

Abstract
Background
T cells undergo autoimmunization following spinal cord injury (SCI) and play both protective and destructive roles during the recovery process. T cell-deficient athymic nude (AN) rats exhibit improved functional recovery when compared to immunocompetent Sprague–Dawley (SD) rats following spinal cord transection.
Methods
In the present study, we evaluated locomotor recovery in SD and AN rats following moderate spinal cord contusion. To explain variable locomotor outcome, we assessed whole-genome expression using RNA sequencing, in the acute (1 week post-injury) and chronic (8 weeks post-injury) phases of recovery.
Results
Athymic nude rats demonstrated greater locomotor function than SD rats only at 1 week post-injury, coinciding with peak T cell infiltration in immunocompetent rats. Genetic markers for T cells and helper T cells were acutely enriched in SD rats, while AN rats expressed genes for Th2 cells, cytotoxic T cells, NK cells, mast cells, IL-1a, and IL-6 at higher levels. Acute enrichment of cell death-related genes suggested that SD rats undergo secondary tissue damage from T cells. Additionally, SD rats exhibited increased acute expression of voltage-gated potassium (Kv) channel-related genes. However, AN rats demonstrated greater chronic expression of cell death-associated genes and less expression of axon-related genes. Immunostaining for macrophage markers revealed no T cell-dependent difference in the acute macrophage infiltrate.
Conclusions
We put forth a model in which T cells facilitate early tissue damage, demyelination, and Kv channel dysregulation in SD rats following contusion SCI. However, compensatory features of the immune response in AN rats cause delayed tissue death and limit long-term recovery. T cell inhibition combined with other neuroprotective treatment may thus be a promising therapeutic avenue.
Electronic supplementary material
The online version of this article (doi:10.1186/s12868-015-0212-0) contains supplementary material, which is available to authorized users.
Keywords: Spinal cord injury, Inflammation, Locomotor function, Neuronal cell death, Axonal regeneration
Background
The role of the immune system in spinal cord injury (SCI) is complex, controversial, and relevant to the pathophysiology and treatment of SCI. The literature supports both positive and negative effects of the immune system in functional recovery after SCI [1, 2]. T cells, which help coordinate the immune response, have received substantial attention for their role in SCI.
Systemic spread of central nervous system (CNS) antigens after SCI leads to the formation of autoimmune T cells [3, 4] that migrate to the injury site, reaching peak accumulation at 1 week post-injury [5, 6]. Helper T cells are responsible for macrophage recruitment and activation, which has been shown to produce focal axonal injury and demyelination [7, 8]. These cells also cause formation of autoimmune B cells that produce antibodies to CNS proteins [9]. Meanwhile, autoimmune cytotoxic T cells have been demonstrated to produce significant white matter pathology [10, 11]. Interestingly, while myelin-reactive T cells are associated with tissue loss and functional impairment [12, 13], these same cells have been found to secrete neuroprotective factors and prevent secondary damage to the spinal cord via “protective autoimmunity” [14–16].
T cell suppression appears to improve recovery from SCI. T cell inhibitors including cyclosporine [17, 18], fingolimod [19], and integrin inhibitors [20, 21] have been shown to improve neurological function after SCI, though their benefit may depend upon injury severity [22]. T cell-deficient athymic nude (AN) rats demonstrate superior locomotor function after transection injury compared to immunocompetent Sprague–Dawley (SD) rats [23]. The effects of T cell deficiency in nervous system trauma are not limited to SCI; T and B cell-deficient RAG1−/− mice suffer less damage from traumatic brain injury [24].
In order to characterize the whole-genome response to SCI, researchers have employed microarrays [25, 26] and RNA sequencing (RNA-seq) [27]. Gene transcripts associated with inflammation, cell death and survival, tissue and vascular repair, and nervous system function and development are most highly represented in the injury epicenter [25–29]. The only study to examine gene expression differences between AN and SD rats [25] employed microarray analysis—a more limited technique than RNA-seq—and found only 80 genes that differed between the two strains.
In the present study, we assessed (1) locomotor recovery and (2) gene expression in the injury epicenter, following SCI, in both SD and AN rats. We focused on the acute (1 week post-injury, when the T cell response is maximal) and chronic (8 weeks post-injury) phases of recovery. Based on previously findings after spinal cord transection, we hypothesized that AN rats would show sustained superior locomotor function after injury compared to SD rats. We subsequently used RNA-seq to identify gene expression differences between the two strains that could explain variation in locomotor function.
Methods
Spinal cord injury and postoperative care
All studies were approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC). Age-matched adult (16–18 week old) female SD (Hsd:SD, N = 15) and AN (Hsd:RH-rnu/rnu, N = 15) rats (Charles River, Wilmington, MA, USA) received moderate contusion injury using the Infinite Horizon impactor (Precision Systems, Lexington, KY, USA). Briefly, animals were anesthetized with isoflurane and underwent T8/T9 laminectomy followed by spinal cord impact at a force of 200 kdyn. Animals received buprenorphine (0.05 mg/kg SC q12 h) for 3 days after surgery to moderate acute pain, and amoxicillin (14 g/L in drinking water) for 7 days after surgery to prevent urinary tract infection as part of standard postoperative care. Urine was expressed manually twice daily for the first 3 days after surgery and daily thereafter until the return of independent urination. Animals were assigned to either the acute (1 week, N = 5 per strain) or chronic (8 weeks, N = 10 per strain) recovery phase groups.
Functional assessment
Locomotion was assessed by two observers using the Basso, Beattie, and Bresnahan (BBB) scale [30] on the second postoperative day and at the end of each week following surgery. Animals were on site for at least 1 week prior to SCI to acclimate to the environment and handling by the observers. Raters were trained by a researcher (A.P.) with extensive experience using the BBB scale. BBB scores were compared between strains via repeated-measures analysis of variance.
Tissue preparation
At 1 or 8 weeks after injury, animals were anesthetized with ketamine (100 mg/kg IP) and xylazine (10 mg/kg IP). For RNA-seq analysis, animals underwent transcardial perfusion with phosphate buffered saline (PBS), and a 1-mm spinal cord segment through the injury site was harvested and flash-frozen in liquid nitrogen. For histological analysis, animals underwent transcardial perfusion with 4 % paraformaldehyde. The spinal cord was removed and cryoprotected in 30 % sucrose.
Histology
Spinal cord segments from some animals sacrificed 1 week post-injury (acute recovery group) were frozen in OCT compound and sectioned in the parasagittal plane at a thickness of 10 μm. Representative sections for each spinal cord were selected from the mid-sagittal area and immunostained. The following antibodies were used: anti-CD68 (1:500, AbD Serotec, Raleigh, NC, USA) for macrophages, anti-CD16 (1:100, Abcam, Cambridge, United Kingdom) for M1 macrophages, anti-CD163 (1:50, AbD Serotec) for M2 macrophages, donkey anti-rabbit IgG (1:1000, ThermoFisher Scientific, Carlsbad, CA, USA), and donkey anti-mouse IgG (1:1000, Abcam). Negative controls were obtained by omission of the primary antibody. Immunofluorescent photomicrographs were taken from a 1-mm segment centered on the lesion epicenter.
Immunofluorescence images were analyzed using ImageJ (National Institutes of Health, Bethesda, MD, USA). Threshold values for excluding background fluorescence were determined as the 95th percentile of pixel intensity averaged across all negative control images for each immunostain. The Analyze Particle function was then used to determine the percent of tissue area stained. Percent area was compared between AN and SD groups via the two-tailed Student’s t test using an alpha value of 0.05.
RNA extraction and sequencing
RNA extraction, cDNA library creation, and RNA-seq were performed with the assistance of the University of Minnesota Biomedical Genomics Center staff. RNA was extracted from tissue samples using the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands). RNA quantity and quality were assessed with the Aligent 2100 Bioanalyzer (Aligent Technologies, Santa Clara, CA, USA). The polyadenylated coding mRNA in each extract (N = 3 for each strain–time point combination) was isolated and reverse transcribed using random primers. The resulting paired-end cDNA libraries were subsequently sequenced using the HiSeq 2000 (Illumina, San Diego, CA, USA). For each sample, at least 40 million paired-end reads of 51 base pairs were performed.
Read mapping and transcript identification
Analysis of RNA-seq data was carried out using the Galaxy software platform [31–33] hosted by the Minnesota Supercomputing Institute. Reads first underwent quality control assessment with the FastQC tool and adapter sequence contamination removal with the CutAdapt tool. Because all base positions had first-quartile Phred scores greater than 20, trimming with FastQ Trimmer was not needed. Reads were subsequently mapped to the Rattus norvegicus RN4 reference genome with TopHat (version 1.5.0) [34] using an empirically determined insertion size of 210 base pairs. The mapped reads were assembled into transcripts with Cufflinks (version 0.0.6) [35] using quartile normalization. Transcriptional datasets for each time point were pooled using CuffMerge, and differences between strains at each time point were identified with CuffDiff. Gene expression differences with a Q value (false discovery rate-adjusted P value) less than 0.05 were considered to be statistically significant.
Immune and neural marker genes
To measure the presence and activity of both immune and neural cells, we first identified a variety of genetic markers for different cell types belonging to the innate immune system, adaptive immune system, and CNS as follows: dendritic cell (CD83), macrophage (ITGAM, CD68, CD14), M1 macrophage (CD86, RBPJ), M2 macrophage (ARG1, CMAF), mast cell (FCER1A, ENPP3, KIT), granulocyte (CEACAM3, LY6C), NK cell (NCR3, KLRA1, KLRA2), T cell (CD3d, CD3e, CD3g), helper T cell (CD4), Th1 cell (IL12Rb1, CXCR3, CCR5), Th2 cell (IL1R1, IL2R2, IL2RL1, CXCR4, CCR3, CCR4, CCR7, CCR8, TNFRSF8), cytotoxic T cell (CD8a, CD8b, GZMB, TIA1), B cell (CD19, CD20), neuron (NEFH, RBFOX3), oligodendrocyte (APC, MBP, PLP), and astrocyte (GFAP). For each cell type, the marker with the greatest abundance (fragments per kilobase of exon per million fragments mapped, FPKM) summed over the four conditions (strain–time point combinations) was used as the final cell marker.
Broad patterns of differential gene expression
In order to broadly understand strain-dependent differences in biological processes in response to injury, we categorized a subgroup of genes using gene set enrichment analysis (GSEA). Specifically, genes differentially expressed between strains in the acute phase only were examined via GSEA. First, rat genome database (RGD) gene identifiers were converted to gene symbols using the DAVID Gene ID Conversion Tool (http://david.abcc.ncifcrf.gov). Gene symbols for differentially expressed genes were then entered into the GSEA utility (http://www.broadinstitute.org/gsea) and mapped to the Canonical Pathways gene set collection. The 20 most highly enriched gene sets were identified.
Investigation of specific pathophysiological processes
To investigate differences in specific processes (e.g. cell death) between strains, we identified appropriate gene ontology (GO) terms related to those processes (refer to “Results” section for a list of all GO terms used). Along with these GO terms, we entered genes differentially expressed in the acute and/or chronic phases into the AmiGO Slimmer tool (http://amigo1.geneontology.org). This allowed us to compare gene expression in processes of interest between strains at different time points.
Results
Locomotor recovery
A total of 15 AN and 15 SD rats underwent contusive SCI at a force of 200 kdyn. The BBB locomotor scores of AN and SD rats differed (P = 0.02) at 1 week following injury, at which point the mean BBB scores (±1 standard error) were 8.5 ± 0.5 and 5.5 ± 0.9, respectively (Fig. 1a). The BBB scores of the two strains did not differ at any other time point.
Fig. 1.
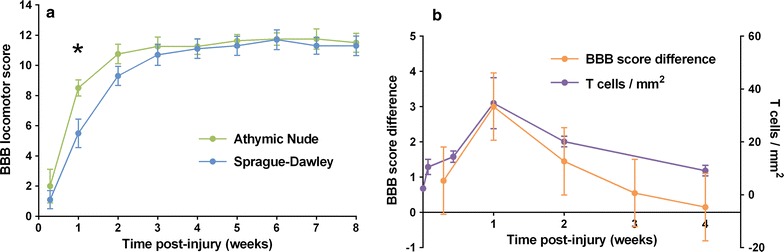
Variation in locomotor recovery between AN and SD rats. a Mean BBB locomotor score over time following SCI. Asterisk indicates P < 0.05. N = 8 for AN rats; N = 10 for SD rats. Error bars represent ±1 standard error. b Locomotor difference and T cell infiltration. Mean BBB score difference between AN and SD rats (left axis) and T cell density (T cells/mm2) in injury epicenter of SD rat (right axis). T cell data is used with permission [5]. Error bars represent ±1 standard error
Figure 1b shows the time course of the difference between BBB scores of AN and SD rats as well as the density of the T cell infiltrate in the SD rat injury epicenter (data used with permission) [5] over the first 4 weeks after injury. The locomotor advantage of AN rats over SD rats at 1 week coincides with the maximal T cell infiltrate, and both functional difference and T cell density decrease gradually thereafter.
RNA-seq experimental design
Based upon the locomotor difference between AN and SD rats in the acute (1 week post-injury) but not chronic (8 weeks post-injury) phase of recovery, we took a multi-step approach to analysis of RNA-seq data. First, to gain a broad understanding of the transcriptional basis of the acute locomotor difference, we compared the acute- and chronic-phase differential expression profiles to identify genes that were differentially expressed in the acute phase only. We then identified physiological pathways that were highly represented among these genes. Second, to investigate specific pathophysiological processes involved in SCI, we identified genes that were differentially expressed—in the acute and/or chronic phase—and associated with select GO terms.
Public data availability
RNA-seq data (raw and processed files) are available in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE62760.
Quality control
RNA samples sent for sequencing had 1.8–10 ng of RNA at a concentration of 92–100 ng/µL and an RNA integrity number (RIN) of 9.7–10. For each sample, 40.0–51.6 million reads 51 base pairs in length were sequenced. Both paired read sets for every sample had a per-base first-quartile Phred quality score greater than 30 for all bases, indicating a base measurement error less than 0.1 %. Expression ranges were highly consistent between samples (Fig. 2).
Fig. 2.
Expression ranges for individual tissue samples. Box-and-whisker plot of log(FPKM) for each animal (3-digit identification number). Whiskers indicate 1st to 99th percentile; values outside this range are not represented. Note that the expression ranges were highly consistent between samples
Differential gene expression
A total of 14,911 genes were identified by RNA-seq: 14,565 in the acute phase and 14,567 in the chronic phase. Read mapping statistics for each sample are summarized in Table 1. Statistical significance for differential gene expression was determined based upon the Q value (false discovery rate-adjusted P value). Magnitude of differential expression was quantified as fold change (FC), equal to AN FKPM divided by SD FKPM. Volcano plots of statistical significance (−log10Q) versus difference magnitude (log2FC) reveal that the magnitude of differential gene expression was larger in the acute phase (Fig. 3). The median absolute value of log2FC for differentially expressed genes was 1.27 in the acute phase, compared to 0.99 in the chronic phase (P < 0.001 for Mann–Whitney U test).
Table 1.
Read mapping from RNA-seq output
| Phase | Strain | Animal | Total reads | Mapped reads | % Mapped | Genes |
|---|---|---|---|---|---|---|
| Acute | AN | 714 | 51,634,511 | 50,136,688 | 97.1 | 13,574 |
| 715 | 44,269,036 | 43,001,258 | 97.1 | 13,471 | ||
| 717 | 44,842,419 | 43,542,173 | 97.1 | 13,559 | ||
| SD | 618 | 48,194,423 | 46,090,448 | 95.6 | 14,035 | |
| 619 | 39,885,927 | 38,511,046 | 96.6 | 13,610 | ||
| 620 | 45,447,598 | 43,769,395 | 96.3 | 13,745 | ||
| Chronic | AN | 707 | 44,446,497 | 42,956,572 | 96.6 | 13,763 |
| 712 | 45,858,389 | 44,358,919 | 96.7 | 13,653 | ||
| 713 | 43,186,929 | 41,819,751 | 96.8 | 13,632 | ||
| SD | 605 | 49,095,638 | 47,230,803 | 96.2 | 13,861 | |
| 606 | 45,671,290 | 43,873,662 | 96.1 | 13,849 | ||
| 608 | 45,194,438 | 43,242,894 | 95.7 | 13,926 |
Fig. 3.
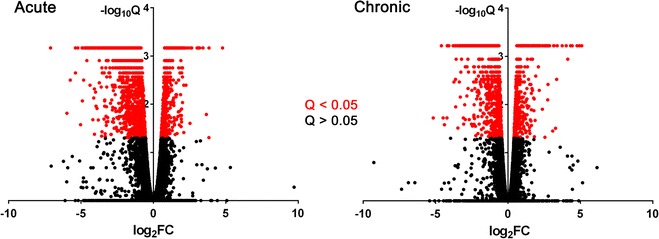
Volcano plots for differential gene expression in the acute and chronic phases of SCI. Colors represent Q < 0.05 (red) and Q > 0.05 (black) for differential expression testing. The overall magnitude of differential gene expression was greater in the acute phase, as evidenced by a larger median absolute value of log2FC (1.27 acute versus 0.99 chronic; P < 0.001) for differentially expressed genes. FC fold change (AN FKPM divided by SD FKPM). Q, false discovery rate-adjusted P value
Of the 14,911 genes identified by RNA-seq, 11,888 displayed no differential expression (Fig. 4a). Among differentially expressed genes, 999 differed in the acute phase only, 888 in the chronic phase only, and 1136 in both acute and chronic phases. Most genes that were differentially expressed in both acute and chronic phases were greater expressed in SD rats in both phases, indicated by log2FC values less than zero (Fig. 4b). Forty-two genes were greater expressed in SD rats in one phase and AN rats in the other. A complete list of the identified genes, expression data, and results of significance testing is provided in Additional file 1: Table S1 (as well as in the Gene Expression Omnibus; see above).
Fig. 4.
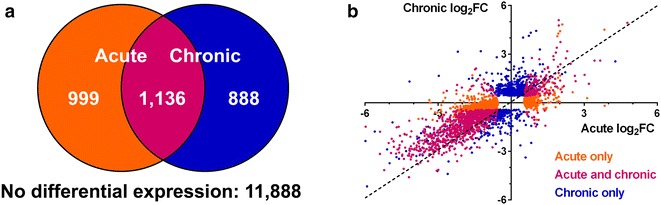
Differential gene expression in the acute and chronic phases of SCI. a Venn diagram depicting number of differentially expressed genes over time. b Plot of gene log2FC values in the acute (horizontal axis) and chronic (vertical axis) phases. Colors represent the phase in which genes are differentially expressed. The dashed line (y = x) has a slope of 1. FC fold change (AN FKPM divided by SD FKPM)
Immune and neural marker genes
The highest-abundance immune and neural cell marker genes were identified and compared between strains in the acute and chronic phases (Table 2). Genes corresponding to T cells (CD3g) and helper T cells (CD4) were enriched in SD rats (relative to AN rats) in the acute phase only. In AN rats, Th1 cell marker CXCR4 was enriched in AN rats in both the acute and chronic phases, while cytotoxic T cell marker CD8a was enriched in the acute phase only. Gene markers for NK cells (KLRA1) and mast cells (KIT) were enriched in AN rats in the chronic phase only. Expression of markers for dendritic cells (CD83), macrophages (all: CD68; M1: CD86; M2: CMAF), granulocytes (Ly6C), Th1 cells (CCR5), and B cells (CD19) did not vary by strain. The neurofilament gene NEFH was enriched in AN rats in the chronic phase; oligodendrocyte (PLP) and astrocyte (GFAP) marker genes were not differentially expressed.
Table 2.
Analysis of cell types via marker genes
| System | Cell type | Marker | Acute | Chronic | ||||
|---|---|---|---|---|---|---|---|---|
| Greater | log2FC | Q | Greater | log2FC | Q | |||
| Innate immune system | Dendritic cell | CD83 | 0.14 | 0.77 | −0.02 | 0.98 | ||
| Macrophage | CD68 | 0.66 | 0.12 | 0.47 | 0.07 | |||
| M1 macrophage | CD86 | 0.50 | 0.16 | −0.18 | 0.68 | |||
| M2 macrophage | CMAF | 0.46 | 0.12 | 0.32 | 0.18 | |||
| Mast cell | KIT | −0.05 | 0.95 | AN | 0.57 | 0.01 | ||
| Granulocyte | Ly6C | 0.19 | 0.91 | 0.26 | 0.87 | |||
| NK cell | KLRA1 | 0.63 | 0.15 | AN | 1.20 | <0.01 | ||
| Adaptive immune system | T cell | CD3g | SD | −2.37 | 0.02 | −0.88 | 0.17 | |
| Helper T cell | CD4 | SD | −1.20 | <0.01 | −0.25 | 0.36 | ||
| Th1 cell | CCR5 | −0.12 | 0.83 | −0.14 | 0.72 | |||
| Th2 cell | CXCR4 | AN | 0.65 | 0.02 | AN | 0.49 | 0.01 | |
| Cytotoxic T cell | CD8a | AN | 1.39 | <0.01 | −0.37 | 0.16 | ||
| B cell | CD19 | −1.29 | 1.00 | 0.74 | 1.00 | |||
| Central nervous system | Neuron | NEFH | −3.31 | 0.05 | SD | −1.90 | <0.01 | |
| Oligodendrocyte | PLP | −1.27 | 0.30 | −0.21 | 0.82 | |||
| Astrocyte | GFAP | −1.48 | 0.34 | 0.00 | 1.00 | |||
FC fold change (AN FKPM divided by SD FKPM); Q, false discovery rate-adjusted P value
Gene set enrichment analysis
The 20 most highly enriched gene sets were identified for genes differentially expressed in the acute phase only (Table 3). We then manually classified these gene sets into five themes: immune system (7 sets), neurons and axons (5 sets), GPCR signaling (3 sets), cell transport (3 sets), and other (2 sets). For ease of interpretation, percentage of genes with FC >1 in the acute period—meaning genes acutely enriched in AN rats—is reported below for all gene sets.
Table 3.
Gene set enrichment analysis (GSEA) of genes differentially expressed in the acute phase only
| Themea | Gene set | Genes | % Acutely enriched | −log(Q)b | |
|---|---|---|---|---|---|
| AN (FC >1) | SD (FC <1) | ||||
| Immune system | Cell adhesion molecules | 25 | 32 | 68 | Infinite |
| Cell surface interactions at the vascular wall | 17 | 71 | 29 | 10.4 | |
| Hematopoietic cell lineage | 14 | 64 | 36 | 7.6 | |
| Hemostasis | 45 | 67 | 33 | Infinite | |
| Immune system | 51 | 76 | 24 | 10.2 | |
| Leukocyte trans-endothelial migration | 17 | 59 | 41 | 8.7 | |
| Platelet activation, signaling, and aggregation | 21 | 62 | 38 | 8.1 | |
| Neurons and axons | Axon guidance (Kegg gene set) | 17 | 12 | 88 | 8.1 |
| Axon guidance (Reactome gene set) | 29 | 17 | 83 | 12.8 | |
| Neuroactive ligand-receptor interaction | 23 | 22 | 78 | 7.6 | |
| Neuronal system | 32 | 16 | 84 | Infinite | |
| Transmission across chemical synapses | 24 | 17 | 83 | 11.5 | |
| GPCR signaling | Class A1 rhodopsin-like receptors | 24 | 46 | 54 | 7.3 |
| GPCR ligand binding | 32 | 41 | 59 | 9.9 | |
| GPCR signaling | 45 | 42 | 58 | 7.6 | |
| Cell transport | Solute carrier-mediated transmembrane transport | 28 | 36 | 64 | 12.4 |
| Transmembrane transport of small molecules | 44 | 32 | 68 | Infinite | |
| Transport of inorganic cations/anions and amino acids/oligopeptides | 19 | 32 | 68 | 12.4 | |
| Other | Cell cycle | 17 | 94 | 6 | 8.2 |
| Developmental biology | 32 | 22 | 78 | 10.2 | |
FC fold change (AN FKPM divided by SD FKPM), Q false discovery rate-adjusted P value
aThe 20 most highly enriched gene sets have been manually grouped for conceptual purposes
bThe value of −log(Q) is given for the GSEA Q value for each gene set
Most genes belonging to immune system gene sets were greater expressed in AN rats (59–71 % with acute FC >1), with the exception of cell adhesion molecules (32 % with acute FC >1). Examination of genes belonging to the “immune system” gene set revealed that genes for cytokines IL-1a (acute FC = 2.24, Q = 0.01; chronic FC = 1.05, Q = 0.95) and IL-6 (acute FC = 3.86, Q = 0.01; chronic FC = 1.29, Q = 1.00) were among the most AN-enriched immune system-related genes in the acute phase. Genes belonging to neurons and axon gene sets were greater expressed in SD rats (12–22 % with acute FC >1). Genes belonging to GPCR signaling and cell transport gene sets were more frequently enriched in SD rats (41–46 % and 32–36 % with acute FC >1, respectively). The “other” theme consisted of the “cell cycle” and “developmental biology” gene sets. Cell cycle genes were nearly all enriched in AN rats (94 % with acute FC >1), while developmental biology genes were predominantly enriched in SD rats (22 % with acute FC >1).
Cell death-associated genes
We identified cell death-associated genes differentially expressed in the acute and/or chronic phases (Table 4). Genes associated with the cell death parent GO term were acutely enriched in SD rats (39 % with acute FC >1) and chronically enriched in AN rats (57 % with chronic FC >1). Programmed cell death (202 genes in acute phase and 179 genes in chronic phase) was more highly represented than necrotic cell death (7 genes in acute phase and 6 genes in chronic phase). Of note, however, necrosis-associated genes were enriched in AN rats in both the acute (86 % with acute FC >1) and chronic (83 % with chronic FC >1) phases. Genes associated with regulation of cell death were acutely enriched in SD rats (39 % with acute FC >1) and chronically enriched in AN rats (58 % with chronic FC >1). Neuron death-associated genes were enriched in SD rats in both acute (21 % with acute FC >1) and chronic (41 % with chronic FC >1) phases, though less so in the chronic phase. For cell and neuron death at a given time point, the strain with greater expression of death-associated genes also had greater expression of both positive and negative death regulation-associated genes.
Table 4.
Differential expression within gene ontologies of interest
| Identifier | Gene ontology | Acute | Chronic | ||||
|---|---|---|---|---|---|---|---|
| Genes | % Enriched | Genes | % Enriched | ||||
| AN (FC >1) | SD (FC <1) | AN (FC >1) | SD (FC <1) | ||||
| GO:0008219 | Cell death | 221 | 39 | 61 | 196 | 57 | 43 |
| Type of cell death | |||||||
| GO:0012501 | Programmed | 202 | 39 | 61 | 179 | 56 | 44 |
| GO:0070265 | Necrotic | 7 | 86 | 14 | 6 | 83 | 17 |
| GO:0010941 | Regulation of cell death | 181 | 39 | 61 | 151 | 58 | 42 |
| GO:0010942 | Positive regulation | 60 | 42 | 58 | 49 | 65 | 35 |
| GO:0060548 | Negative regulation | 100 | 38 | 62 | 86 | 53 | 47 |
| GO:0070997 | Neuron death | 47 | 21 | 79 | 37 | 41 | 59 |
| GO:1901216 | Positive regulation | 11 | 9 | 91 | 5 | 40 | 60 |
| GO:1901215 | Negative regulation | 28 | 21 | 79 | 26 | 42 | 58 |
| Voltage-gated potassium channel | |||||||
| GO:0005249 | Activity | 27 | 4 | 96 | 23 | 4 | 96 |
| GO:0008076 | Complex | 22 | 5 | 95 | 15 | 7 | 93 |
| GO:0045163 | Clustering | 1 | 0 | 100 | 0 | n/a | n/a |
FC fold change (AN FKPM divided by SD FKPM)
Voltage-gated potassium channel-related genes
We identified differentially expressed genes associated with voltage-gated potassium (Kv) channels (Table 4). Genes related to Kv channel activity were enriched in SD rats in both the acute (4 % with acute FC >1) and chronic (4 % with chronic FC >1) phases. Similarly, genes related to the Kv channel complex were enriched in SD rats in both the acute (5 % with acute FC >1) and chronic (7 % with chronic FC >1) phases. One gene (CNTN2) related to Kv channel clustering was differentially expressed. CNTN2 was differentially expressed in the acute phase (acute FC = 0.44, Q < 0.01) but not in the chronic phase (chronic FC = 0.98, Q = 0.97).
Histologic assessment of acute macrophage infiltrate
To corroborate our gene expression data, we stained spinal cord tissue obtained 1 week post-injury for macrophages (anti-CD68) as well as the M1 (anti-CD16) and M2 (anti-CD163) macrophage subtypes. Figure 5 depicts the results of fluorescence immunostaining. Percentage of tissue area stained did not vary by strain for any macrophage marker (P = 0.19 for CD68, P = 0.85 for CD16, and P = 0.71 for CD163).
Fig. 5.
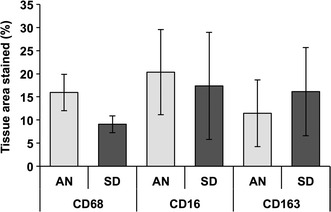
Histologic assessment of macrophage infiltrate at the acute time point (1 week post-injury). The percentage of tissue area positively stained for macrophages (CD68), M1 macrophages (CD16), or M2 macrophages (CD163) was compared between AN and SD rats (N = 4 rats per strain). No significant differences were observed (P = 0.19 for CD68, P = 0.85 for CD16, and P = 0.71 for CD163). Error bars represent ±1 standard error
Discussion
In this study, we found that T cell-deficient AN rats possess greater locomotor function than immunocompetent SD rats at 1 week post-SCI, but not at other time points. We examined differential gene expression between AN and SD rats using RNA-seq to clarify the physiological and cellular basis for this functional difference. A prior study of gene expression in AN and SD rats after SCI found only 80 genes whose expression differed by strain [25]. In contrast, we identified 3023 differentially expressed genes in the acute (1 week post-injury) and/or chronic (8 weeks post-injury) phases of SCI. The only published report of functional recovery after SCI in AN and SD rats found sustained locomotor advantage in AN rats following spinal cord transection [23]. We too observed functional superiority in AN rats after contusion SCI; however, this advantage was only seen at 1 week post-injury.
Effect of T cell deficiency on locomotor recovery
Athymic Nude rats have previously been found to achieve greater locomotor function than SD rats after spinal cord transection [23]. Whereas SD rats only recovered slight movement of hindlimb joints, AN rats on average regained extensive movement in one or more hindlimb joints. Based upon this finding, we initially hypothesized that AN rats would demonstrate superior locomotor function over the entire recovery period following moderate spinal cord contusion. In our study, AN rats achieved greater BBB locomotor scores (8.5 ± 0.5) than SD rats (5.5 ± 0.9) at 1 week post-injury, with weight-supported stepping observed in some AN rats but no SD rats. However, this functional advantage was transient. The BBB score difference followed a similar time course to the magnitude of the T cell infiltration in immunocompetent SD rats [5]. Also in line with difference in functional outcome, greater magnitude of differential gene expression was observed in the acute phase of recovery than in the chronic phase.
Discrepancy between our results and those of the aforementioned study are likely due to substantial differences in the mechanism and severity of injury. The authors of the prior study conceded that axonal regrowth through the transection site was implausible and attributed prolonged functional difference to sparing of local neuronal connections and central pattern generators [23]. In contusion injury, longitudinal white matter tracts are partially spared [25, 27] and undergo some long-distance regeneration in addition to local sprouting [36, 37]. Variation in the extent and function of surviving tissue may help to account for the difference in locomotor recovery between AN and SD rats.
Mechanistic basis of altered locomotor recovery
The pathophysiology of SCI is complex, and the processes of secondary injury, wound healing, and functional recovery are multifactorial [38]. Consequently, comprehensive analysis of these processes is difficult, despite the use of broad-spectrum techniques such as RNA-seq. We have therefore focused on specific, known pathophysiological aspects of SCI that may plausibly be related to T cell function.
The differential recovery course between SD and AN rats in this study has two possible explanations which are not mutually exclusive. One hypothesis is that while AN rats recover function more quickly after SCI, they are limited in their ultimate recovery in the same ways as SD rats. For example, while the absence of T cells may contribute to tissue sparing in the acute phase of SCI, that tissue may be gradually lost nonetheless. A second hypothesis is that AN rats are less vulnerable to temporary causes of paralysis after SCI, but functional differences disappear once those effects have subsided. These causes may include edema, metabolic dysfunction, and Kv channel dysregulation [38].
In our analysis, we compared components of the immune response between AN and SD rats. We then investigated strain-dependent differences in cell death, tissue repair, and Kv channel function. Observed trends in gene expression were consistent with involvement of all of these processes in differential recovery from SCI. We propose a model in which T cells contribute to early tissue damage, demyelination, and Kv channel dysregulation in SD rats following contusion SCI, but delayed tissue death limits the long-term recovery of AN rats.
Immune response to SCI in AN rats
The immune response differed substantially between AN and SD rats. Among genes differentially expressed in the acute phase only, immune system-related genes were most highly represented. As expected, T cell and helper T cell marker genes were greater expressed in SD rats in the acute phase, when the T cell response is maximal [5, 6]. However, other T cell subsets appeared to be more numerous in AN rats. Cytotoxic T cell marker CD8a was enriched in AN rats in the acute phase, while Th2 cell marker CXCR4 was enriched in AN rats in both acute and chronic phases. Shift from Th1 to Th2 has been associated with anti-inflammatory processes [39], so Th2 enrichment in AN rats may facilitate locomotion in the acute period.
The presence and functionality of T cells in AN rats remains an open topic of research. The absence of a functional thymus leads to production of fewer, less mature “T-like cells” [40]. Culture of T-like cells from AN rats yields cytotoxic T cells that recognize foreign antigens in vitro but not in vivo [41, 42]. T-like cells accumulate over time, reaching half of the normal T cell level by 8–12 months of age [43]. Rats in the present study, however, were injured at 4 months of age.
Elevated CD8a and CXCR4 expression in AN rats conceivably might not reflect an overabundance of cytotoxic T cells and Th2 cells. CD8a is also expressed on NK cells [44] and CNS-infiltrating macrophages [45]. However, while CD8a was acutely enriched in AN rats, NK cell marker KLRA1 was enriched in AN rats in the chronic phase only, and macrophage marker CD68 was not differentially expressed. CXCR4 is involved in CNS development [46], but development-related genes were predominantly enriched in SD rats. Thus, increased CD8a and CXCR4 expression is consistent with greater cytotoxic T cell and Th2 cell infiltration in AN rats. The basis of these differences is unclear but may relate to complex T cell abnormalities in AN rats.
Highly enriched expression of macrophage-secreted cytokines IL-1a and IL-6 in AN rats may reflect compensation for the deficient T cell response. NK cells, which are actually more numerous and effective in AN rats compared to normal littermates [47], appeared to be abundant in the chronic injury site of AN rats and may similarly compensate for T cell deficiency. Conversely, chronic enrichment of mast cells in AN rats may have aided recovery, as mast cells have been found to reduce inflammation following SCI [48]. The role of mast cells in CNS injury is controversial, however, as mast cells have been reported to exacerbate brain injury in ischemic stroke [49] and intracerebral hemorrhage [50].
Macrophages, dendritic cells, granulocytes, and B cells play important roles in SCI [2, 51], but their respective marker genes did not vary between AN and SD rats. Macrophage recruitment and activation is driven by helper T cells, and prior research has found reduced macrophage infiltrate in AN rats compared to SD rats at 11 weeks post-injury [23]. We therefore performed fluorescent immunostaining for macrophage (CD68) and macrophage subtype (M1: CD16; M2: CD163) markers in the acute injury phase (1 week post-SCI) [52, 53]. We found no strain-dependent differences in the presence of any of these markers, consistent with our gene expression findings. It should be noted that histological differentiation of M1 and M2 macrophage subtypes is difficult [54] and is often done using double-staining methods [55], which were not feasible in our case owing to technical limitations.
Cell death and tissue repair
Our findings suggest that while SD rats appear to suffer greater acute neuronal death, delayed cell death was more common in AN rats. In the acute phase, 61 % of cell death-associated genes and 79 % of neuron death-associated genes were enriched in SD rats; in the chronic phase, 57 % of cell death-associated genes were enriched in AN rats. Only 41 % of neuron death-associated genes were chronically enriched in AN rats; this discrepancy suggests that delayed cell death in AN rats may involve less loss of neurons than oligodendrocytes and other cells.
The predominant form of cell death appears to have been apoptosis rather than necrosis. Of note, necrosis-associated genes were enriched in AN rats, suggesting greater non-immune-mediated cell death. This is consistent with a past report of reduced apoptosis 24 h after traumatic brain injury in mice without functional T cells compared with immunocompetent mice [24].
Kv channel dysregulation
Demyelination of intact axons uncovers Kv channels located in the juxtaparanodal and internodal regions [56, 57]. Exposure of these channels leads to decreased axonal excitability and creates a conduction block [58, 59]. This problem is compounded by increased Kv expression [58] and redistribution of Kv channels along axons [57, 58].
Conceivably, if T cell activity causes increased cell death and demyelination, Kv channel dysregulation may contribute to the transient locomotor superiority of AN rats after contusion SCI. Consistent with this hypothesis, we found that nearly all differentially expressed Kv channel-related genes were enriched in SD rats in both the acute and chronic phases. Notably, the Kv channel clustering gene CNTN2 was greater expressed in SD rats in the acute phase only. CNTN2 (contactin-2, also known as TAG-1) is necessary to confine Kv channels to the juxtaparanodal region [60]. Expression of CNTN2 has been found to increase after SCI, and CNTN2 dysfunction is associated with worse motor recovery and decreased axonal regrowth following injury [61]. Thus, increased expression of CNTN2 and Kv channel-related genes may occur in response to greater early demyelination in SD rats. Delayed demyelination (e.g. due to oligodendrocyte death—see above) in AN rats may explain the absence of chronic differential expression of CNTN2.
Other contributing factors
Despite greater acute tissue damage, SD rats may have undergone greater axonal regrowth. Neurofilament expression was strongly elevated in SD rats in the acute (FC = −3.31, Q = 0.05) and chronic (FC = −1.90, Q < 0.01) phases. Axon guidance-related genes were highlighted in GSEA and were highly enriched in SD rats. Axon guidance refers to migration of the axon growth cone, and like increased neurofilament synthesis, enrichment of axon guidance-related genes suggests increased axonal repair in SD rats. Axonal tracing studies are necessary to test this hypothesis. Local sprouting and long-distance axonal regeneration are known to occur after contusion SCI [36, 37]. However, these changes are believed to produce little if any functional improvement [38] and are therefore unlikely to play a major role here.
Edema is a prominent feature of acute SCI. Vascular injury and blood–brain barrier disruption lead to vasogenic edema, which contributes to early spinal cord dysfunction [38]. While not measured in the present study, the volume of edematous spinal cord tissue—identified on MRI as the T2-hyperintense region—has been found to be identical in AN and SD rats throughout recovery from SCI [25]. Therefore, it seems unlikely that edema is responsible for strain-dependent variation in locomotor recovery.
As previously stated, we did not assess all pathophysiological processes involved in SCI. Reactive oxygen species production, glutamate-mediated excitotoxicity, and microvascular dysfunction were less well suited to gene expression-based analysis than cell death, tissue repair, and Kv channel dysregulation. While the potential relationship of these unexamined processes to T cell activity is unclear, they may nevertheless contribute to the effects of T cells on functional recovery.
Differences between the genomes of AN and SD rats warrant consideration as well. The AN rat is a homozygous rnu/rnu mutant derived from a colony of hooded rats [62, 63]. To our knowledge, there is no published comparison of the AN and SD genomes. Functional SCI outcomes are known to vary between common laboratory rat strains [64, 65], raising the possibility that non-T cell-related genes may account for the locomotor difference observed in the present study. However, the albino SD rat demonstrates higher BBB locomotor scores after SCI compared to the hooded Wistar and Long-Evans rats [64]. This suggests that if background genomic inequality is present, AN rats are more likely at a disadvantage—rather than an advantage—compared to SD rats.
Conclusions
Taken together, these findings implicate an altered immune response with relevant pro- and anti-inflammatory elements in the altered course of locomotor recovery of AN rats. Less damage by T cells—coupled with anti-inflammatory effects of Th2 cells (albeit of questionable functionality) and mast cells—may have led to temporary tissue preservation. The absence of superior long-term functional recovery may be due to compensatory NK cell overabundance and macrophage overproduction of inflammatory cytokines IL-1a and IL-6. We posit that T cells promote early tissue damage, demyelination, and Kv channel dysregulation after contusion SCI. However, compensatory features of the immune response (e.g. higher NK cell activity and increased IL-1a and IL-6 production) may cause delayed tissue death in T cell-deficient AN rats, limiting long-term recovery.
By nature, this work constitutes a broad-spectrum analysis of the role of T cell deficiency in SCI. While we have identified many promising correlations between gene expression and locomotor recovery, these relationships require extensive confirmatory studies. Several factors involved in the pathology of SCI were not investigated in this study and may contribute to the effect of T cells on functional recovery. The mechanistic basis for certain aspects of the AN immune response (particularly increased Th2 and cytotoxic T cell involvement) remains unclear, and may serve as a basis for subsequent experiments. Many studies—particularly systematic, quantitative immunohistochemistry—are thus required to corroborate and expand upon our findings.
Beyond helping to further elucidate the role of T cells in recovery from spinal cord trauma, our findings may inform future investigation of therapy for SCI. We have proposed that the immune consequences of T cell deficiency permit sparing of spinal cord tissue that is eventually lost in both immunodeficiency and immunocompetence. Combination therapy with T cell inhibitors and other neuroprotective treatment may allow continued survival of these vulnerable tissues and produce sustained functional recovery.
Authors’ contributions
Conceived and designed the experiments: DS CM JM JRD WCL AMP. Performed the experiments: DS CM JM CD RM. Analyzed the data: DS. Wrote the paper: DS WCL AMP. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by private philanthropy through the University of Minnesota Foundation (UMF). Additionally, research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- AN rat
athymic nude rat
- FC
fold change (AN FKPM divided by SD FKPM)
- FPKM
fragments per kilobase of exon per million fragments mapped
- GO
gene ontology
- GSEA
gene set enrichment analysis
- KV channel
voltage-gated potassium channel
- SCI
spinal cord injury
- SD rat
Sprague–Dawley rat
Additional file
RNA-seq data (raw and processed files) are available in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE62760.
10.1186/s12868-015-0212-0 Complete list of identified genes, expression values, and significance testing.
Contributor Information
David Satzer, Email: satz0002@umn.edu.
Catherine Miller, Email: mill5459@umn.edu.
Jacob Maxon, Email: maxon004@umn.edu.
Joseph Voth, Email: vothx042@umn.edu.
Christina DiBartolomeo, Email: dibar002@umn.edu.
Rebecca Mahoney, Email: mahon212@umn.edu.
James R. Dutton, Email: dutto015@umn.edu
Walter C. Low, Email: lowwalt@umn.edu
Ann M. Parr, Phone: 612-625-4102, Email: amparr@umn.edu
References
- 1.Chan CC. Inflammation: beneficial or detrimental after spinal cord injury? Recent Pat CNS Drug Discov. 2008;3(3):189–199. doi: 10.2174/157488908786242434. [DOI] [PubMed] [Google Scholar]
- 2.Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209(2):378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popovich PG, Stokes BT, Whitacre CC. Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res. 1996;45(4):349–363. doi: 10.1002/(SICI)1097-4547(19960815)45:4<349::AID-JNR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 4.Jones TB, Basso DM, Sodhi A, Pan JZ, Hart RP, MacCallum RC, Lee S, Whitacre CC, Popovich PG. Pathological CNS autoimmune disease triggered by traumatic spinal cord injury: implications for autoimmune vaccine therapy. J Neurosci. 2002;22(7):2690–2700. doi: 10.1523/JNEUROSCI.22-07-02690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague–Dawley and Lewis rats. J Comp Neurol. 1997;377(3):443–464. doi: 10.1002/(SICI)1096-9861(19970120)377:3<443::AID-CNE10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 6.Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol. 2003;462(2):223–240. doi: 10.1002/cne.10736. [DOI] [PubMed] [Google Scholar]
- 7.Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med (Berl) 1997;75(3):165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 8.Schroeter M, Jander S. T-cell cytokines in injury-induced neural damage and repair. Neuromol Med. 2005;7(3):183–195. doi: 10.1385/NMM:7:3:183. [DOI] [PubMed] [Google Scholar]
- 9.Ankeny DP, Lucin KM, Sanders VM, McGaughy VM, Popovich PG. Spinal cord injury triggers systemic autoimmunity: evidence for chronic B lymphocyte activation and lupus-like autoantibody synthesis. J Neurochem. 2006;99(4):1073–1087. doi: 10.1111/j.1471-4159.2006.04147.x. [DOI] [PubMed] [Google Scholar]
- 10.Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166(12):7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- 11.Deb C, Lafrance-Corey RG, Schmalstieg WF, Sauer BM, Wang H, German CL, Windebank AJ, Rodriguez M, Howe CL. CD8 + T cells cause disability and axon loss in a mouse model of multiple sclerosis. PLoS One. 2010;5(8):e12478. doi: 10.1371/journal.pone.0012478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones TB, Ankeny DP, Guan Z, McGaughy V, Fisher LC, Basso DM, Popovich PG. Passive or active immunization with myelin basic protein impairs neurological function and exacerbates neuropathology after spinal cord injury in rats. J Neurosci. 2004;24(15):3752–3761. doi: 10.1523/JNEUROSCI.0406-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones TB, Hart RP, Popovich PG. Molecular control of physiological and pathological T-cell recruitment after mouse spinal cord injury. J Neurosci. 2005;25(28):6576–6583. doi: 10.1523/JNEUROSCI.0305-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz M, Kipnis J. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med. 2001;7(6):252–258. doi: 10.1016/S1471-4914(01)01993-1. [DOI] [PubMed] [Google Scholar]
- 15.Ishii H, Jin X, Ueno M, Tanabe S, Kubo T, Serada S, Naka T, Yamashita T. Adoptive transfer of Th1-conditioned lymphocytes promotes axonal remodeling and functional recovery after spinal cord injury. Cell Death Dis. 2012;3:e363. doi: 10.1038/cddis.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishii H, Tanabe S, Ueno M, Kubo T, Kayama H, Serada S, Fujimoto M, Takeda K, Naka T, Yamashita T. IFN-gamma-dependent secretion of IL-10 from Th1 cells and microglia/macrophages contributes to functional recovery after spinal cord injury. Cell Death Dis. 2013;4:e710. doi: 10.1038/cddis.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ibarra A, Correa D, Willms K, Merchant MT, Guizar-Sahagun G, Grijalva I, Madrazo I. Effects of cyclosporin-A on immune response, tissue protection and motor function of rats subjected to spinal cord injury. Brain Res. 2003;979(1–2):165–178. doi: 10.1016/S0006-8993(03)02898-1. [DOI] [PubMed] [Google Scholar]
- 18.Lu HZ, Wang YX, Zhou JS, Wang FC, Hu JG. Cyclosporin A increases recovery after spinal cord injury but does not improve myelination by oligodendrocyte progenitor cell transplantation. BMC Neurosci. 2010;11:127. doi: 10.1186/1471-2202-11-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norimatsu Y, Ohmori T, Kimura A, Madoiwa S, Mimuro J, Seichi A, Yatomi Y, Hoshino Y, Sakata Y. FTY720 improves functional recovery after spinal cord injury by primarily nonimmunomodulatory mechanisms. Am J Pathol. 2012;180(4):1625–1635. doi: 10.1016/j.ajpath.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Bao F, Chen Y, Schneider KA, Weaver LC. An integrin inhibiting molecule decreases oxidative damage and improves neurological function after spinal cord injury. Exp Neurol. 2008;214(2):160–167. doi: 10.1016/j.expneurol.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Fleming JC, Bao F, Chen Y, Hamilton EF, Relton JK, Weaver LC. Alpha4beta1 integrin blockade after spinal cord injury decreases damage and improves neurological function. Exp Neurol. 2008;214(2):147–159. doi: 10.1016/j.expneurol.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 22.Ibarra A, Hernandez E, Lomeli J, Pineda D, Buenrostro M, Martinon S, Garcia E, Flores N, Guizar-Sahagun G, Correa D, Madrazo I. Cyclosporin-A enhances non-functional axonal growing after complete spinal cord transection. Brain Res. 2007;1149:200–209. doi: 10.1016/j.brainres.2007.02.056. [DOI] [PubMed] [Google Scholar]
- 23.Potas JR, Zheng Y, Moussa C, Venn M, Gorrie CA, Deng C, Waite PM. Augmented locomotor recovery after spinal cord injury in the athymic nude rat. J Neurotrauma. 2006;23(5):660–673. doi: 10.1089/neu.2006.23.660. [DOI] [PubMed] [Google Scholar]
- 24.Fee D, Crumbaugh A, Jacques T, Herdrich B, Sewell D, Auerbach D, Piaskowski S, Hart MN, Sandor M, Fabry Z. Activated/effector CD4+ T cells exacerbate acute damage in the central nervous system following traumatic injury. J Neuroimmunol. 2003;136(1–2):54–66. doi: 10.1016/S0165-5728(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 25.Velardo MJ, Burger C, Williams PR, Baker HV, Lopez MC, Mareci TH, White TE, Muzyczka N, Reier PJ. Patterns of gene expression reveal a temporally orchestrated wound healing response in the injured spinal cord. J Neurosci. 2004;24(39):8562–8576. doi: 10.1523/JNEUROSCI.3316-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chamankhah M, Eftekharpour E, Karimi-Abdolrezaee S, Boutros PC, San-Marina S, Fehlings MG. Genome-wide gene expression profiling of stress response in a spinal cord clip compression injury model. BMC Genom. 2013;14:583. doi: 10.1186/1471-2164-14-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen K, Deng S, Lu H, Zheng Y, Yang G, Kim D, Cao Q, Wu JQ. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS One. 2013;8(8):e72567. doi: 10.1371/journal.pone.0072567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen XJ, Mou XQ, Zou YG, Peng ZY, Yang JX. Screening of key genes associated with contused rat spinal cord with DNA microarray. Eur Rev Med Pharmacol Sci. 2013;17(21):2949–2955. [PubMed] [Google Scholar]
- 29.Hui SP, Sengupta D, Lee SG, Sen T, Kundu S, Mathavan S, Ghosh S. Genome Wide Expression Profiling during Spinal Cord Regeneration Identifies Comprehensive Cellular Responses in Zebrafish. PLoS One. 2014;9(1):e84212. doi: 10.1371/journal.pone.0084212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12(1):1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- 31.Goecks J, Nekrutenko A, Taylor J, Galaxy Team. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86-2010-11-8-r86. Epub 2010 Aug 25. [DOI] [PMC free article] [PubMed]
- 32.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010; Chapter 19:Unit 19.10.1-21. [DOI] [PMC free article] [PubMed]
- 33.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beattie MS, Bresnahan JC, Komon J, Tovar CA, Van Meter M, Anderson DK, Faden AI, Hsu CY, Noble LJ, Salzman S, Young W. Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol. 1997;148(2):453–463. doi: 10.1006/exnr.1997.6695. [DOI] [PubMed] [Google Scholar]
- 37.Hill CE, Beattie MS, Bresnahan JC. Degeneration and sprouting of identified descending supraspinal axons after contusive spinal cord injury in the rat. Exp Neurol. 2001;171(1):153–169. doi: 10.1006/exnr.2001.7734. [DOI] [PubMed] [Google Scholar]
- 38.Rowland JW, Hawryluk GW, Kwon B, Fehlings MG. Current status of acute spinal cord injury pathophysiology and emerging therapies: promise on the horizon. Neurosurg Focus. 2008;25(5):E2. doi: 10.3171/FOC.2008.25.11.E2. [DOI] [PubMed] [Google Scholar]
- 39.Romagnani S. T-cell subsets (Th1 versus Th2) Ann Allergy Asthma Immunol. 2000;85(1):9–18. doi: 10.1016/S1081-1206(10)62426-X. [DOI] [PubMed] [Google Scholar]
- 40.Rolstad B. The athymic nude rat: an animal experimental model to reveal novel aspects of innate immune responses? Immunol Rev. 2001;184:136–144. doi: 10.1034/j.1600-065x.2001.1840113.x. [DOI] [PubMed] [Google Scholar]
- 41.Whitby EH, Sparshott SM, Bell EB. Allograft rejection in athymic nude rats by transferred T-cell subsets. I. The response of naive CD4+ and CD8+ thoracic duct lymphocytes to complete allogeneic incompatibilities. Immunology. 1990;69(1):78–84. [PMC free article] [PubMed] [Google Scholar]
- 42.Sarawar SR, Yang CP, Bell EB. T-cell receptor-bearing cells from athymic nude rats respond to alloantigen in vitro but are defective in vivo. Immunology. 1991;73(3):334–341. [PMC free article] [PubMed] [Google Scholar]
- 43.Helgeland L, Brandtzaeg P, Rolstad B, Vaage JT. Sequential development of intraepithelial gamma delta and alpha beta T lymphocytes expressing CD8 alpha beta in neonatal rat intestine: requirement for the thymus. Immunology. 1997;92(4):447–456. doi: 10.1046/j.1365-2567.1997.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torres-Nagel N, Kraus E, Brown MH, Tiefenthaler G, Mitnacht R, Williams AF, Hunig T. Differential thymus dependence of rat CD8 isoform expression. Eur J Immunol. 1992;22(11):2841–2848. doi: 10.1002/eji.1830221113. [DOI] [PubMed] [Google Scholar]
- 45.Popovich PG, van Rooijen N, Hickey WF, Preidis G, McGaughy V. Hematogenous macrophages express CD8 and distribute to regions of lesion cavitation after spinal cord injury. Exp Neurol. 2003;182(2):275–287. doi: 10.1016/S0014-4886(03)00120-1. [DOI] [PubMed] [Google Scholar]
- 46.Klein RS, Rubin JB. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol. 2004;25(6):306–314. doi: 10.1016/j.it.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 47.Lovik G, Vaage JT, Naper C, Benestad HB, Rolstad B. Recruitment of alloreactive natural killer cells to the rat peritoneum by a transfected cell line secreting rat recombinant interleukin-2. J Immunol Methods. 1995;179(1):59–69. doi: 10.1016/0022-1759(94)00270-7. [DOI] [PubMed] [Google Scholar]
- 48.Nelissen S, Vangansewinkel T, Geurts N, Geboes L, Lemmens E, Vidal PM, Lemmens S, Willems L, Boato F, Dooley D, Pehl D, Pejler G, Maurer M, Metz M, Hendrix S. Mast cells protect from post-traumatic spinal cord damage in mice by degrading inflammation-associated cytokines via mouse mast cell protease 4. Neurobiol Dis. 2014;62:260–272. doi: 10.1016/j.nbd.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 49.Jin Y, Silverman AJ, Vannucci SJ. Mast cells are early responders after hypoxia-ischemia in immature rat brain. Stroke. 2009;40(9):3107–3112. doi: 10.1161/STROKEAHA.109.549691. [DOI] [PubMed] [Google Scholar]
- 50.Strbian D, Tatlisumak T, Ramadan UA, Lindsberg PJ. Mast cell blocking reduces brain edema and hematoma volume and improves outcome after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27(4):795–802. doi: 10.1038/sj.jcbfm.9600387. [DOI] [PubMed] [Google Scholar]
- 51.Ankeny DP, Popovich PG. Mechanisms and implications of adaptive immune responses after traumatic spinal cord injury. Neuroscience. 2009;158(3):1112–1121. doi: 10.1016/j.neuroscience.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moestrup SK, Moller HJ. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med. 2004;36(5):347–354. doi: 10.1080/07853890410033171. [DOI] [PubMed] [Google Scholar]
- 53.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29(43):13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ambarus CA, Krausz S, van Eijk M, Hamann J, Radstake TR, Reedquist KA, Tak PP, Baeten DL. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J Immunol Methods. 2012;375(1–2):196–206. doi: 10.1016/j.jim.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 55.Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS One. 2013;8(11):e80908. doi: 10.1371/journal.pone.0080908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sherratt RM, Bostock H, Sears TA. Effects of 4-aminopyridine on normal and demyelinated mammalian nerve fibres. Nature. 1980;283(5747):570–572. doi: 10.1038/283570a0. [DOI] [PubMed] [Google Scholar]
- 57.Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, Shrager P. Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci. 1998;18(1):36–47. doi: 10.1523/JNEUROSCI.18-01-00036.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nashmi R, Fehlings MG. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. 2001;104(1):235–251. doi: 10.1016/S0306-4522(01)00009-4. [DOI] [PubMed] [Google Scholar]
- 59.Nashmi R, Fehlings MG. Mechanisms of axonal dysfunction after spinal cord injury: with an emphasis on the role of voltage-gated potassium channels. Brain Res Brain Res Rev. 2001;38(1–2):165–191. doi: 10.1016/S0165-0173(01)00134-5. [DOI] [PubMed] [Google Scholar]
- 60.Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162(6):1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin JF, Pan HC, Ma LP, Shen YQ, Schachner M. The cell neural adhesion molecule contactin-2 (TAG-1) is beneficial for functional recovery after spinal cord injury in adult zebrafish. PLoS One. 2012;7(12):e52376. doi: 10.1371/journal.pone.0052376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Festing MF, May D, Connors TA, Lovell D, Sparrow S. An athymic nude mutation in the rat. Nature. 1978;274(5669):365–366. doi: 10.1038/274365a0. [DOI] [PubMed] [Google Scholar]
- 63.Cash JM, Remmers EF, Goldmuntz EA, Crofford LJ, Zha H, Hansen CT, Wilder RL. Genetic mapping of the athymic nude (RNU) locus in the rat to a region on chromosome 10. Mamm Genome. 1993;4(1):37–42. doi: 10.1007/BF00364661. [DOI] [PubMed] [Google Scholar]
- 64.Mills CD, Hains BC, Johnson KM, Hulsebosch CE. Strain and model differences in behavioral outcomes after spinal cord injury in rat. J Neurotrauma. 2001;18(8):743–756. doi: 10.1089/089771501316919111. [DOI] [PubMed] [Google Scholar]
- 65.Webb AA, Gowribai K, Muir GD. Fischer (F-344) rats have different morphology, sensorimotor and locomotor abilities compared to Lewis, Long-Evans, Sprague–Dawley and Wistar rats. Behav Brain Res. 2003;144(1–2):143–156. doi: 10.1016/S0166-4328(03)00076-7. [DOI] [PubMed] [Google Scholar]