type of neuronal migration disorder is lissencephaly, “smooth brain”. It is a brain malformation characterized by the absence of gyri and sulci [7, 33]. Most individuals with this condition also present with microcephaly (small head). Although the symptoms vary, they often feature seizures, intellectual disability, developmental delays, poor motor function, difficulties with feeding, and swelling in the extremities.
LIS1 and DCX
Mutations in lissencephaly 1 (LIS1) and doublecortin (DCX) have been shown to cause type I lissencephaly (Table 1). This disorder is often associated with axon outgrowth and guidance defects such as agenesis of the corpus callosum [34]. Neuronal positioning and further differentiation may coordinate to develop the pathogenesis of lissencephaly. Classic lissencephaly (type I) includes isolated lissencephaly and subcortical band heterotopia (“double cortex”) which are caused by DCX mutations [33, 35]. In addition, heterozygous mutation of Lis1 in mice has been shown to impair normal neuron positioning and synaptogenesis in the amygdala [36]. Interestingly, there is a skewed sex ratio in subcortical band heterotopia and isolated lissencephaly. Females with a mutation affecting one copy of the DCX gene usually develop subcortical band heterotopia while males with one DCX gene mutation show isolated lissencephaly [37–41]. Males with subcortical band heterotopia or females with isolated lissencephaly are rarely reported [42, 43].
DCX encodes a microtubule-associated protein that stabilizes microtubules and causes bundling [44–46]. This is an important molecule in neuron migration and neurite growth in the developing brain [47–49]. DCX is expressed in neuronal precursor cells and immature neurons during brain development and in the adult hippocampus. More importantly, DCX is associated with the neuronal migration disorders, lissencephaly, pachygyria, and subcortical band heterotopia [37, 38, 41, 50–52]. Mutations in DCX prevent neurons from migrating into the cortical plate [45]. Abnormal microtubule functions dependent on DCX appear to underlie lissencephaly because pathological mutations in DCX prevent its product binding and subsequent stabilization of microtubules [53, 54].
Miller-Dieker syndrome is characterized as a congenital brain malformation due to the microdeletion of chromosome 17p13.3 including the LIS1 gene, which can also cause classical lissencephaly [37, 41, 51, 55–58]. LIS1 encodes a dynein-binding protein and controls mitotic spindle orientation in neural cells [59–61]. The most common type of mutation is a deletion of a single copy of the gene, resulting in haploinsufficiency. Individuals with LIS1 mutations have not only lissencephaly, but often show other pathological features including corpus callosum hypoplasia and ventricle enlargement [58, 62]. These anatomical abnormalities correlate with the critical roles of LIS1 in neuronal migration and axon formation [57, 58, 63]. In contrast to lissencephaly caused by mutations in DCX, LIS1 mutations preferentially affect the parieto-occipital cortex [37, 41, 64]. Mutations in LIS1 and DCX account for approximately 85% of patients with the classic form of lissencephaly [37, 41, 65].
YWHAE
Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein, Epsilon (YWHAE) is another gene that encodes a microtubule-associated protein and is located just 1 Mb away from LIS1 on chromosome 17p. YWHAE also participates in the LIS1 pathway, and homozygous deletion of mouse Ywhae leads to neuronal migration defects. Large deletions of the 17p13.3 region (which contains both YWHAE and LIS1) causes Miller-Dieker syndrome, and patients with this deletion display more severe neuronal migration defects than those observed in LIS1 mutant heterozygote-caused lissencephaly [54].
TUBA1A and TUBB2
Tubulin Alpha 1a (TUBA1A) and Tubulin Beta 2 (TUBB2) encode critical structural subunits of microtubules that are enriched during brain development [66]. TUBA1A mutations are identified in 1 % of classic lissencephaly and 30% of lissencephaly with cerebellar hypoplasia [67–69]. Meanwhile, TUBB2 mutations are associated with symmetric polymicrogyria and pachygyria [70]. Guanosine triphosphate (GTP) contributes to microtubule assembly by binding to soluble tubulin heterodimers [71]. Mutations in these tubulin genes prevent microtubule polymerization. For example, the S140G mutation reduces the protein capacities of GTP binding and native heterodimer formation, thus preventing polymerization of microtubules and neuronal migration in mice [72]. In contrast to TUBA1A and TUBB2, TUBB3 is important in axon guidance and microtubule dynamics, but dispensable for neuronal migration [73].
ARX
Aristaless related homeobox (ARX) is a homeobox-containing gene expressed in the nervous system during development [74–76]. ARX mutations are associated with an X-linked lissencephaly syndrome with infantile spasms as well as abnormal genitalia [77–79]. Mutations that cause lissencephaly often lead to premature truncation or alter the DNA binding domain of the protein (homeodomain) [80, 81]. Studies using human brain samples and animal models have revealed that ARX is important in proliferation of radial and intermediate neural progenitors, and migration of excitatory cortical neurons [75, 79, 80, 82]. It also critically controls the migration and further differentiation of inhibitory GABAergic interneurons [79, 80, 82–86]. This is consistent with the fact that ARX is expressed in the ganglionic eminence and cortical ventricular zone where interneuron and pyramidal neural progenitors reside, respectively [79]. Furthermore, ARX overexpression promotes the development of tangentially migrating interneurons [82, 86]. However, some mutations disrupt neuronal excitability without affecting neuronal migration or the cortical lamination pattern in the brain [87].
RELN
Reelin (RELN) and its cellular receptor very-low-density-lipoprotein receptor (VLDLR) are cellular signaling components. RELN is required for neuronal migration in the developing cortex [54, 88–91]. Accordingly, VLDLR critically regulates neuronal migration and positioning in the cerebral cortex [92]. RELN promotes hippocampal dendrite development through the VLDLR-Dab1 pathway as well [93]. Mutations in these genes are known to cause lissencephaly with cerebellar hypoplasia [54, 94–96]. The RELN mutation syndrome appears to be inherited in an autosomal-recessive pattern and these patients appear to be relatively rare [94]. Mutations in VLDLR can cause combinations of ataxia, intellectual disability, and quadrupedal gait [97].
Type II lissencephaly
Type II lissencephaly is often referred to as “cobblestone lissencephaly” because patients typically only have regional agyria. It is associated with Walker-Warburg syndrome, a heterogeneous group of muscular dystrophy-dystroglycanopathy (MDDG) conditions that can be caused by homozygous mutations in the genes FKTN (Fukuyama syndrome), POMT2 and POMGnT1 (muscle-eye-brain disease), as well as POMGNT2, FKRP, LARGE, ISPD, GTDC2, TMEM5, B3GALNT2, SGK196, B3GNT1, and GMPPB [98–101]. In type II lissencephaly, there are no layers present in the cortex. Instead, irregularities in neuronal placement exist. Abnormal glycosylation of matrix proteins in the cerebral cortex is thought to cause these migration defects [102, 103].
Loss of function mutations in SNAP29, which encodes a member of the SNARE protein family, has been shown to cause CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis and keratoderma) syndrome [104]. Brain MRI scans of CEDNIK syndrome patients revealed apparent extensive aberrant neuronal migration, as evidenced by corpus callosum abnormalities and cortical dysplasia, along with pachygyria, polymicrogyria and cobblestone lissencephaly [105]. Migration defects in SNAP29 mutants may be attributed to an impairment in β1-integrin [106].
Polymicrogyria
Polymicrogyria is a neurological condition characterized by an excessive number of small and fused gyri separated by shallow sulci in the cerebral cortex compared to normal cerebral surfaces [6, 70, 107, 108]. Mutations in the TUBB2, GPR56, and WDR62 genes are associated with this condition [70, 109–111]. Polymicrogyria develops between the late stage of neuronal migration and the early point of cortical organization [108, 112]. Patients with polymicrogyria show a layer of intracortical laminar necrosis and subsequent disruption of late cortical lamination. Some cerebral cortices have a molecular layer that does not align along the borders of gyri. Neurons under this layer have a radial distribution without laminar organization [111]. Polymicrogyria most often occurs as an isolated feature. However, it is sometimes shown in multiple genetic syndromes associated with intellectual disability and birth defects including 22q11.2 deletion syndrome, Adams-Oliver syndrome (genetically heterogeneous, caused by mutations in ARHGAP31, RBPJ, DOCK6, EOGT, and NOTCH1), Aicardi syndrome, Galloway-Mowat syndrome, Joubert syndrome, and Zellweger spectrum (peroxisome biogenesis disorders including Zellweger syndrome, neonatal adrenoleukodystrophy, and Refsum disease) [111, 113–116]. The clinical features and etiology of polymicrogyria are heterogeneous. Most patients with polymicrogyria develop epilepsy during their early childhood (4–12 years of age). Seizures are resistant to pharmacological drugs in many cases of polymicrogyria.
TBR2 and PAX6
Pax6, which encodes paired box protein 6, is highly expressed in radial glia, but is downregulated as they transition into intermediate progenitor cells during neurogenesis. This coincides with an upregulation of T-brain gene-2 (TBR2) that persists until intermediate neural progenitor cells differentiate into postmitotic neurons [117]. Mutations in TBR2 and PAX6 have been shown to cause polymicrogyria, due to defects in neuronal migration, differentiation and proliferation of neural progenitors [118–120].
SRPX2
SRPX2 encodes a secreted sushi-repeat containing protein that is expressed in neurons. A rare missense mutation in the SRPX2 gene causes bilateral perisylvian polymicrogyria, though its mechanism in development of this disease remains unknown. SRPX2 is expressed in humans in the fetal and adult brain, whereas in mice, measurable expression does not begin until birth [121]. This poses problems for further studies into the role of SRPX2 in brain development and neuronal migration.
KIAA1279
Homozygous nonsense mutations in the KIAA1279 gene cause Goldberg-Shprintzen syndrome, which is characterized by bilateral generalized polymicrogyria, microcephaly, mental retardation, and an enteric nervous disorder [122]. KIAA1279 encodes a kinesin family member-binding protein, but its role in the pathology of Goldberg-Shprintzen syndrome is still unknown [123, 124]. It was recently shown, however, that KIAA1279 co-localizes with both α-tubulin and F-actin. Relatedly, KIAA1279 is also involved in neurite outgrowth. Inhibition of KIAA1279 expression using siRNA leads to dendritic spine depletion and a decrease in neurite length in neuroblastoma cells, and overexpression of KIAA1279 triggers an increase in dendritic spine and neurite length, compared to controls [123].
RAB3GAP
Rab3 GTPase-activating protein (RAB3GAP) is a heterodimeric complex comprised of a catalytic subunit (RAB3GAP1) and a slightly larger non-catalytic subunit (RAB3GAP2). This complex acts as a guanine-nucleotide exchange factor for the RAB18 protein [125]. RAB18 is also regulated by the GTP-activating protein TBC1D20 [126]. Mutations or dysregulation of RAB18 causes Warburg Micro syndrome, which is characterized by ocular and neurodevelopmental abnormalities, including polymicrogyria, microcephaly, pachygyria, polymicrogyria, and hypoplasia of the corpus calossum. It is unclear by what molecular mechanism RAB18 dysfunction leads to these neurodevelopmental aberrations, but mutations to RAB3GAP1, RAB3GAP2, TBC1D20 and RAB18 are all sufficient to cause these symptoms [125, 126]. It has recently been demonstrated, that TBC1D20 activity fosters extraction of RAB18 from the ER membrane and facilitates its retargeting for the cis-Golgi. In the cis-Golgi, it appears that the RAB3GAP complex recruits and stabilizes the RAB18 protein [126].
Heterotopia
In addition to cortical gyration disorder, dysfunctional neuronal migration can lead to the development of neuronal population in aberrant locations. Periventricular nodular heterotopia is one of these neuronal migration disorders [5–7, 107, 108]. In this case, failed migration leads to the formation of heterotopic neurons along the ventricular surfaces in the brain. Therefore, the neurons are positioned deeper than those found in type I lissencephaly. This malformation can be bilateral or unilateral. Periventricular heterotopia is diagnosed with magnetic resonance imaging (MRI) and seizure symptoms. Affected individuals usually have normal intelligence, although some have mild intellectual disability. Some cases of periventricular heterotopia are associated with dyslexia [127]. For example, a specific reading fluency deficit is identified in a heterogeneous group of patients with periventricular heterotopia who have seizures, heterotopic neurons, and disrupted cortical connectivity [127, 128].
FLNA
The most common genetic cause of periventricular heterotopia is the X-linked dominant inheritance of Filamin A (FLNA) gene mutations [129, 130]. The FLNA gene encodes an F-actin-binding cytoplasmic protein involved in neurogenesis and neuronal migration in the developing brain [131, 132]. FLNA crosslinks actin filaments into the cortical cytoskeleton. FLNA mutations are associated with classical bilateral periventricular nodular heterotopia and account for the majority of X-linked inherited periventricular heterotopias [6, 133, 134]. FLNA regulates neuronal migration in the cerebral cortex [131]. Mutations in the human FLNA gene may also cause connective tissue disorders associated with Ehlers-Danlos syndrome which include extremely flexible joints, stretchable skin, and fragile blood vessels [135]. Unsurprisingly, patients with Ehlers-Danlos syndrome also frequently present with epilepsy and periventricular heterotopia [136].
PVNH3 and PVNH4
In addition to FLNA mutations, duplications and deletions in chromosome 5 which includes Periventricular Nodular Heterotopia 3 (PVNH3) and Periventricular Nodular Heterotopia 5 (PVNH5) have been seen in patients with periventricular heterotopia without mutations in other causative genes [137]. Periventricular nodular heterotopia is also found in individuals with other conditions, including Ehlers-Danlos syndrome [135].
FMR1
CGG trinucleotide repeat expansion of the FMR1 gene causes fragile X syndrome in humans and has also been shown to lead to periventricular heterotopia. This may indicate a role for the FMR1 protein in neuronal migration [138].
ARFGEF2
ADP-ribosylation factor guanine exchange factor 2 (ARFGEF2) encodes a protein kinase A-anchoring protein that regulates GDP-GTP conversion of ADP-ribosylation factor [139, 140]. Via mediation of Filamin A signaling, ARFGEF2 is involved in neuronal migration through the regulation of vesicle trafficking. Mutations in ARFGEF2 also cause bilateral periventricular nodular heterotopia, as well as putaminal hyperintensity and microcephaly [131, 141].
LRP2
Low density lipoprotein-related protein 2 (LRP2) encodes megalin, a multiligand receptor. Mutations to LRP2 cause Donnai-Barrow syndrome, which is associated with several neurological and cranial abnormalities, including periventricular nodular heterotopia [142]. Megalin facilitates the endocytosis of sonic hedgehog (Shh) in embryonic neuroepithelium [143]. Furthermore, megalin has been shown to bind and sequester Shh in the forebrain, and mediate Shh-Ptch endocytosis [144]. This key interaction with Shh signaling in the developing brain could explain the aberrant neuronal positioning observed in patients with LRP2 mutations.
Focal cortical dysplasia
Focal cortical dysplasia is a rare lamination abnormality in the cerebral cortex characterized by focal cortical thickening or thinning, focal atrophy, or blurring of the gray-white junction [6, 145]. Focal cortical dysplasia is the most common cause of medically refractory epilepsy in the pediatric population [145]. Defective regulation of neuronal migration or cell death is speculated to cause focal cortical dysplasia [146, 147]. There are three types of focal cortical dysplasia [34, 145, 148, 149]. Type I focal cortical dysplasia is found in the temporal lobe of the brain. This type is late onset, thus often seen in adults. Patients with this condition show mild symptoms. Type II focal cortical dysplasia, however, is mostly found in children and the clinical symptoms are more severe. There are more extensive changes outside the temporal lobe with predilection for the frontal lobes. Type III focal cortical dysplasia occurs in combination with hippocampal sclerosis, epilepsy-associated tumors, vascular malformation, or epileptogenic lesions. Studies have suggested that mutations in the TSC1 (Tuberous Sclerosis 1) gene is associated with the formation of focal dysplasia [145, 150, 151]. Changes in Wnt and Notch signaling components that control proper neuronal migration are also found in focal cortical dysplasia [145, 152].
Hemimegalencephaly
Hemimegalencephaly is implicated in neuronal positioning abnormality. Hemimegalencephaly features one side of the brain that is abnormally larger than the other [6, 108, 153]. The unusual enlargement of the brain causes seizures and intellectual disability [154]. This condition is thought to take place when neurons are abnormally organized due to defective migration in the developing cerebral cortex because the enlarged hemisphere usually shows focal or diffused regions of polymicrogyria, pachygyria, and heterotopia [155–159]. However, whether abnormal neuronal migration during development causes hemimegalencephaly is unclear. Using exome sequencing, recent studies have identified de novo germline and somatic mutations of PI3K-AKT-mTOR components (PIK3CA, AKT3, and MTOR genes) in patients with hemimegalencephaly [160–164]. Thus, hemimegalencephaly may be a genetically mosaic disease caused by abnormal PI3K-AKT-mTOR signaling. In addition to its role in neuronal migration, PI3K-AKT-mTOR signaling critically regulates neural progenitor proliferation and neurogenesis [32, 165–168].
Conclusions
Recent advances in neurogenetics and brain imaging have revealed genes responsible for neuronal migration disorders. Efforts have been made to characterize the functions of the causative genes and develop appropriate animal models. Still, research that overcomes these disorders is only in the beginning stage of work. Further human genetic analysis and neurobiological studies should expand our understanding of the pathogenesis of neuronal migration disorders, which will help to develop therapeutic strategies for these disorders in the future.
Acknowledgements
We thank Drs. Robert Norgren and Shelley Smith for valuable advice and comments on the manuscript. This work was supported by an award from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R01NS091220 and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under award number P20GM103471 to WYK.
Abbreviations
- AKT
RAC-alpha serine/threonine-protein kinase
- ARGEF2
ADP-ribosylation factor guanine exchange factor 2
- ARHGAP31
rho GTPase Activating Protein 31
- ARX
Aristaless-related homeobox
- B3GALNT2
Beta-1,3-N-acetylgalactosaminyltransferase 2
- B3GNT1
Beta-1,3-N-acetylglucosaminyltransferase 1
- CEDNIK
Cerebral dysgenesis-neuropathy-ichthyosis-palmoplantar keratoderma
- CP
Cortical plate
- DCX
Doublecortin
- DOCK6
Dedicator of cytokinesis 6
- EOGT
EGF domain-specific O-linked N-Acetylglucosamine (GlcNAc) transferase
- FKRP
Fukutin related protein
- FKTN
Fukutin
- FLNA
Filamin A
- FMR1
Fragile X mental retardation 1
- GMPPB
GDP-mannose pyrophosphorylase B
- GPR56
G protein-coupled receptor 56
- GTDC2
Glycosyltransferase-like domain containing 2
- GTP
Guanosine triphosphate
- ILS
Isolated lissencephaly sequence
- ISPD
Isoprenoid synthase domain containing
- IZ
Intermediate zone
- KIAA1279
KIF1 binding protein (KIF1BP)
- LARGE
Like-glycosyltransferase
- LCH
Lissencephaly with cerebellar hypoplasia
- LGE
Lateral ganglionic eminence
- LIS1
Lissencephaly 1
- LRP2
Low density lipoprotein receptor-related protein 2
- MEB
Muscle-eye-brain disease
- MRI
Magnetic resonance imaging
- mTOR
Mechanistic target of rapamycin
- MGE
Medial ganglionic eminence
- MZ
Marginal zone
- PAX6
Paired box 6
- PH
Periventricular heterotopia
- PI3K
Phosphatidylinositol-4, 5-bisphosphate 3-kinase
- POMGnT1
Protein O-linked mannose N-acetylglucosaminyltransferase 1
- POMGnT2
Protein O-linked mannose N-acetylglucosaminyltransferase 2
- POMT2
Protein-O-mannosyltransferase 2
- PVNH3
Periventricular nodular heterotopia 3
- PVNH4
Periventricular nodular heterotopia 4
- RAB18
RAB18, member RAS oncogene family
- RAB3GAP
Rab3 GTPase activating protein
- RBPJ
Recombination signal binding protein for immunoglobulin kappa J region
- RELN
Reelin
- RGP
Radial glial progenitors
- RMS
Rostral migratory stream
- SBH
Subcortical band heterotopia
- SGK196
Protein-O-mannose kinase (POMK)
- SNAP29
Synaptosomal-associated protein, 29kDa
- SNARE
Soluble NSF Attachment Protein Receptor
- SRPX2
Sushi-repeat containing protein, X-linked 2
- SVZ
Subventricular zone
- TBC1D20
TBC1 domain family, member 20
- TBR2
T-brain gene-2
- TMEM5
Transmembrane protein 5
- TSC1
Tuberous sclerosis 1
- TSC2
Tuberous sclerosis 2
- TUBA1A
Tubulin, alpha 1a
- TUBB2
Tubulin, beta 2
- TUBB3
Tubulin, beta 3
- VLDR
Very-low-density-lipoprotein receptor
- VZ
Ventricular zone
- WBSCR16
Williams-Beuren syndrome chromosome region 16
- WDR62
WD repeat domain 62
- WWS
Walker-Warburg syndrome
- YWHAE
Tyrosine 3-monooxygenase/Tryptophan 5-Monooxygenase Activation Protein, Epsilon
Footnotes
Jeffrey J. Moffat and Minhan Ka contributed equally to this work.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
JM, MK, EJ, and WK analyzed the published studies and wrote the paper. WK conceived the study. All authors read and approved the final manuscript.
Contributor Information
Jeffrey J. Moffat, Email: jeffrey.moffat@unmc.edu
Minhan Ka, Email: minhan.ka@unmc.edu.
Eui-Man Jung, Email: euiman.jung@unmc.edu.
Woo-Yang Kim, Email: wooyang.kim@unmc.edu.
References
- 1.Martini FJ, Valiente M, Lopez Bendito G, Szabo G, Moya F, Valdeolmillos M, et al. Biased selection of leading process branches mediates chemotaxis during tangential neuronal migration. Development. 2009;136(1):41–50. doi: 10.1242/dev.025502. [DOI] [PubMed] [Google Scholar]
- 2.Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23(8):352–9. doi: 10.1016/S0166-2236(00)01607-6. [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10(10):981–91. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- 4.Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119(6):755–70. doi: 10.1007/s00401-010-0655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu JS. Molecular genetics of neuronal migration disorders. Curr Neurol Neurosci Rep. 2011;11(2):171–8. doi: 10.1007/s11910-010-0176-5. [DOI] [PubMed] [Google Scholar]
- 6.Guerrini R, Parrini E. Neuronal migration disorders. Neurobiol Dis. 2010;38(2):154–66. doi: 10.1016/j.nbd.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135(Pt 5):1348–69. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rakic P. Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci. 2009;10(10):724–35. doi: 10.1038/nrn2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci. 2013;14(11):755–69. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franco SJ, Muller U. Shaping our minds: stem and progenitor cell diversity in the mammalian neocortex. Neuron. 2013;77(1):19–34. doi: 10.1016/j.neuron.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Q, Wang X. Neuronal stem cells in the central nervous system and in human diseases. Protein Cell. 2012;3(4):262–70. doi: 10.1007/s13238-012-2930-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang BL. Molecular genetic determinants of human brain size. Biochem Biophys Res Commun. 2006;345(3):911–6. doi: 10.1016/j.bbrc.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 13.Dobyns WB, Stratton RF, Greenberg F. Syndromes with lissencephaly. I: Miller-Dieker and Norman-Roberts syndromes and isolated lissencephaly. Am J Med Genet. 1984;18(3):509–26. doi: 10.1002/ajmg.1320180320. [DOI] [PubMed] [Google Scholar]
- 14.Chanas-Sacre G, Rogister B, Moonen G, Leprince P. Radial glia phenotype: origin, regulation, and transdifferentiation. J Neurosci Res. 2000;61(4):357–63. doi: 10.1002/1097-4547(20000815)61:4<357::AID-JNR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409(6821):714–20. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- 16.Hartfuss E, Galli R, Heins N, Gotz M. Characterization of CNS precursor subtypes and radial glia. Dev Biol. 2001;229(1):15–30. doi: 10.1006/dbio.2000.9962. [DOI] [PubMed] [Google Scholar]
- 17.Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145(1):61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 18.Tan SS, Kalloniatis M, Sturm K, Tam PP, Reese BE, Faulkner-Jones B. Separate progenitors for radial and tangential cell dispersion during development of the cerebral neocortex. Neuron. 1998;21(2):295–304. doi: 10.1016/S0896-6273(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 19.Evsyukova I, Plestant C, Anton ES. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu Rev Cell Dev Biol. 2013;29:299–353. doi: 10.1146/annurev-cellbio-101512-122400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sultan KT, Brown KN, Shi SH. Production and organization of neocortical interneurons. Front Cell Neurosci. 2013;7:221. doi: 10.3389/fncel.2013.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128(3):353–63. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- 22.Rakic P. Neuronal migration and contact guidance in the primate telencephalon. Postgrad Med J. 1978;54(Suppl 1):25–40. [PubMed] [Google Scholar]
- 23.Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8(6):427–37. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 24.Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci. 2003;23(2):622–31. doi: 10.1523/JNEUROSCI.23-02-00622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartolini G, Ciceri G, Marin O. Integration of GABAergic interneurons into cortical cell assemblies: lessons from embryos and adults. Neuron. 2013;79(5):849–64. doi: 10.1016/j.neuron.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 26.Lodato S, Rouaux C, Quast KB, Jantrachotechatchawan C, Studer M, Hensch TK, et al. Excitatory projection neuron subtypes control the distribution of local inhibitory interneurons in the cerebral cortex. Neuron. 2011;69(4):763–79. doi: 10.1016/j.neuron.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RA, Beyer RP, et al. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc Natl Acad Sci U S A. 2010;107(29):13129–34. doi: 10.1073/pnas.1002285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghashghaei HT, Lai C, Anton ES. Neuronal migration in the adult brain: are we there yet? Nat Rev Neurosci. 2007;8(2):141–51. doi: 10.1038/nrn2074. [DOI] [PubMed] [Google Scholar]
- 29.Taupin P, Gage FH. Adult neurogenesis and neural stem cells of the central nervous system in mammals. J Neurosci Res. 2002;69(6):745–9. doi: 10.1002/jnr.10378. [DOI] [PubMed] [Google Scholar]
- 30.Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11(1):173–89. doi: 10.1016/0896-6273(93)90281-U. [DOI] [PubMed] [Google Scholar]
- 31.Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. 2004;427(6976):740–4. doi: 10.1038/nature02301. [DOI] [PubMed] [Google Scholar]
- 32.Malagelada C, Lopez-Toledano MA, Willett RT, Jin ZH, Shelanski ML, Greene LA. RTP801/REDD1 regulates the timing of cortical neurogenesis and neuron migration. J Neurosci. 2011;31(9):3186–96. doi: 10.1523/JNEUROSCI.4011-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobyns WB, Truwit CL, Ross ME, Matsumoto N, Pilz DT, Ledbetter DH, et al. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology. 1999;53(2):270–7. doi: 10.1212/WNL.53.2.270. [DOI] [PubMed] [Google Scholar]
- 34.Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52(1):158–74. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hehr U, Uyanik G, Aigner L, Couillard-Despres S, Winkler J. DCX-Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R). Seattle (WA): University of Washington; 1993. [PubMed]
- 36.Toba S, Tamura Y, Kumamoto K, Yamada M, Takao K, Hattori S, et al. Post-natal treatment by a blood–brain-barrier permeable calpain inhibitor, SNJ1945 rescued defective function in lissencephaly. Sci Rep. 2013;3:1224. doi: 10.1038/srep01224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilz DT, Matsumoto N, Minnerath S, Mills P, Gleeson JG, Allen KM, et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet. 1998;7(13):2029–37. doi: 10.1093/hmg/7.13.2029. [DOI] [PubMed] [Google Scholar]
- 38.des Portes V, Francis F, Pinard JM, Desguerre I, Moutard ML, Snoeck I, et al. doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH) Hum Mol Genet. 1998;7(7):1063–70. doi: 10.1093/hmg/7.7.1063. [DOI] [PubMed] [Google Scholar]
- 39.Gleeson JG, Minnerath SR, Fox JW, Allen KM, Luo RF, Hong SE, et al. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol. 1999;45(2):146–53. doi: 10.1002/1531-8249(199902)45:2<146::AID-ANA3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 40.Kato M, Kimura T, Lin C, Ito A, Kodama S, Morikawa T, et al. A novel mutation of the doublecortin gene in Japanese patients with X-linked lissencephaly and subcortical band heterotopia. Hum Genet. 1999;104(4):341–4. doi: 10.1007/s004390050963. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto N, Pilz DT, Fantes JA, Kittikamron K, Ledbetter DH. Isolation of BAC clones spanning the Xq22.3 translocation breakpoint in a lissencephaly patient with a de novo X;2 translocation. J Med Genet. 1998;35(10):829–32. doi: 10.1136/jmg.35.10.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pilz DT, Kuc J, Matsumoto N, Bodurtha J, Bernadi B, Tassinari CA, et al. Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet. 1999;8(9):1757–60. doi: 10.1093/hmg/8.9.1757. [DOI] [PubMed] [Google Scholar]
- 43.Dobyns WB, Elias ER, Newlin AC, Pagon RA, Ledbetter DH. Causal heterogeneity in isolated lissencephaly. Neurology. 1992;42(7):1375–88. doi: 10.1212/WNL.42.7.1375. [DOI] [PubMed] [Google Scholar]
- 44.Horesh D, Sapir T, Francis F, Wolf SG, Caspi M, Elbaum M, et al. Doublecortin, a stabilizer of microtubules. Hum Mol Genet. 1999;8(9):1599–610. doi: 10.1093/hmg/8.9.1599. [DOI] [PubMed] [Google Scholar]
- 45.Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23(2):257–71. doi: 10.1016/S0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 46.Moores CA, Perderiset M, Francis F, Chelly J, Houdusse A, Milligan RA. Mechanism of microtubule stabilization by doublecortin. Mol Cell. 2004;14(6):833–9. doi: 10.1016/j.molcel.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49(1):41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka T, Koizumi H, Gleeson JG. The doublecortin and doublecortin-like kinase 1 genes cooperate in murine hippocampal development. Cereb Cortex. 2006;16(Suppl 1):i69–73. doi: 10.1093/cercor/bhk005. [DOI] [PubMed] [Google Scholar]
- 49.Pramparo T, Youn YH, Yingling J, Hirotsune S, Wynshaw-Boris A. Novel embryonic neuronal migration and proliferation defects in Dcx mutant mice are exacerbated by Lis1 reduction. J Neurosci. 2010;30(8):3002–12. doi: 10.1523/JNEUROSCI.4851-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92(1):63–72. doi: 10.1016/S0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 51.Wynshaw-Boris A, Pramparo T, Youn YH, Hirotsune S. Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin Cell Dev Biol. 2010;21(8):823–30. doi: 10.1016/j.semcdb.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jang MA, Woo HI, Kim JW, Lee J, Ki CS. Identification of DCX gene mutation in lissencephaly spectrum with subcortical band heterotopia using whole exome sequencing. Pediatr Neurol. 2013;48(5):411–4. doi: 10.1016/j.pediatrneurol.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 53.Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG. Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem. 2000;275(44):34442–50. doi: 10.1074/jbc.M007078200. [DOI] [PubMed] [Google Scholar]
- 54.Moon HM, Wynshaw-Boris A. Cytoskeleton in action: lissencephaly, a neuronal migration disorder. Wiley Interdiscip Rev Dev Biol. 2013;2(2):229–45. doi: 10.1002/wdev.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pilz DT, Macha ME, Precht KS, Smith AC, Dobyns WB, Ledbetter DH. Fluorescence in situ hybridization analysis with LIS1 specific probes reveals a high deletion mutation rate in isolated lissencephaly sequence. Genet Med. 1998;1(1):29–33. doi: 10.1097/00125817-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 56.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364(6439):717–21. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 57.Wynshaw-Boris A. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin Genet. 2007;72(4):296–304. doi: 10.1111/j.1399-0004.2007.00888.x. [DOI] [PubMed] [Google Scholar]
- 58.Reiner O, Sapir T. LIS1 functions in normal development and disease. Curr Opin Neurobiol. 2013;23(6):951–6. doi: 10.1016/j.conb.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Siller KH, Doe CQ. Lis1/dynactin regulates metaphase spindle orientation in Drosophila neuroblasts. Dev Biol. 2008;319(1):1–9. doi: 10.1016/j.ydbio.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yingling J, Youn YH, Darling D, Toyo-Oka K, Pramparo T, Hirotsune S, et al. Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell. 2008;132(3):474–86. doi: 10.1016/j.cell.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moon HM, Youn YH, Pemble H, Yingling J, Wittmann T, Wynshaw-Boris A. LIS1 controls mitosis and mitotic spindle organization via the LIS1-NDEL1-dynein complex. Hum Mol Genet. 2014;23(2):449–66. doi: 10.1093/hmg/ddt436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saillour Y, Carion N, Quelin C, Leger PL, Boddaert N, Elie C, et al. LIS1-related isolated lissencephaly: spectrum of mutations and relationships with malformation severity. Arch Neurol. 2009;66(8):1007–15. doi: 10.1001/archneurol.2009.149. [DOI] [PubMed] [Google Scholar]
- 63.Grabham PW, Seale GE, Bennecib M, Goldberg DJ, Vallee RB. Cytoplasmic dynein and LIS1 are required for microtubule advance during growth cone remodeling and fast axonal outgrowth. J Neurosci. 2007;27(21):5823–34. doi: 10.1523/JNEUROSCI.1135-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pang T, Atefy R, Sheen V. Malformations of cortical development. Neurologist. 2008;14(3):181–91. doi: 10.1097/NRL.0b013e31816606b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cardoso C, Leventer RJ, Matsumoto N, Kuc JA, Ramocki MB, Mewborn SK, et al. The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum Mol Genet. 2000;9(20):3019–28. doi: 10.1093/hmg/9.20.3019. [DOI] [PubMed] [Google Scholar]
- 66.Jaglin XH, Poirier K, Saillour Y, Buhler E, Tian G, Bahi-Buisson N, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009;41(6):746–52. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar RA, Pilz DT, Babatz TD, Cushion TD, Harvey K, Topf M, et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet. 2010;19(14):2817–27. doi: 10.1093/hmg/ddq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sohal AP, Montgomery T, Mitra D, Ramesh V. TUBA1A mutation-associated lissencephaly: case report and review of the literature. Pediatr Neurol. 2012;46(2):127–31. doi: 10.1016/j.pediatrneurol.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 69.Hikita N, Hattori H, Kato M, Sakuma S, Morotomi Y, Ishida H, et al. A case of TUBA1A mutation presenting with lissencephaly and Hirschsprung disease. Brain Dev. 2014;36(2):159–62. doi: 10.1016/j.braindev.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 70.Guerrini R, Mei D, Cordelli DM, Pucatti D, Franzoni E, Parrini E. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur J Hum Genet. 2012;20(9):995–8. doi: 10.1038/ejhg.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arai T, Kaziro Y. Role of GTP in the assembly of microtubules. J Biochem. 1977;82(4):1063–71. doi: 10.1093/oxfordjournals.jbchem.a131777. [DOI] [PubMed] [Google Scholar]
- 72.Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, Cleak J, et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128(1):45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140(1):74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miura H, Yanazawa M, Kato K, Kitamura K. Expression of a novel aristaless related homeobox gene ‘Arx’ in the vertebrate telencephalon, diencephalon and floor plate. Mech Dev. 1997;65(1–2):99–109. doi: 10.1016/S0925-4773(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 75.Colasante G, Simonet JC, Calogero R, Crispi S, Sessa A, Cho G, et al. ARX Regulates Cortical Intermediate Progenitor Cell Expansion and Upper Layer Neuron Formation Through Repression of Cdkn1c. Cereb Cortex. 2013. doi:10.1093/cercor/bht222 [DOI] [PMC free article] [PubMed]
- 76.Simonet JC, Sunnen CN, Wu J, Golden JA, Marsh ED. Conditional Loss of Arx From the Developing Dorsal Telencephalon Results in Behavioral Phenotypes Resembling Mild Human ARX Mutations. Cereb Cortex. 2014. doi:10.1093/cercor/bhu090 [DOI] [PMC free article] [PubMed]
- 77.Abedini SS, Kahrizi K, Behjati F, Banihashemi S, Ghasemi Firoozabadi S, Najmabadi H. Mutational screening of ARX gene in Iranian families with X-linked intellectual disability. Arch Iran Med. 2012;15(6):361–5. [PubMed] [Google Scholar]
- 78.Charzewska A, Nawara M, Jakubiuk-Tomaszuk A, Obersztyn E, Hoffman-Zacharska D, Elert E, et al. Expanding the phenotype associated with missense mutations of the ARX gene. Am J Med Genet A. 2013;161A(7):1813–6. doi: 10.1002/ajmg.a.36003. [DOI] [PubMed] [Google Scholar]
- 79.Kato M, Dobyns WB. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet. 2003;12(Spec No 1):R89–96. doi: 10.1093/hmg/ddg086. [DOI] [PubMed] [Google Scholar]
- 80.Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32(3):359–69. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- 81.Shoubridge C, Fullston T, Gecz J. ARX spectrum disorders: making inroads into the molecular pathology. Hum Mutat. 2010;31(8):889–900. doi: 10.1002/humu.21288. [DOI] [PubMed] [Google Scholar]
- 82.Friocourt G, Kanatani S, Tabata H, Yozu M, Takahashi T, Antypa M, et al. Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J Neurosci. 2008;28(22):5794–805. doi: 10.1523/JNEUROSCI.1067-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132(Pt 6):1563–76. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vogt D, Hunt RF, Mandal S, Sandberg M, Silberberg SN, Nagasawa T, et al. Lhx6 directly regulates Arx and CXCR7 to determine cortical interneuron fate and laminar position. Neuron. 2014;82(2):350–64. doi: 10.1016/j.neuron.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Olivetti PR, Noebels JL. Interneuron, interrupted: molecular pathogenesis of ARX mutations and X-linked infantile spasms. Curr Opin Neurobiol. 2012;22(5):859–65. doi: 10.1016/j.conb.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Friocourt G, Parnavelas JG. Identification of Arx targets unveils new candidates for controlling cortical interneuron migration and differentiation. Front Cell Neurosci. 2011;5:28. doi: 10.3389/fncel.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beguin S, Crepel V, Aniksztejn L, Becq H, Pelosi B, Pallesi-Pocachard E, et al. An epilepsy-related ARX polyalanine expansion modifies glutamatergic neurons excitability and morphology without affecting GABAergic neurons development. Cereb Cortex. 2013;23(6):1484–94. doi: 10.1093/cercor/bhs138. [DOI] [PubMed] [Google Scholar]
- 88.D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24(2):471–9. doi: 10.1016/S0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 89.Frotscher M. Cajal-Retzius cells, Reelin, and the formation of layers. Curr Opin Neurobiol. 1998;8(5):570–5. doi: 10.1016/S0959-4388(98)80082-2. [DOI] [PubMed] [Google Scholar]
- 90.Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4(6):496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- 91.Forster E, Jossin Y, Zhao S, Chai X, Frotscher M, Goffinet AM. Recent progress in understanding the role of Reelin in radial neuronal migration, with specific emphasis on the dentate gyrus. Eur J Neurosci. 2006;23(4):901–9. doi: 10.1111/j.1460-9568.2006.04612.x. [DOI] [PubMed] [Google Scholar]
- 92.Sharaf A, Bock HH, Spittau B, Bouche E, Krieglstein K. ApoER2 and VLDLr are required for mediating reelin signalling pathway for normal migration and positioning of mesencephalic dopaminergic neurons. PLoS One. 2013;8(8):e71091. doi: 10.1371/journal.pone.0071091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Niu S, Renfro A, Quattrocchi CC, Sheldon M, D'Arcangelo G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron. 2004;41(1):71–84. doi: 10.1016/S0896-6273(03)00819-5. [DOI] [PubMed] [Google Scholar]
- 94.Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26(1):93–6. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- 95.Ozcelik T, Akarsu N, Uz E, Caglayan S, Gulsuner S, Onat OE, et al. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc Natl Acad Sci U S A. 2008;105(11):4232–6. doi: 10.1073/pnas.0710010105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Folsom TD, Fatemi SH. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology. 2013;68:122–35. doi: 10.1016/j.neuropharm.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boycott KM, Bonnemann C, Herz J, Neuert S, Beaulieu C, Scott JN, et al. Mutations in VLDLR as a cause for autosomal recessive cerebellar ataxia with mental retardation (dysequilibrium syndrome) J Child Neurol. 2009;24(10):1310–5. doi: 10.1177/0883073809332696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Palomero-Dominguez MA, Ramos-Fernandez JM, Dominguez-Santurino F, Ruiz-Gomez M. Type II lissencephaly: presentation of intermediate form. Rev Neurol. 1998;27(158):594–6. [PubMed] [Google Scholar]
- 99.Dobyns WB, Kirkpatrick JB, Hittner HM, Roberts RM, Kretzer FL. Syndromes with lissencephaly. II: Walker-Warburg and cerebro-oculo-muscular syndromes and a new syndrome with type II lissencephaly. Am J Med Genet. 1985;22(1):157–95. doi: 10.1002/ajmg.1320220118. [DOI] [PubMed] [Google Scholar]
- 100.Aida N. Fukuyama congenital muscular dystrophy: a neuroradiologic review. J Magn Reson Imaging. 1998;8(2):317–26. doi: 10.1002/jmri.1880080211. [DOI] [PubMed] [Google Scholar]
- 101.Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet J Rare Dis. 2006;1:29. doi: 10.1186/1750-1172-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Herman TE, Siegel MJ. Miller--Dieker syndrome, type 1 lissencephaly. J Perinatol. 2008;28(4):313–5. doi: 10.1038/sj.jp.7211920. [DOI] [PubMed] [Google Scholar]
- 103.Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1(5):717–24. doi: 10.1016/S1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 104.Fuchs-Telem D, Stewart H, Rapaport D, Nousbeck J, Gat A, Gini M, et al. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br J Dermatol. 2011;164(3):610–6. doi: 10.1111/j.1365-2133.2010.10133.x. [DOI] [PubMed] [Google Scholar]
- 105.Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, Rapaport D, Goldsher D, Indelman M, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet. 2005;77(2):242–51. doi: 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rapaport D, Lugassy Y, Sprecher E, Horowitz M. Loss of SNAP29 impairs endocytic recycling and cell motility. PLoS One. 2010;5(3):e9759. doi: 10.1371/journal.pone.0009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guerrini R, Filippi T. Neuronal migration disorders, genetics, and epileptogenesis. J Child Neurol. 2005;20(4):287–99. doi: 10.1177/08830738050200040401. [DOI] [PubMed] [Google Scholar]
- 108.Verrotti A, Spalice A, Ursitti F, Papetti L, Mariani R, Castronovo A, et al. New trends in neuronal migration disorders. Eur J Paediatr Neurol. 2010;14(1):1–12. doi: 10.1016/j.ejpn.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 109.Murdock DR, Clark GD, Bainbridge MN, Newsham I, Wu YQ, Muzny DM, et al. Whole-exome sequencing identifies compound heterozygous mutations in WDR62 in siblings with recurrent polymicrogyria. Am J Med Genet A. 2011;155A(9):2071–7. doi: 10.1002/ajmg.a.34165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bhat V, Girimaji SC, Mohan G, Arvinda HR, Singhmar P, Duvvari MR, et al. Mutations in WDR62, encoding a centrosomal and nuclear protein, in Indian primary microcephaly families with cortical malformations. Clin Genet. 2011;80(6):532–40. doi: 10.1111/j.1399-0004.2011.01686.x. [DOI] [PubMed] [Google Scholar]
- 111.Jansen A, Andermann E. Genetics of the polymicrogyria syndromes. J Med Genet. 2005;42(5):369–78. doi: 10.1136/jmg.2004.023952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57(12):2168–78. doi: 10.1212/WNL.57.12.2168. [DOI] [PubMed] [Google Scholar]
- 113.Guerreiro MM, Andermann E, Guerrini R, Dobyns WB, Kuzniecky R, Silver K, et al. Familial perisylvian polymicrogyria: a new familial syndrome of cortical maldevelopment. Ann Neurol. 2000;48(1):39–48. doi: 10.1002/1531-8249(200007)48:1<39::AID-ANA7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 114.Chang BS, Piao X, Giannini C, Cascino GD, Scheffer I, Woods CG, et al. Bilateral generalized polymicrogyria (BGP): a distinct syndrome of cortical malformation. Neurology. 2004;62(10):1722–8. doi: 10.1212/01.WNL.0000125187.52952.E9. [DOI] [PubMed] [Google Scholar]
- 115.Dobyns WB, Mirzaa G, Christian SL, Petras K, Roseberry J, Clark GD, et al. Consistent chromosome abnormalities identify novel polymicrogyria loci in 1p36.3, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2. Am J Med Genet A. 2008;146A(13):1637–54. doi: 10.1002/ajmg.a.32293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Robin NH, Taylor CJ, McDonald-McGinn DM, Zackai EH, Bingham P, Collins KJ, et al. Polymicrogyria and deletion 22q11.2 syndrome: window to the etiology of a common cortical malformation. Am J Med Genet A. 2006;140(22):2416–25. doi: 10.1002/ajmg.a.31443. [DOI] [PubMed] [Google Scholar]
- 117.Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25(1):247–51. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mitchell TN, Free SL, Williamson KA, Stevens JM, Churchill AJ, Hanson IM, et al. Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann Neurol. 2003;53(5):658–63. doi: 10.1002/ana.10576. [DOI] [PubMed] [Google Scholar]
- 119.Baala L, Briault S, Etchevers HC, Laumonnier F, Natiq A, Amiel J, et al. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet. 2007;39(4):454–6. doi: 10.1038/ng1993. [DOI] [PubMed] [Google Scholar]
- 120.Manuel MN, Mi D, Mason JO, Price DJ. Regulation of cerebral cortical neurogenesis by the Pax6 transcription factor. Front Cell Neurosci. 2015;9:70. doi: 10.3389/fncel.2015.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Roll P, Rudolf G, Pereira S, Royer B, Scheffer IE, Massacrier A, et al. SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet. 2006;15(7):1195–207. doi: 10.1093/hmg/ddl035. [DOI] [PubMed] [Google Scholar]
- 122.Brooks AS, Bertoli-Avella AM, Burzynski GM, Breedveld GJ, Osinga J, Boven LG, et al. Homozygous nonsense mutations in KIAA1279 are associated with malformations of the central and enteric nervous systems. Am J Hum Genet. 2005;77(1):120–6. doi: 10.1086/431244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Drevillon L, Megarbane A, Demeer B, Matar C, Benit P, Briand-Suleau A, et al. KBP-cytoskeleton interactions underlie developmental anomalies in Goldberg-Shprintzen syndrome. Hum Mol Genet. 2013;22(12):2387–99. doi: 10.1093/hmg/ddt083. [DOI] [PubMed] [Google Scholar]
- 124.Dafsari HS, Byrne S, Lin JP, Pitt M, Jongbloed JD, Flinter F, et al. Goldberg-Shprintzen megacolon syndrome with associated sensory motor axonal neuropathy. Am J Med Genet A. 2015;167(6):1300–4. doi: 10.1002/ajmg.a.36873. [DOI] [PubMed] [Google Scholar]
- 125.Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K, et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet. 2005;37(3):221–3. doi: 10.1038/ng1517. [DOI] [PubMed] [Google Scholar]
- 126.Handley MT, Carpanini SM, Mali GR, Sidjanin DJ, Aligianis IA, Jackson IJ, et al. Warburg Micro syndrome is caused by RAB18 deficiency or dysregulation. Open Biol. 2015;5(6):150047. doi: 10.1098/rsob.150047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reinstein E, Chang BS, Robertson SP, Rimoin DL, Katzir T. Filamin A mutation associated with normal reading skills and dyslexia in a family with periventricular heterotopia. Am J Med Genet A. 2012;158A(8):1897–901. doi: 10.1002/ajmg.a.35455. [DOI] [PubMed] [Google Scholar]
- 128.Chang BS, Katzir T, Liu T, Corriveau K, Barzillai M, Apse KA, et al. A structural basis for reading fluency: white matter defects in a genetic brain malformation. Neurology. 2007;69(23):2146–54. doi: 10.1212/01.wnl.0000286365.41070.54. [DOI] [PubMed] [Google Scholar]
- 129.Robertson SP. Filamin A: phenotypic diversity. Curr Opin Genet Dev. 2005;15(3):301–7. doi: 10.1016/j.gde.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 130.Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21(6):1315–25. doi: 10.1016/S0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- 131.Zhang J, Neal J, Lian G, Hu J, Lu J, Sheen V. Filamin A regulates neuronal migration through brefeldin A-inhibited guanine exchange factor 2-dependent Arf1 activation. J Neurosci. 2013;33(40):15735–46. doi: 10.1523/JNEUROSCI.1939-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, et al. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation. 2004;110(4):444–51. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- 133.Parrini E, Ramazzotti A, Dobyns WB, Mei D, Moro F, Veggiotti P, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain. 2006;129(Pt 7):1892–906. doi: 10.1093/brain/awl125. [DOI] [PubMed] [Google Scholar]
- 134.Sheen VL, Dixon PH, Fox JW, Hong SE, Kinton L, Sisodiya SM, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10(17):1775–83. doi: 10.1093/hmg/10.17.1775. [DOI] [PubMed] [Google Scholar]
- 135.Gomez-Garre P, Serratosa JM. Gene symbol: FLNA. Disease: Ehlers-Danlos syndrome and periventricular nodular heterotopia. Hum Genet. 2005;118(3–4):545. [PubMed] [Google Scholar]
- 136.Verrotti A, Monacelli D, Castagnino M, Villa MP, Parisi P. Ehlers-Danlos syndrome: a cause of epilepsy and periventricular heterotopia. Seizure. 2014;23(10):819–24. doi: 10.1016/j.seizure.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 137.Sheen VL, Wheless JW, Bodell A, Braverman E, Cotter PD, Rauen KA, et al. Periventricular heterotopia associated with chromosome 5p anomalies. Neurology. 2003;60(6):1033–6. doi: 10.1212/01.WNL.0000052689.03214.61. [DOI] [PubMed] [Google Scholar]
- 138.Moro F, Pisano T, Bernardina BD, Polli R, Murgia A, Zoccante L, et al. Periventricular heterotopia in fragile X syndrome. Neurology. 2006;67(4):713–5. doi: 10.1212/01.wnl.0000230223.51595.99. [DOI] [PubMed] [Google Scholar]
- 139.Togawa A, Morinaga N, Ogasawara M, Moss J, Vaughan M. Purification and cloning of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP-ribosylation factors. J Biol Chem. 1999;274(18):12308–15. doi: 10.1074/jbc.274.18.12308. [DOI] [PubMed] [Google Scholar]
- 140.Sheen VL. Filamin A, mediated Big2 dependent endocytosis: From apical abscission to periventricular heterotopia. Tissue Barriers. 2014;2:e29431. doi: 10.4161/tisb.29431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, Hill RS, et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36(1):69–76. doi: 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- 142.Kantarci S, Al-Gazali L, Hill RS, Donnai D, Black GC, Bieth E, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39(8):957–9. doi: 10.1038/ng2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McCarthy RA, Barth JL, Chintalapudi MR, Knaak C, Argraves WS. Megalin functions as an endocytic sonic hedgehog receptor. J Biol Chem. 2002;277(28):25660–7. doi: 10.1074/jbc.M201933200. [DOI] [PubMed] [Google Scholar]
- 144.Christ A, Christa A, Kur E, Lioubinski O, Bachmann S, Willnow TE, et al. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev Cell. 2012;22(2):268–78. doi: 10.1016/j.devcel.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 145.Kabat J, Krol P. Focal cortical dysplasia - review. Pol J Radiol. 2012;77(2):35–43. doi: 10.12659/PJR.882968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ying Z, Gonzalez-Martinez J, Tilelli C, Bingaman W, Najm I. Expression of neural stem cell surface marker CD133 in balloon cells of human focal cortical dysplasia. Epilepsia. 2005;46(11):1716–23. doi: 10.1111/j.1528-1167.2005.00276.x. [DOI] [PubMed] [Google Scholar]
- 147.Najm IM, Tilelli CQ, Oghlakian R. Pathophysiological mechanisms of focal cortical dysplasia: a critical review of human tissue studies and animal models. Epilepsia. 2007;48(Suppl 2):21–32. doi: 10.1111/j.1528-1167.2007.01064.x. [DOI] [PubMed] [Google Scholar]
- 148.Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary-Schaefer N, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62(6 Suppl 3):S2–8. doi: 10.1212/01.WNL.0000114507.30388.7E. [DOI] [PubMed] [Google Scholar]
- 149.Taylor DC, Falconer MA, Bruton CJ, Corsellis JA. Focal dysplasia of the cerebral cortex in epilepsy. J Neurol Neurosurg Psychiatry. 1971;34(4):369–87. doi: 10.1136/jnnp.34.4.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fassunke J, Blumcke I, Lahl R, Elger CE, Schramm J, Merkelbach-Bruse S, et al. Analysis of chromosomal instability in focal cortical dysplasia of Taylor’s balloon cell type. Acta Neuropathol. 2004;108(2):129–34. doi: 10.1007/s00401-004-0874-7. [DOI] [PubMed] [Google Scholar]
- 151.Becker AJ, Urbach H, Scheffler B, Baden T, Normann S, Lahl R, et al. Focal cortical dysplasia of Taylor’s balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to tuberous sclerosis. Ann Neurol. 2002;52(1):29–37. doi: 10.1002/ana.10251. [DOI] [PubMed] [Google Scholar]
- 152.Cotter D, Honavar M, Lovestone S, Raymond L, Kerwin R, Anderton B, et al. Disturbance of Notch-1 and Wnt signalling proteins in neuroglial balloon cells and abnormal large neurons in focal cortical dysplasia in human cortex. Acta Neuropathol. 1999;98(5):465–72. doi: 10.1007/s004010051111. [DOI] [PubMed] [Google Scholar]
- 153.Sato N, Yagishita A, Oba H, Miki Y, Nakata Y, Yamashita F, et al. Hemimegalencephaly: a study of abnormalities occurring outside the involved hemisphere. AJNR Am J Neuroradiol. 2007;28(4):678–82. [PMC free article] [PubMed] [Google Scholar]
- 154.Hengst M, Tucke J, Zerres K, Blaum M, Hausler M. Megalencephaly, mega corpus callosum, and complete lack of motor development: delineation of a rare syndrome. Am J Med Genet A. 2010;152A(9):2360–4. doi: 10.1002/ajmg.a.33577. [DOI] [PubMed] [Google Scholar]
- 155.Woo CL, Chuang SH, Becker LE, Jay V, Otsubo H, Rutka JT, et al. Radiologic-pathologic correlation in focal cortical dysplasia and hemimegalencephaly in 18 children. Pediatr Neurol. 2001;25(4):295–303. doi: 10.1016/S0887-8994(01)00318-6. [DOI] [PubMed] [Google Scholar]
- 156.Flores-Sarnat L. Hemimegalencephaly: part 1. Genetic, clinical, and imaging aspects. J Child Neurol. 2002;17(5):373–84. doi: 10.1177/088307380201700512. [DOI] [PubMed] [Google Scholar]
- 157.D'Agostino MD, Bastos A, Piras C, Bernasconi A, Grisar T, Tsur VG, et al. Posterior quadrantic dysplasia or hemi-hemimegalencephaly: a characteristic brain malformation. Neurology. 2004;62(12):2214–20. doi: 10.1212/01.WNL.0000130459.91445.91. [DOI] [PubMed] [Google Scholar]
- 158.Barkovich AJ, Chuang SH. Unilateral megalencephaly: correlation of MR imaging and pathologic characteristics. AJNR Am J Neuroradiol. 1990;11(3):523–31. [PMC free article] [PubMed] [Google Scholar]
- 159.Takashima S, Chan F, Becker LE, Kuruta H. Aberrant neuronal development in hemimegalencephaly: immunohistochemical and Golgi studies. Pediatr Neurol. 1991;7(4):275–80. doi: 10.1016/0887-8994(91)90045-M. [DOI] [PubMed] [Google Scholar]
- 160.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44(8):941–5. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74(1):41–8. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44(8):934–40. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14(5):307–20. doi: 10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- 164.Baek ST, Gibbs EM, Gleeson JG, Mathern GW. Hemimegalencephaly, a paradigm for somatic postzygotic neurodevelopmental disorders. Curr Opin Neurol. 2013;26(2):122–7. doi: 10.1097/WCO.0b013e32835ef373. [DOI] [PubMed] [Google Scholar]
- 165.Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294(5549):2186–9. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 166.Segarra J, Balenci L, Drenth T, Maina F, Lamballe F. Combined signaling through ERK, PI3K/AKT, and RAC1/p38 is required for met-triggered cortical neuron migration. J Biol Chem. 2006;281(8):4771–8. doi: 10.1074/jbc.M508298200. [DOI] [PubMed] [Google Scholar]
- 167.Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, et al. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci. 2012;32(17):5880–90. doi: 10.1523/JNEUROSCI.5462-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell. 2011;9(5):447–62. doi: 10.1016/j.stem.2011.09.008. [DOI] [PubMed] [Google Scholar]