

Abstract
Background
Thrombocytopenia-absent radius syndrome (TAR; MIM 274000) is a rare autosomal recessive disorder combining specific skeletal abnormalities with a reduced platelet count. TAR syndrome has been associated with the compound inheritance of an interstitial microdeletion in 1q21.1 and a low frequency noncoding RBM8A SNP.
Results
Here, we report on a patient with scapulo-humeral hypoplasia, bilateral radio-ulnar agenesis with intact thumbs, bilateral proximal positioning of the first metacarpal, bilateral fifth finger clinodactyly, bilateral radial deviation of the hands, and thrombocytopenia. Molecular studies showed compound heterozygosity for the 1q21.1 microdeletion and the RBM8A rs139428292 variant in hemizygous state, inherited from the father and the mother, respectively. A second aborted fetus presented TAR features and 1q21.1 microdeletion.
Discussion
The complex inheritance pattern resulted in reduced expression of Y14, the protein encoded by RBM8A, and a component of the core exon-junction complex (EJC) in platelets. Further studies are needed to explain how Y14 insufficiency and subsequent defects of the EJC could cause the skeletal, haematological and additional features of TAR syndrome. In this study, we discuss other factors that could influence the overall phenotype of patients affected by TAR syndrome.
Conclusion
In this study, we discuss other factors that could influence the overall phenotype of patients affected by TAR syndrome.
Keywords: 1q21.1 microdeletion, TAR syndrome, Array-CGH, RBM8A SNPs
Background
Since the introduction of array-CGH analysis, the reported frequency of genomic imbalances associated with specific phenotypes has dramatically increased. Copy number variations (CNV) with incomplete penetrance and variable expressivity have been described in various disorders [1–5]. The association between genomic imbalances and pathological traits can be variable, and additional genetic variations could contribute to the phenotype.
In addition, several other possibilities as epigenetic phenomena, expression or regulatory variation among genes in the vicinity of the unbalanced region, the unmasking of recessive alleles and the possibility of a “two-hit” model, as proposed by Girirajan et al. [6], may account for the phenotypic variability of some genomic diseases.
Human chromosome 1 is rich in segmental duplications, particularly within the pericentromeric region [7–10]. This fact may result in the susceptibility of this region to both pathological and non-pathological CNVs that might have an evolutionary significance.
TAR syndrome (Thrombocytopenia -Absent-Radius) syndrome (MIM 274000) is characterized by thrombocytopenia that may be episodic, congenital skeletal deformities including bilateral absence of radius, shortening and deformity of the ulnae, and occasionally absence of all the long bones in the arm. The fingers and thumbs are always present, while other skeletal anomalies are frequent [11].
A chromosome 1q21.1 microdeletion was identified in 30 patients affected by TAR syndrome [12]. This microdeletion is mediated by Low Copy Repeats (LCRs) that can be at the basis of recurrent DNA rearrangements such as deletions, duplications and inversions through chromosome or chromatid misalignment followed by non-allelic homologous recombination (NAHR) [13–15].
TAR syndrome has a complex pattern of inheritance associated with a minimal common interstitial microdeletion of 200 Kb on chromosome 1q21.1. In several cases, it is inherited from an unaffected parent, while in others it is originated de novo and the presence of a 1q21.1 microdeletion is necessary but not sufficient to cause the phenotype.
Recently, it has been shown that compound inheritance of a rare null allele and one of the two low-frequency noncoding SNPs (rs139428292 or rs201779890) in RBM8A are crucial for TAR syndrome [16].
Here, we describe the clinical, cytogenetic and molecular features of a 4-month-old boy with TAR syndrome due to co-segregation of 1q21.1 microdeletion and rs139428292. An aborted fetus in the same family presented the same phenotypic features and 1q21.1 microdeletion. We discuss here whether other factors could influence the overall phenotype of TAR syndrome.
Case report
The family tree is depicted in Fig. 1.
Fig. 1.
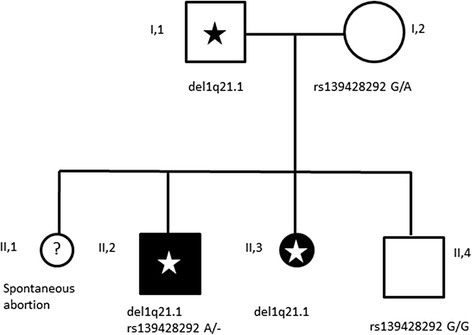
Genealogical tree of the family with TAR syndrome
Patient 1
The child (II-2) (Fig. 2a) is the first male child of apparently healthy nonconsanguineous parents. The mother and the father were 33 and 45 years old respectively at the time of his birth. Fetal movements were poor. Routine ultrasound examination was normal until 23 weeks of gestation when bilateral radial agenesis was demonstrated. The child was born post-term at 43 weeks of gestation by normal vaginal delivery. Birth weight was 2810 g (10th-25th centile). He was admitted to our institute at 4 months of age. Physical examination showed good nutritional status, forehead and right cheek telangiectasia, scapulo-humeral hypoplasia, bilateral radio-ulnar agenesis with intact thumbs, bilateral proximal positioning of the first metacarpal, bilateral fifth finger clinodactyly and bilateral radial deviation of the hands. X-ray confirmed all these skeletal findings. The child also presented thrombocytopenia (40.000/mmc), PTT (43.7 s). The phenotypic features were characteristic of TAR syndrome (MIM 274000).
Fig. 2.
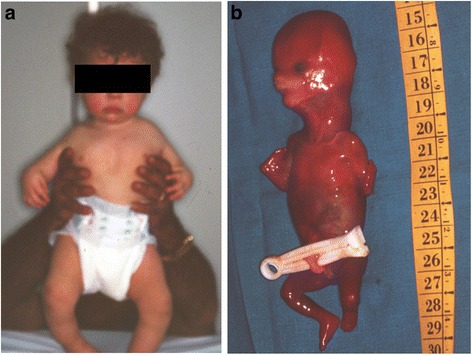
a The proband (II-2); bilateral absence of radius with thumb conservation and bilateral genu varum. b The fetus (II-3); bilateral radial aplasia
Patient 2 (Fetus)
During the third pregnancy (II-3) (Fig. 1b), ultrasound examination was performed at 11 weeks of gestation, suggesting the presence of upper limb anomalies. This finding was confirmed at 15th week of gestation. The couple opted for termination of pregnancy. The chromosomal analysis of amniotic cells (400 bands) excluded any visible abnormality. Post-mortem examination of the fetus demonstrated bilateral radial agenesis (Fig. 2b). The mother had a previous pregnancy, which ended in spontaneous abortion at the 2nd month of gestation (II-1). There was no exposure to alcohol, smoking or infections during pregnancy. A subsequent pregnancy resulted in the delivery of a healthy child (II-4).
Results
All patients and their parents showed a normal karyotype. Array-CGH analysis was performed on the available members of the family. An identical 1q21.1 microdeletion (~539 Kb) [arr 1q21.1(145,291,711-145,831,389)x1] was identified in the child with phenotypic features of TAR syndrome (II, 2) as showed in Fig. 3a. The microdeletion was confirmed by FISH (Fig. 3b). The same microdeletion was also present in his apparently normal father (I, 1). An elective abortion was performed because of ultrasound findings of upper limb anomalies, strongly suggestive of TAR syndrome (II, 3). FISH analysis by BAC RP11-105E14 (chr1:145,474,158-145,636,051) on archived specimens confirmed the presence of the “TAR microdeletion” (Fig. 3c). In the same family, one spontaneous abortion (II, 1) was reported but unfortunately not investigated, as biological specimens were not available. The deleted region contains 12 MIM genes: NBPF20 (MIM 614007; neuroblastoma breakpoint family, member 20), NBPF10 (MIM 614000; neuroblastoma breakpoint family, member 10), NBPF9 (MIM 613999; neuroblastoma breakpoint family, member 9), HFE2 (MIM 602390; hemochromatosis type 2A), TXNIP (MIM 606599; thioredoxin-interacting protein), RBM8A (MIM 605313; RNA-binding motif protein 8A), GNRHR2 (MIM 612875; gonadotropin-releasing hormone receptor 2), PEX11B (MIM 603867; peroxisome biogenesis factor 11B), ITGA10 (MIM 604042; integrin, alpha-10), PIAS3 (MIM 605987; protein inhibitor of activated STAT3), CD160 (MIM 604463; CD160 antigen), PDZK1 (MIM 603831; PDZ domain-containing 1). It is interesting to note the presence of the LIX1L (Gene ID: 128077; Lix1 homolog (chicken) like) gene (Fig. 3d).
Fig. 3.
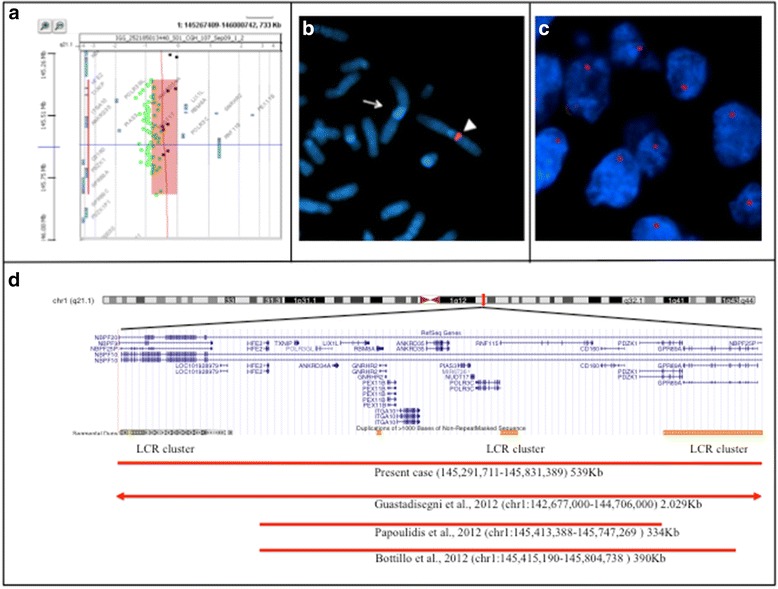
Results of array-CGH and FISH analyses. a Array-CGH analysis shows a ~539 Kb microdeletion at 1q21.1 band inherited from the father. b FISH confirmation of a hemizygous interstitial 1q21.1 deletion using a BAC probe RP11-105E14 (chr1:145,474,158-145,636,051) (red). c Interphase FISH with RP11-105E14 (chr1:145,474,158-145,636,051) (red) on deparaffinized fetal tissue from patient II, 3. Nuclei show a unique red signal indicating the presence of the deletion. d Overview of the 1q21.1 region and its genes and LCRs contents, according to the UCSC Genome Browser (GRCh37/hg19 assembly). The bars indicate the deleted region (red) in our patient and the deleted regions in patients reported by Guastadisegni et al. [28], Papoulidis et al. [30] and Bottillo et al. [29].
The family were analysed for the rs139428292 (G > A) and the rs201779890 (G > C) SNPs of RBM8A gene by direct sequencing. The analysis of rs139428292 showed that the patient harboured the minor (A) allele, which was inherited from his healthy mother. The father and the healthy brother were both homozygous for the major (G) allele. All family members carried the major (G) allele of rs201779890 in a homozygous state (Fig. 4).
Fig. 4.
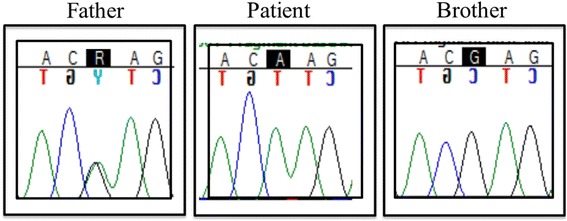
Results of Sanger sequencing of the rs139428292 variant (G > A) obtained from the patient and his unaffected mother and his unaffected brother
Discussion
We describe an identical 1q21.1 microdeletion in affected and non-affected members of a family with TAR syndrome. The same rearrangement was firstly described in 30 patients affected by Thrombocytopenia-Absent-Radius (TAR) syndrome (MIM 274000), a rare malformation syndrome characterized by hypo-megakaryocytic thrombocytopenia and bilateral absence of the radius in the presence of both thumbs [12]. The microdeletion was inherited from either the unaffected mother or the unaffected father in 75 % of cases. Recently, it has been proposed to consider this syndrome as a complex trait disease requiring at least two genetic changes: the rare microdeletion and another, relatively frequent, genetic variation that acts as a modifier of the other one. The family described in this study was referred to our centre for TAR syndrome and was investigated by array-CGH. A 1q21.1 microdeletion of ~539 Kb overlapping the TAR critical region, inherited from the phenotypically normal father, was identified in the two affected cases (II, 2 and II, 3). The identification of the interstitial microdeletion in affected offspring and the paternal inheritance are in agreement with the findings of Klopocki and collaborators [12]. The presence of 1q21.1 microdeletion is necessary but not sufficient to cause the phenotype, since, in the majority of cases, TAR traits can develop only in the presence of a second modifier as the low-frequency regulatory SNP reported by Albers et al. [16].
In fact, a study identified two rare single nucleotide polymorphisms (SNPs) in the regulatory region of the RBM8A gene that are involved in TAR syndrome through the reduction of the expression of the RBM8A-encoded Y14 protein [16]. The first allele (rs139428292 G > A), which is located in the 5′ untranslated region (UTR) of the gene, was demonstrated to have a minor allele frequency (MAF) of 3.05 %, and the second allele (rs201779890 G > C), located in the first intron of the gene, exhibited a MAF of 0.42 %, in 7504 healthy individuals from Cambridge BioResource (Cambridge, UK) [16, 17]. Our patient had inherited a low-frequency 5′UTR SNP (rs139428292 G > A) from his mother and the 1q21.1 microdeletion from his father.
A number of patients with TAR syndrome were reported, but in almost all these cases the precise coordinates of the deleted regions were not available and in others no mutation analysis of RBM8A SNP had been performed [18–27].
At our knowledge, in only three TAR cases with 1q21.1 deletion, analysed by array-CGH, the precise coordinates of the deletion has been reported [28–30] (Fig. 3d). Two are prenatal cases and one postnatal. All have the classical TAR features (thrombocytopenia, upper limbs and hands anomalies); two have inherited the low-frequency 5′ UTR SNP (rs139428292 G > A) and the minimal overlapping 1q21.1 deletion region ranging from 145,415,190 to 145,747,269 [29, 30].
Particularly, the child affected by TAR syndrome associated with Langerhans cell histiocytosis described by Guastadisegni et al. [28] showed a larger deletion (2.029 Kb) and a significant downregulation of the commonly deleted genes. The mainly implicated gene in the syndrome is RBM8A, a gene encoding the exon-junction complex subunit member Y14. Y14 is a small protein with an RNA-binding domain and one of the four components of EJC, which is involved in basic cellular functions such as nuclear export and subcellular localization of specific transcripts, translational enhancement, nonsense-mediated RNA decay and splicing [31]. It has a crucial role during embryonic developmental [32]. The level of Y14 was found to be significantly lower in the platelets of TAR patients. It is noteworthy, that Y14 may regulate the expression of genes involved in the proliferation of hematopoietic cells. However, it is not clear how a deficiency in Y14 exerts its effects at a cellular level and in particular how it affects the production of megakaryocytes and platelets. A possible explanation for this observation could be that deficiency in EJC could have an influence on the defective cell signalling in megakaryocyte. Similarly, no relation has been found between RBM8A deletion and TAR skeletal anomalies. To provide an explanation for the skeletal abnormalities observed in TAR syndrome, Albers et al., [24] assumed that, in addition to a tissue-dependent effect, it is possible that the regulatory SNPs had developmental stage–dependent consequences keeping as an example the Mecom gene encoding Evi1 that is expressed in a transient manner in emerging limb buds in mouse [33].
Albers et al., [24] explained that TAR phenotype could be also influenced from other factors such as environmental factors altering gene expression, incomplete penetrance or additional modifier alleles. We speculate that other genes in the 1q21.1 region other than RBM8A could influence the phenotype of TAR syndrome. It is interesting to note that, among the several genes with a known function located within the region, the PIAS3 gene could be indicated as the most conspicuous candidate for thrombocytopenia and the Lix1L gene, known be transiently expressed during chick hind-limb development, could be proposed as the candidate gene for limb abnormalities [34–36].
Conclusions
In conclusion, we reported on a new familial case of TAR syndrome in a child and in a fetus carrying a 1q21.1 microdeletion and the low-frequency 5′UTR SNP (rs139428292 G > A). We also focused on two genes (PIAS3 and Lix1L), contained in the deleted region, which could play a role in determining some important aspects of the phenotype of this syndrome. Obviously, further in-depth studies are needed to clarify the possible role of these genes.
Methods
Cytogenetics, fluorescent in situ hybridization and array-CGH analyses
Karyotypes were performed on peripheral blood of patients and their parents. Fluorescent in situ hybridization (FISH) analysis was performed following the manufacturer’s instructions (Vysis, Abbott Molecular, Illinois, U.S.A.). BAC clone was selected from the human library RPCI-11 according to the UCSC Human Genome Assembly (GRCh37/hg19). Array-CGH using the Human CGH Kit 244 K (Agilent Technologies, Palo Alto, CA, U.S.A.) covering the whole genome with a 8.9 Kb overall median probe spacing was performed following the manufacturer’s protocol.
FISH analysis on paraffin-embedded fetal tissues
To prepare paraffin-embedded tissue sections fixed on positively charged slides we cut 4–5 mμ thick paraffin sections using a microtome. Floating sections were mounted on positively charged slides. Slides were treated by Paraffin Pretreatment Kit (Abbott Molecular Inc., IL, USA) to deparaffinise specimens. Then, the slides were treated with protease and hybridization with BAC probes was performed according to the appropriate Vysis protocol.
Genotyping of 5′ UTR and intronic SNP of the RBM8A gene
The genotypes of the rs139428292 and the rs201779890 SNPs were obtained. Genomic DNA was isolated from peripheral blood leukocytes using a standard protocol. The genomic regions encompassing the two SNPs were PCR amplified, purified and then sequenced on both strands using the BigDye dideoxy-terminator chemistry on an ABI 3100 DNA sequencer (Applied Biosystems, Foster City, CA). Primers used for both amplification and sequencing were the following: rs139428292 (Fw: CCTTTCCCCTCTGCGACA; Rv: CCCAGCCTCGTGAAGATCTA) and rs201779890 (Fw: TAGATCTTCACGAGGCTGGG; Rv: GGGGCGGAATCTCTAATCCA).
Ethics Statement
The current study was performed using peripheral blood of the members of the family treated at the Istituto Giannina Gaslini, Genova, Italy. The parents of the patient gave written informed consent allowing molecular and genetic studies. We didn’t request approval by Review Board of our institution, because our study request only classical and molecular cytogenetic analyses. For cytogenetics analyses are sufficient only written informed consent of the parents (DM 21 dicembre 2007). The informed consents of the parents were previously authorized by the Review Board of our institution. We didn’t conduct research outside our country of residence. We didn’t approach the local authorities before beginning work on this study. The full name of the ethics committee of our institution is Comitato di Etica per la Ricerca Scientifica Biomedica, per la Buona Pratica Clinica e per la Sperimentazione dei Farmaci.
Consent
Written informed consent was obtained from the patient's parents for the publication of this report and any accompanying images.
Acknowledgments
We thank the patient’s parents for their kind participation and support. We thanks Marco Bertorello and Corrado Torello for technical supports. This work was supported by “Cinque per mille dell’IRPEF- Finanziamento della ricerca sanitaria” and “Finanziamento Ricerca Corrente, Ministero Salute (contributo per la ricerca intramurale).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. All authors have been involved in drafting the manuscript and revising it critically for important intellectual content. All authors read and approved the final manuscript.
References
- 1.Kirov G. A The role of copy number variation in schizophrenia. Expert Rev Neurother. 2010;10:25–32. doi: 10.1586/ern.09.133. [DOI] [PubMed] [Google Scholar]
- 2.Hannes FD, Sharp AJ, Mefford HC, de Ravel T, Ruivenkamp CA, Breuning MH, et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2009;46:223–32. doi: 10.1136/jmg.2007.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vassos E, Collier DA, Holden S, Patch C, Rujescu D, St Clair D, Lewis CM. Penetrance for copy number variants associated with schizophrenia. Hum Mol Genet. 2010;19:3477–81. doi: 10.1093/hmg/ddq259. [DOI] [PubMed] [Google Scholar]
- 5.Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–5. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–9. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–17. doi: 10.1101/gr.GR-1871R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–7. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 9.Cheung J, Estivill X, Khaja R, MacDonald JR, Lau K, Tsui LC, Scherer SW. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 2003;4:R25. doi: 10.1186/gb-2003-4-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.She X, Jiang Z, Clark RA, Liu G, Cheng Z, Tuzun E, et al. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature. 2004;431:927–30. doi: 10.1038/nature03062. [DOI] [PubMed] [Google Scholar]
- 11.Hall J, Levin J, Kuhn J, Ottenheimer E. Berkum Kv, McKusick V. Thrombocytopenia with absent radius (TAR) Medicine. 1969;48:411–39. doi: 10.1097/00005792-196948060-00001. [DOI] [PubMed] [Google Scholar]
- 12.Klopocki E, Schulze H, Strauss G, et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007;80:232–40. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–22. doi: 10.1016/S0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 14.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/S0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 15.Lupski JR, Stankiewicz P. Genomic disorders: the genomic basis of disease. Totowa: Humana Press; 2006. [DOI] [PubMed] [Google Scholar]
- 16.Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012;44:435–9. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soranzo N, Spector TD, Mangino M, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182–90. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonsi L, Marchionni C, Alviano F, Lanzoni G, Franchina M, et al. Thrombocytopenia with absent radii (TAR) syndrome: from hemopoietic progenitor to mesenchymal stromal cell disease? Exp Hematol. 2009;37:1–7. doi: 10.1016/j.exphem.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Giordano P, Cecinati V, Grassi M, Giordani L, De Mattia D, Santoro N. Langerhans cell histiocytosis in a pediatric patient with thrombocytopenia-absent radius syndrome and 1q21.1 deletion: case report and proposal of a rapid molecular diagnosis of 1q21.1 deletion. Immunopharmacol Immunotoxicol. 2011;33:754–8. doi: 10.3109/08923973.2011.557077. [DOI] [PubMed] [Google Scholar]
- 20.Houeijeh A, Andrieux J, Saugier-Veber P, David A, Goldenberg A, Bonneau D, et al. Thrombocytopenia-absent radius (TAR) syndrome: a clinical genetic series of 14 further cases. Impact of the associated 1q21.1 deletion on the genetic counselling. Eur J Med Genet. 2011;54:e471–7. doi: 10.1016/j.ejmg.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Toriello HV. Thrombocytopenia-absent radius syndrome. Semin Thromb Hemost. 2011;37:707–12. doi: 10.1055/s-0031-1291381. [DOI] [PubMed] [Google Scholar]
- 22.Rosenfeld JA, Traylor RN, Schaefer GB, McPherson EW, Ballif BC, Klopocki E, et al. 1q21.1 Study Group. Proximal microdeletions and microduplications of 1q21.1 contribute to variable abnormal phenotypes. Eur J Hum Genet. 2012;20:754–61. doi: 10.1038/ejhg.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012;44:435–9. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albers CA, Newbury-Ecob R, Ouwehand WH, Ghevaert C. New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr Opin Genet Dev. 2013;23:316–23. doi: 10.1016/j.gde.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 25.Baken L, Groenenberg IA, Hoogeboom AJ, Koning AH, Exalto N. First-trimester diagnosis of thrombocytopenia-absent radius syndrome using virtual reality. Clin Dysmorphol. 2014;23:71–3. doi: 10.1097/MCD.0000000000000029. [DOI] [PubMed] [Google Scholar]
- 26.Yassaee VR, Hashemi-Gorji F, Soltani Z, Poorhosseini SM. A new approach for molecular diagnosis of TAR syndrome. Clin Biochem. 2014;47:835–9. doi: 10.1016/j.clinbiochem.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 27.Kumar C, Sharma D, Pandita A, Bhalerao S. Thrombocytopenia absent radius syndrome with Tetralogy of Fallot: a rare association. Int Med Case Rep J. 2015;8:81–5. doi: 10.2147/IMCRJ.S81770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guastadisegni MC, Roberto R, L'Abbate A, Palumbo O, Carella M, Giordani L, et al. Thrombocytopenia-absent-radius syndrome in a child showing a larger 1q21.1 deletion than the one in his healthy mother, and a significant downregulation of the commonly deleted genes. Eur J Med Genet. 2012;55:120–3. doi: 10.1016/j.ejmg.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 29.Bottillo I, Castori M, De Bernardo C, Fabbri R, Grammatico B, Preziosi N, et al. Prenatal diagnosis and post-mortem examination in a fetus with thrombocytopenia-absent radius (TAR) syndrome due to compound heterozygosity for a 1q21.1 microdeletion and a RBM8A hypomorphic allele: a case report. BMC Res Notes. 2013;22:376. doi: 10.1186/1756-0500-6-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papoulidis I, Oikonomidou E, Orru S, Siomou E, Kontodiou M, Eleftheriades M, et al. Prenatal detection of TAR syndrome in a fetus with compound inheritance of an RBM8A SNP and a 334 Kb deletion: a case report. Mol Med Rep. 2014;9:163–5. doi: 10.3892/mmr.2013.1788. [DOI] [PubMed] [Google Scholar]
- 31.Chuang TW, Lee KM, Tarn WY. Function and Pathological Implications of Exon Junction Complex Factor Y14. Biomol. 2015;5:343–55. doi: 10.3390/biom5020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haremaki T, Sridharan J, Dvora S, Weinstein DC. Regulation of vertebrate embryogenesis by the exon junction complex core component Eif4a3. Dev Dyn. 2010;239:1977–87. doi: 10.1002/dvdy.22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perkins AS, Mercer JA, Jenkins NA, Copeland NG. Patterns of Evi-1 expression in embryonic and adult tissues suggest that Evi-1 plays an important regulatory role in mouse development. Development. 1991;111:479–87. doi: 10.1242/dev.111.2.479. [DOI] [PubMed] [Google Scholar]
- 34.Rodel B, Tavassoli K, Karsunky H, Schmidt T, Bachmann M, Schaper F, et al. The zinc finger protein Gfi-1 can enhance STAT3 signaling by interacting with the STAT3 inhibitor PIAS3. EMBO J. 2000;19:5845–55. doi: 10.1093/emboj/19.21.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsiao HH, Liu YC, Yang MY, et al. Decreased expression of PIAS1 and PIAS3 in essential thrombocythemia patients. Genet Mol Res. 2013;12:5617–22. doi: 10.4238/2013.November.18.10. [DOI] [PubMed] [Google Scholar]
- 36.Swindell EC, Moeller C, Thaller C, Eichele G. Cloning and expression analysis of chicken Lix1, a founding member of a novel gene family. Mech Dev. 2001;109:405–8. doi: 10.1016/S0925-4773(01)00535-4. [DOI] [PubMed] [Google Scholar]