

Abstract
Objective:
To track the effects of fingolimod, an approved drug for multiple sclerosis (MS), on the activation of myeloid cells from the periphery to the CNS.
Methods:
In vitro and ex vivo immunologic studies coupled with flow cytometry were performed to evaluate the action of fingolimod on lipopolysaccharide (LPS)–induced expression of activation markers in human monocytes from healthy participants, participants with untreated MS, and participants with fingolimod-treated MS. In vivo administration of fingolimod during experimental autoimmune encephalomyelitis (EAE) was established to verify the activation state of splenic, CNS infiltrating, and CNS resident myeloid cells ex vivo at flow cytometer.
Results:
We found that in vitro exposure of human monocytes to fingolimod inhibited LPS-induced CD25 and CD150 expression and tumor necrosis factor–α (TNF-α) secretion without altering immune cell survival. Further, EAE treatment with fingolimod led to reduced amounts of TNF-α produced by myeloid cells in vivo in the spleen and CNS. Finally, while displaying normal induction of CD25 and CD150 levels at high LPS concentration, monocytes from patients with fingolimod-treated MS showed significantly higher activation threshold at suboptimal LPS stimulation than controls.
Conclusions:
The inhibition of myeloid cell activation may be part of the immunosuppressive action of fingolimod and take place in the periphery and in the CNS.
Multiple sclerosis (MS) is a chronic disorder of the CNS characterized by inflammatory, demyelinating, and neurodegenerative processes causing neurologic disability.1 Among the approved MS treatments, fingolimod is a small molecule acting as structural analogue of sphingosine, a metabolite of the cell membrane lipid sphingomyelin.2 In vivo fingolimod is phosphorylated to fingolimod-phosphate, which binds 4 of the 5 known sphingosine-1-phosphate (S1P) receptors. Among them, S1P1 is involved in regulation of lymphocyte egress from lymphoid tissues into the circulation. After binding, fingolimod-phosphate induces S1P1 internalization and degradation, thus preventing immune cell trafficking toward the CNS.3 Clinical trials indicate that fingolimod represents a valid therapy for relapsing-remitting MS, as it reduces relapse rate and disability progression.4–6 In the animal model of disease, experimental autoimmune encephalomyelitis (EAE), treatment with fingolimod causes significant clinical amelioration associated with reduced CNS inflammation and astrogliosis.7,8
Owing to the expression of S1P receptors,9–13 monocytes, macrophages, and microglia may represent a target for fingolimod in MS. Here we provide evidence that (1) in vitro exposure to fingolimod significantly inhibits reactivity of human monocytes; (2) oral administration of fingolimod to EAE mice attenuates the inflammatory phenotype of myeloid cells in the spleen and CNS; (3) monocytes from fingolimod-treated patients with MS display impaired activation compared with monocytes from control participants. These data suggest that, in addition to the well-known sequestration of the immune cells within lymphoid organs, fingolimod may induce immunosuppression by altering the reactivity of circulating, CNS-infiltrating, and CNS-resident myeloid cells.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was approved by the local ethics committee and written informed consent was obtained from all study participants.
All procedures involving animals were authorized by the local institutional ethical animal committee and by the Italian General Direction for Animal Health at the Ministry for Health.
Human participants and blood sampling.
Blood was drawn from patients with fingolimod‐treated MS, patients with untreated MS, and healthy individuals. Patients with MS were adults of European ancestry with a relapsing-remitting course of the disease according to McDonald criteria14 and did not have any other acute or chronic inflammatory or autoimmune disorders. At sampling, all patients with MS were in the remission phase of disease. The untreated MS group included 7 patients (5 women, 2 men; mean age 38.9 ± 9.2 years; mean Expanded Disability Status Scale [EDSS] score 1.4 ± 0.3). Five of these patients with MS had not received any immunosuppressive or immunomodulatory therapy before sampling, whereas 2 patients, who had been treated with steroids at relapse and one of whom had previously received immunomodulatory drugs, were free from any therapy in the 18 months preceding blood sampling for this study. Patients with fingolimod‐treated MS (n = 11, 6 women, 5 men; mean age 39.8 ± 9.8 years; mean EDSS 2.1 ± 1) had received 0.5 mg fingolimod daily with a mean treatment duration of 18 ± 0.7 months. Before starting fingolimod therapy, 8 patients with MS had been treated with immunomodulatory drugs, whereas 3 patients with MS had not received any immunosuppressive or immunomodulatory therapy. Blood sampling was performed at least 3 months from treatment with steroids. The group of healthy donors included 8 participants (5 women, 3 men; mean age 30.5 ± 4.9 years).
Isolation and analysis of human peripheral blood mononuclear cells.
Peripheral blood mononuclear cells (PBMC) were isolated from anticoagulated whole blood using discontinuous density gradient as previously described,15 and immediately processed for the in vitro assays as detailed in the supplemental data at Neurology.org/nn.
EAE induction and treatment.
EAE was induced in 7-week-old female C57BL/6N mice (Harlan Laboratories, Udine, Italy) and animals were assessed for clinical signs of EAE as previously described.15,16 Fingolimod (FTY720, 3 mg/kg body weight, same batch used in reference 8; Selleckchem, DBA Italia, Milan, Italy) was given by oral gavage once daily starting 3 days after the appearance of the first clinical sign of disease. Control mice received administration of vehicle (0.9% NaCl physiologic solution). Experimenters were blind to treatment regimen of EAE mice.
Isolation and analysis of immune cells from spleen and CNS of EAE mice.
At day 33 after immunization, both vehicle- and fingolimod-treated mice were anaesthetized and perfused with phosphate-buffered saline. Splenocytes and CNS cells were then isolated and analyzed by flow cytometry as detailed in the supplemental data.
Statistical analysis.
Normality of the distribution was assessed by Kolmogorov-Smirnov statistics and significance was measured by Student t test with homeostatic variance in case of normal distribution or by Mann-Whitney U test in case of non-normal distribution. For statistical evaluation of EAE clinical score, linear regression analysis with 95% confidence interval was used. All p values were 2-sided and subjected to a significance level of 0.05.
RESULTS
In vitro exposure to fingolimod inhibits monocyte activation.
To evaluate fingolimod action on human myeloid cells, we performed in vitro immunologic assays where PBMC from healthy donors were preincubated with increasing fingolimod concentrations and then stimulated with a saturating dose of the monocyte activator lipopolysaccharide (LPS) for 18 hours (100 ng/mL17). First, cell staining for a vital dye (7-AAD) was evaluated at flow cytometer. After gating on monocytes in forward scatter vs side scatter plot, we compared the frequencies of 7-AAD+ cells under the different conditions. As shown in figure 1, the percentage of dead cells was low in both unstimulated and stimulated cultures, and did not change in cultures exposed to fingolimod (figure 1), indicating that fingolimod was not toxic to monocytes at any dose. We then analyzed the LPS-mediated induction of specific activation markers on viable monocytes in the same cultures. A previous study of our group defined that human monocytes respond to LPS stimulation via upregulation of a series of activation markers, including CD25 and CD150.18 Accordingly, we observed that stimulation with LPS for 18 hours effectively induced surface expression of CD25 and CD150 on human monocytes (middle panels in figure 2, A and C, pale purple bars in figure 2, B and D). Notably, preincubation with fingolimod reduced the percentage of CD25- and CD150-positive cells at all compound doses (third panels in figure 2, A and C, light purple, purple, and dark purple bars in figure 2, B and D). Previous publications of our group characterized the dose response of human monocytes to LPS and showed that induction of activation markers starts at 150 pg/mL LPS and reaches a plateau at LPS concentrations >1 ng/mL.17,18 When repeating the immunologic assays using nonsaturating LPS (600 pg/mL), fingolimod-mediated inhibition of monocyte activation was stronger at 600 pg/mL LPS than at 100 ng/mL LPS (figure 2E). Further, the drug decreased LPS-mediated tumor necrosis factor–α (TNF-α) production as detected in culture supernatants by ELISA (figure 3).
Figure 1. Fingolimod does not alter human monocyte viability.

Peripheral blood mononuclear cells from healthy donors were briefly exposed to increasing concentrations of fingolimod (0.1 μM, 1 μM, and 10 μM) or vehicle, and left in culture for 18 hours, eventually in the presence of 100 ng/mL lipopolysaccharide (LPS). Dead cells were stained with the vital dye 7-AAD and measured at flow cytometer. Monocytes were gated in forward vs side scatter plot. Graph depicts the frequency of cell death in unstimulated or LPS-stimulated cultures after exposure to fingolimod. Data were obtained from 3 independent experiments. Bars represent SEM.
Figure 2. Fingolimod impairs induction of activation markers on human monocytes.

Peripheral blood mononuclear cells from healthy donors were briefly exposed to increasing concentrations of fingolimod (0.1 μM, 1 μM, and 10 μM) or vehicle, and left in culture for 18 hours, eventually in the presence of 100 ng/mL lipopolysaccharide (LPS). Monocyte activation was monitored by staining for 2 surface markers, CD25 (A, B) and CD150 (C, D). Gates on monocytes in forward vs side scatterplot and then on viable (7-AAD negative) cells were applied. Thresholds were set on relative isotype controls. (A, C) Representative CD25 and CD150 stainings in unstimulated (left panels), LPS-stimulated (middle panels), or LPS-stimulated cultures exposed to 1 μM fingolimod (right panels). (B, D) Frequency of CD25 and CD150 expressing monocytes in unstimulated and LPS-stimulated cultures in the absence or presence of increasing concentrations of fingolimod. Data were obtained from 5 independent experiments. (E) Graphs represent fingolimod-mediated inhibition of activation markers induced at 2 LPS doses. Data were obtained from 2 to 5 experiments. Bars represent SEM. *p < 0.05; **p < 0.01. FSC = forward scatter.
Figure 3. Fingolimod inhibits TNF-α secretion by human monocytes.
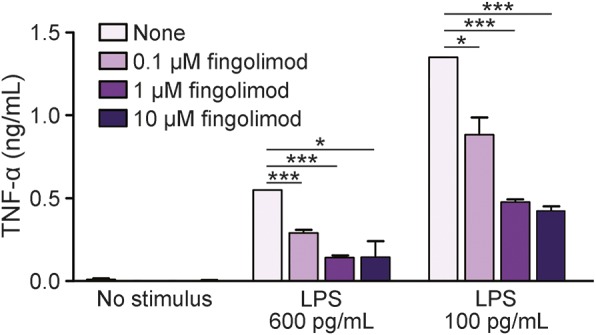
Peripheral blood mononuclear cells from healthy donors were briefly exposed to increasing concentrations of fingolimod (0.1 μM, 1 μM, and 10 μM) or vehicle, and left in culture for 18 hours, eventually in the presence of 600 pg/mL or 100 ng/mL lipopolysaccharide (LPS). Supernatants were collected and tumor necrosis factor–α (TNF-α) levels were detected by ELISA assay. Data of a representative experiment are shown. Bars represent SEM. *p < 0.05; ***p < 0.001.
Overall, these in vitro data indicate that fingolimod may impair responsiveness of human monocytes to activation and that its efficacy may depend on the strength of activating stimuli.
Treatment of EAE with fingolimod reduces TNF-α production by myeloid cells in the periphery and in the CNS.
We performed EAE experiments to evaluate whether fingolimod modulates activation of myeloid cells in vivo during neuroinflammation. Administration of fingolimod at disease onset is effective in ameliorating EAE, as shown in a recent publication of our group.8 For the current study, we started treatment 3 days after disease onset to isolate sufficient numbers of immune cells from each EAE mouse for phenotypical analyses. EAE was induced in C57BL/6 wild-type mice with myelin oligodendrocyte glycoprotein (MOG)35-55 peptide, and a group of animals was treated with 3 mg/kg/d fingolimod by oral administration, while the control group received vehicle. Daily observation of the clinical state revealed a milder chronic phase in fingolimod-treated mice compared to vehicle-treated group (figure 4A). In fact, clinical curves synchronized for time from immunization or for treatment duration were significantly different in the 2 groups (figure 4A). On day 33 postimmunization, spleen cells from both vehicle- and fingolimod-treated EAE mice were isolated, briefly reactivated in vitro with LPS, stained for surface lineage markers CD45 and CD11b and for intracellular TNF-α, and analyzed by flow cytometry (figure 4, B and C). Interestingly, whereas about 10% of CD45+ CD11b+ macrophages of vehicle-treated EAE mice produced TNF-α, only 5% of spleen macrophages isolated from fingolimod-treated EAE mice stained positive for the inflammatory cytokine (p < 0.05; figure 4, B and C). Similar experiments were performed on myeloid cells isolated from the CNS of EAE mice, and CNS-infiltrating macrophages and resident microglia, defined as CD45high CD11b+ and CD45low CD11b+ Ly6C− cells, respectively, were analyzed separately (figure 4D, left panels). Representative dot plots for TNF-α staining of cells from vehicle- and fingolimod-treated EAE mice are shown in the middle and right panels in figure 4D. Similarly to spleen myeloid cells and in line with the mild clinical amelioration, macrophages and microglia in the CNS of fingolimod-treated EAE mice showed about 30% reduction in the percentage of TNF-α-positive cells compared with cells from vehicle-treated mice (8.1% vs 5.7% and 13.5% vs 9.7% for macrophages or microglia, respectively, in vehicle-treated vs fingolimod-treated mice, p < 0.05 figure 4E).
Figure 4. Therapeutic administration of fingolimod during EAE reduces myeloid cell activation in the spleen and CNS.

(A) Clinical expression of MOG 35-55 peptide-induced experimental autoimmune encephalomyelitis (EAE) in fingolimod-treated (n = 13, black dots) or vehicle-treated (n = 12, white dots) mice. Drug or vehicle were administered daily starting 3 days after disease onset. Left graph shows mean clinical scores from day of immunization, right graph depicts mean clinical score according to treatment duration. (B) Representative stainings for tumor necrosis factor–α (TNF-α) production by CD45+ CD11b+ spleen myeloid cells from vehicle- and fingolimod-treated EAE mice. (C) Frequency of TNF-α–producing myeloid cells in vehicle-treated (n = 5) and fingolimod-treated EAE (n = 4) mice. (D) Representative stainings for TNF-α production by CNS myeloid cells from vehicle-treated and fingolimod-treated EAE mice. Left panels show gating strategy. R′: CD45hiCD11b+ blood-borne CNS infiltrating myeloid cells. Gate R″: CD45lowCD11b+Ly6C− CNS resident microglia. Middle and right panels depict representative stainings for TNF-α production by CNS infiltrating (upper) or resident (lower) myeloid cells from vehicle-treated and fingolimod-treated EAE mice. (E) Frequency of TNF-α–producing CNS myeloid cells in vehicle-treated (n = 5) and fingolimod-treated EAE mice (n = 5). Representative animals of the mean EAE clinical score (A) were analyzed in C and E. Bars represent SEM. *p < 0.05; ***p < 0.001.
Taken together, these results show that therapy of experimental MS with fingolimod attenuates TNF-α synthesis by myeloid cells both in periphery and in the CNS.
Treatment of MS with fingolimod impairs monocyte reactivity.
Taking into consideration the anti-inflammatory effect exerted by fingolimod treatment on peripheral myeloid cells in the EAE model, we set up ex vivo immunologic assays to monitor monocyte activation in patients with fingolimod-treated MS and compared it to that in controls. We recruited 11 fingolimod-treated patients, 7 patients with untreated MS, and 8 healthy participants, isolated PBMC, and assessed LPS-induced expression of CD25 and CD150 on CD14+ monocytes for each individual. Note that in these experiments immune cells were not exposed to fingolimod in vitro. Figure 5A shows that the percentage of CD25 and CD150 induced by saturating LPS concentrations (100 ng/mL) on monocytes from patients with fingolimod-treated MS was not significantly different from that present in cells from healthy participants or patients with untreated MS. On the contrary, when cells were stimulated with low/intermediate LPS concentrations, monocytes from patients with fingolimod-treated MS were resistant to activation compared with cells from control participants, as the fraction of CD25- or CD150-positive monocytes in treated patients was much lower than in controls. Figure 5B shows representative stainings for the activation markers at 600 pg/mL LPS in the 3 groups, and figure 5C depicts the LPS-induced activation curves for CD25 and CD150 for each participant and the group means. Despite interindividual variability, a lower proportion of monocytes upregulated CD25 and CD150 in patients with fingolimod-treated MS in the low LPS dose range. For example, the percentage of monocytes expressing CD25 at 600 pg/mL LPS in the fingolimod-treated MS group was 1.7%, compared to 12.5% and 15.2% of monocytes from patients with untreated MS and healthy participants, respectively (p < 0.001; figure 5C, mean curves). The 2 control groups did not differ apart from a lower percentage of CD150-positive monocytes at 600 pg/mL and at 100 ng/mL LPS in patients with untreated MS compared to healthy controls. These results indicate that treatment with fingolimod raises the activation threshold of human monocytes in vivo.
Figure 5. Treatment with fingolimod raises the activation threshold of monocytes in MS.

Peripheral blood mononuclear cells from 8 healthy donors, 7 patients with untreated multiple sclerosis (MS), and 11 patients with fingolimod-treated MS were briefly stimulated ex vivo with lipopolysaccharide (LPS). Monocytes were gated by forward vs side scatterplot and CD14-positive staining. Gate on viable (7-AAD negative) cells was then applied. Monocyte activation was monitored by staining for the 2 surface markers CD25 and CD150. Thresholds were set on relative isotype controls. (A) Frequency of CD25-positive (upper graph) and CD150-positive (lower graph) monocytes cultured ex vivo with 100 ng/mL LPS; each dot represents a single participant; black bars represent mean values for each group. (B) CD25 (upper) and CD150 (lower) stainings in cultures stimulated with 600 pg/mL LPS from a representative healthy control (left), a patient with untreated MS (middle), and a patient with fingolimod-treated MS (right). (C) Frequency of CD25 and CD150 positive monocytes at increasing LPS concentrations. Dose response curves are shown for each participant. Right panels show the means for each group. *p < 0.05; ***p < 0.001. FSC = forward scatter.
DISCUSSION
In this study, we provide evidence for an effect of fingolimod on myeloid cells in vitro and in vivo. In vitro exposure of PBMC to the compound impaired responsiveness of human monocytes to LPS stimulation by reducing expression of the activation markers CD25 and CD150 and of the inflammatory cytokine TNF-α. Accordingly, oral administration of fingolimod to EAE mice attenuated the inflammatory phenotype of myeloid cells residing in the periphery and in the CNS. Finally, ex vivo monitoring of monocyte reactivity in fingolimod-treated patients with MS and controls demonstrated that therapy with fingolimod lowered monocyte susceptibility to activation. These results shed new light on the role of S1P signaling pathway in myeloid cells and on the mechanism of action of fingolimod.
S1P is an important bioactive lipid mediator regulating cellular processes like growth, survival, and differentiation.19,20 It is synthesized from sphingosine by the action of sphingosine kinases and degraded by S1P lyase, whose constitutive activity keeps S1P levels low in most tissues with the exception of blood and lymph.21 S1P regulates immune functions by binding specific plasma membrane G protein-coupled receptors (S1P1-5) or acting on intracellular targets. S1P receptors are widely expressed on innate and adaptive immune cells.9–13,22 The well-known function of S1P signaling in immunity regards the regulation of immune cell trafficking, as the different S1P concentration between lymphoid tissues and circulation creates the S1P gradient used by mature T lymphocytes to leave lymphoid organs.21 A few reports in the literature suggest that S1P signaling may regulate immune cell functions like proliferation and cytokine production.22 In myeloid cells, S1P pathway protects from apoptosis, regulates phagosome maturation, and induces cytokine release.23 Thus, our hypothesis was that targeting S1P receptors with fingolimod could alter some myeloid cell functions. Limited information is present in the literature regarding this issue. In vitro studies have shown that exposure to fingolimod may reduce the production of inflammatory cytokines by CD40L-stimulated human monocyte-derived macrophages,12 or LPS-activated mouse monocytes, dendritic cells, or microglia,13,24,25 suggesting that the drug may interfere with myeloid cell activation in the periphery and in the CNS. We verified it with our exploratory in vitro studies, where human monocytes from peripheral blood were briefly exposed to fingolimod and activated with LPS. These data indicate that in vitro blockade of S1P signaling via fingolimod does not alter cell viability but impairs myeloid cell reactivity. In fact, LPS-mediated induction of the activation markers CD25 and CD150 and the release of TNF-α were much lower in fingolimod-treated cultures compared with control cells. This effect was not due to cell death as toxicity induced by the drug was excluded.
Recent evidence by Lewis et al.24 indicates that a single dose of fingolimod is sufficient to cause accumulation of monocytes in rodent spleen and bone marrow, thus reducing myeloid cell number in circulation. Further, it decreases the expression of activation markers on blood monocytes from naive mice and cytokine release by monocytes of LPS-challenged mice.24 A drop in the number of circulating monocytes has been reported in EAE animals treated daily with the drug24; however, the effect of the therapy on myeloid cell activation during neuroinflammation in rodents has not been addressed in any study to our knowledge. Our ex vivo investigations in the EAE model provide evidence for an inhibitory effect of fingolimod on myeloid cells in the lymphoid organs and the CNS. In fact, the fraction of TNF-α-producing splenic, CNS infiltrating, or CNS resident myeloid cells was lower in fingolimod-treated mice than in control mice. Interestingly, intraperitoneal administration of fingolimod to an animal model of atherosclerosis modulates LPS-elicited production of inflammatory mediators by peritoneal macrophages.26 Together with our results, this evidence indicates that, as part of its mechanism of action, fingolimod targets innate immunity in vivo regardless of the administration route. The relevant observation in our study is that this action is not restricted to the periphery but extended to the target organ of experimental neuroinflammation, the CNS, raising the possibility that cerebral innate immunity in patients with MS may be regulated by fingolimod treatment, an issue that deserves further investigation.
Regarding the effects of fingolimod therapy on immune cells in circulation, it reduces the absolute number of B and T lymphocytes27,28 but not of monocytes in peripheral blood of patients with MS,25 so that the percentage of myeloid cells within PBMC is increased in patients with treated MS compared to controls.28 The levels of common surface markers of monocytes (e.g., CD86, HLA-DR) do not change under treatment.25 However, these proteins are not indicative of a functional state as abundantly expressed already in resting cells. In contrast, short ex vivo stimulation assays followed by detection of activation-induced markers may highlight treatment-related functional alterations.17,25 Interestingly, CD1c+ DC and monocytes from patients with fingolimod-treated MS show impaired LPS-mediated production of IL1β but not of other inflammatory mediators.28 The possibility that fingolimod can act on the activation threshold of myeloid cells was not addressed in these experiments, as a single saturating dose of LPS was used. To translate to human our observation that the drug may alter myeloid cell activation in vitro and in the EAE model, we tested monocyte reactivity from patients with treated MS and controls by ex vivo stimulation assays previously developed by our group.17,18 Here we show that therapy of MS with fingolimod strongly raises the activation threshold of human monocytes. The implications for MS are numerous. It is well-known that in MS lesions activated macrophages and microglia outnumber lymphocytes, phagocyte myelin, and release toxic mediators.29,30 Thus the observation that blockade of S1P signaling via fingolimod impairs myeloid activation supports the hypothesis of an anti-inflammatory tissue protective effect exerted by the drug in the CNS. Theoretically, the reduced propensity of myeloid cells to initiate the inflammatory program may have an impact on the activation of T lymphocytes or skew T-cell differentiation from an inflammatory to an anti-inflammatory or regulatory phenotype. Indeed, in vitro treatment of mouse bone marrow–derived DC with fingolimod impairs the ability of the myeloid cells to activate myelin-reactive T lymphocytes.25 In MS, fingolimod affects the number of Th17 cells in blood31 and the frequency of interferon-γ-producing CD4 and CD8 T lymphocytes in short-term T cell lines,31,32 while increasing the frequency of circulating regulatory T lymphocytes.31,32 These findings may be the result of altered T-cell differentiation or of selective retention of distinct immune cell subsets in secondary lymphoid organs. Appropriate studies in the disease model are required to clarify this issue. Overall, we may envisage that treatment with fingolimod suppresses the generation of inflammatory antigen-specific T-cell responses via modulation of myeloid cell activation. Though beneficial for the control of encephalitogenic T cells in MS, this scenario may imply impairment of innate and adaptive immune responses to pathogens. Our ex vivo study clearly shows that at saturating stimulation conditions no difference in monocyte activation can be detected between patients with fingolimod-treated MS and controls. Further, the frequency of T lymphocytes against influenza following vaccination is similar in patients with treated MS and healthy controls.33 However, recent studies indicate that patients with fingolimod-treated MS display reduced humoral responses to vaccination with novel or recall antigens and higher rates of varicella-zoster virus infection compared with placebo recipients.34,35 A rare but serious complication in immunocompromised individuals is the development of progressive multifocal leukoencephalopathy (PML) caused by JC virus reactivation. This event has been associated with natalizumab treatment in patients with MS36 and has been reported in a few patients with MS after switching from natalizumab to fingolimod.37 Whether fingolimod alone alters immune surveillance to JC virus remains under investigation. Up to now, only one PML case has been reported in a patient with fingolimod-treated MS who had not been exposed to natalizumab previously (Novartis; Basel, Switzerland).
We have described a practical immunologic test that measures large and reproducible effects associated with the treatment despite the small sample size. We propose our ex vivo assay for monitoring monocyte function in large cohorts of patients with MS starting fingolimod treatment to correlate the immunologic response with therapeutic efficacy.
Overall, we have demonstrated that blockade of S1P signaling via fingolimod impairs myeloid cell activation in vitro, in EAE, and in MS. This observation extends the current knowledge about the mechanism of action of this drug, and may have important clinical implications, which need to be explored in future studies.
ACKNOWLEDGMENT
The authors thank Eleonora Capitolo, IRCCS San Raffaele Scientific Institute (Milan, Italy), for technical assistance and Amici Centro Sclerosi Multipla (ACESM) for support.
GLOSSARY
- EAE
experimental autoimmune encephalomyelitis
- EDSS
Expanded Disability Status Scale
- LPS
lipopolysaccharide
- MS
multiple sclerosis
- PBMC
peripheral blood mononuclear cells
- PML
progressive multifocal leukoencephalopathy
- S1P
sphingosine-1-phosphate
- TNF-α
tumor necrosis factor–α
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
D.D.M. performed in vitro and EAE experiments, performed statistical analyses, analyzed data, and wrote the article. C.E. performed in vitro and EAE experiments. G.C. performed ex vivo experiments from human subjects. D.F.D. performed cytofluorimetric experiments on mouse tissues. M.M.J., R.M., S.F., M.L., R.M.E., and M.V. enrolled patients with MS and provided clinical information. M.G. and C.G. discussed results and gave feedback on the manuscript. C.F. conceived and designed the experiments, analyzed data, and wrote the article.
STUDY FUNDING
This study was funded by the Italian Ministry for Health. E. Colombo was supported by a FISM (Fondazione Italiana Sclerosi Multipla) research fellowship [Cod. 2013/B/3].
DISCLOSURE
M. Di Dario, E. Colombo, C. Govi, D. De Feo, M.J. Messina, M.A.L. Romeo, and F. Sangalli report no disclosures. L. Moiola received speaker honoraria from Biogen Idec, Merck Serono, and Sanofi Aventis. M. Rodegher is on the scientific advisory board for Novartis and received travel support from Merck Serono, Bayer Schering, Teva, and Sanofi Aventis. G. Martino is an associate editor for Stem Cell Research & Therapy, section editor for Multiple Sclerosis and Related Disorders, is an editorial board member for Current Gene Therapy, Multiple Sclerosis International, Multiple Sclerosis Journal, Neurological Sciences, Regenerative Medicine, The Open Gene Therapy Journal, World Journal of Neurology, and World Journal of Stem Cells, and holds patents/patent applications for Inflammation, Laquinimod for the treatment of cannabinoid receptor type 1–mediated disorders and Laquinimod for the treatment of GABA-mediated disorders. V. Martinelli served on the scientific advisory board for Merck Serono and Genzyme and received travel funding and/or speaker honoraria from Biogen Idec, Merck Serono, Bayer, Teva, Novartis, and Genzyme. G. Comi received personal fees from Novartis, Teva, Sanofi, Genzyme, Merck Serono, Excemed, and Receptos, and is an associate editor for Neurological Sciences. C. Farina is on the editorial board for Journal Advances in Medicine and received research support from Merck-Serono, Italian Ministry for Health, and Fondazione Italiana Sclerosi Multipla. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Compston A, Coles A. Multiple sclerosis. Lancet 2008;372:1502–1517. [DOI] [PubMed] [Google Scholar]
- 2.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008;9:139–150. [DOI] [PubMed] [Google Scholar]
- 3.Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 2010;9:883–897. [DOI] [PubMed] [Google Scholar]
- 4.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med 2006;355:1124–1140. [DOI] [PubMed] [Google Scholar]
- 5.O'Connor P, Comi G, Montalban X, et al. Oral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension study. Neurology 2009;72:73–79. [DOI] [PubMed] [Google Scholar]
- 6.Kappos L, Radue EW, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387–401. [DOI] [PubMed] [Google Scholar]
- 7.Kataoka H, Sugahara K, Shimano K, et al. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol 2005;2:439–448. [PubMed] [Google Scholar]
- 8.Colombo E, Di Dario M, Capitolo E, et al. Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann Neurol 2014;76:325–337. [DOI] [PubMed] [Google Scholar]
- 9.Duong CQ, Bared SM, Abu-Khader A, Buechler C, Schmitz A, Schmitz G. Expression of the lysophospholipid receptor family and investigation of lysophospholipid-mediated responses in human macrophages. Biochim Biophys Acta 2004;1682:112–119. [DOI] [PubMed] [Google Scholar]
- 10.Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res 2008;102:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michaud J, Im DS, Hla T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J Immunol 2010;184:1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durafourt BA, Lambert C, Johnson TA, Blain M, Bar-Or A, Antel JP. Differential responses of human microglia and blood-derived myeloid cells to FTY720. J Neuroimmunol 2011;230:10–16. [DOI] [PubMed] [Google Scholar]
- 13.Noda H, Takeuchi H, Mizuno T, Suzumura A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J Neuroimmunol 2013;256:13–18. [DOI] [PubMed] [Google Scholar]
- 14.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol 2001;50:121–127. [DOI] [PubMed] [Google Scholar]
- 15.Menon R, Di Dario M, Cordiglieri C, et al. Gender-based blood transcriptomes and interactomes in multiple sclerosis: Involvement of SP1 dependent gene transcription. J Autoimmun 2012;38:J144–J155. [DOI] [PubMed] [Google Scholar]
- 16.Colombo E, Cordiglieri C, Melli G, et al. Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med 2012;209:521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber MS, Starck M, Wagenpfeil S, Meinl E, Hohlfeld R, Farina C. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain 2004;127:1370–1378. [DOI] [PubMed] [Google Scholar]
- 18.Farina C, Theil D, Semlinger B, Hohlfeld R, Meinl E. Distinct responses of monocytes to toll-like receptor ligands and inflammatory cytokines. Int Immunol 2004;16:799–809. [DOI] [PubMed] [Google Scholar]
- 19.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 2003;4:397–407. [DOI] [PubMed] [Google Scholar]
- 20.Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol 2011;11:403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature 2014;510:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol 2005;5:560–570. [DOI] [PubMed] [Google Scholar]
- 23.Weigert A, Weis N, Brune B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology 2009;214:748–760. [DOI] [PubMed] [Google Scholar]
- 24.Lewis ND, Haxhinasto SA, Anderson SM, et al. Circulating monocytes are reduced by sphingosine-1-phosphate receptor modulators independently of S1P3. J Immunol 2013;190:3533–3540. [DOI] [PubMed] [Google Scholar]
- 25.Luessi F, Kraus S, Trinschek B, et al. FTY720 (fingolimod) treatment tips the balance towards less immunogenic antigen-presenting cells in patients with multiple sclerosis. Mult Scler. Epub 2015 Mar 2. [DOI] [PubMed]
- 26.Nofer JR, Bot M, Brodde M, et al. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007;115:501–508. [DOI] [PubMed] [Google Scholar]
- 27.Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol 2009;158:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowarik MC, Pellkofer HL, Cepok S, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology 2011;76:1214–1221. [DOI] [PubMed] [Google Scholar]
- 29.Barnett MH, Henderson AP, Prineas JW. The macrophage in MS: Just a scavenger after all? Pathology and pathogenesis of the acute MS lesion. Mult Scler 2006;12:121–132. [DOI] [PubMed] [Google Scholar]
- 30.King IL, Dickendesher TL, Segal BM. Circulating ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009;113:3190–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehling M, Lindberg R, Raulf F, et al. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology 2010;75:403–410. [DOI] [PubMed] [Google Scholar]
- 32.Serpero LD, Filaci G, Parodi A, et al. Fingolimod modulates peripheral effector and regulatory T cells in MS patients. J Neuroimmun Pharmacol 2013;8:1106–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehling M, Hilbert P, Fritz S, et al. Antigen-specific adaptive immune responses in fingolimod-treated multiple sclerosis patients. Ann Neurol 2011;69:408–413. [DOI] [PubMed] [Google Scholar]
- 34.Kappos L, Mehling M, Arroyo R, et al. Randomized trial of vaccination in fingolimod-treated patients with multiple sclerosis. Neurology 2015;84:872–879. [DOI] [PubMed] [Google Scholar]
- 35.Arvin AM, Wolinsky JS, Kappos L, et al. Varicella-zoster virus infections in patients treated with fingolimod: Risk assessment and consensus recommendations for management. JAMA Neurol 2015;72:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med 2012;366:1870–1880. [DOI] [PubMed] [Google Scholar]
- 37.Killestein J, Vennegoor A, van Golde AE, Bourez RL, Wijlens ML, Wattjes MP. PML-IRIS during fingolimod diagnosed after natalizumab discontinuation. Case Rep Neurol Med 2014;2014:307872. [DOI] [PMC free article] [PubMed] [Google Scholar]